
Whole-Genome Resequencing Revealed Selective Signatures for Growth Traits in Hu and Gangba Sheep
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Sampling
2.3. Resequencing Data Processing, and SNP Calling
- Average sequencing depth > 3;
- Maximum deletion rate ≤ 0.9;
- Hardy–Weinberg equilibrium (HWE) < 10−5;
- Minor allele frequencies (MAFs) < 0.05.
2.4. Selective Scanning Detection
2.5. Functional Enrichment Analysis
3. Results
3.1. Phenotype Value Data Statistics
3.2. Sequencing Data Statistics
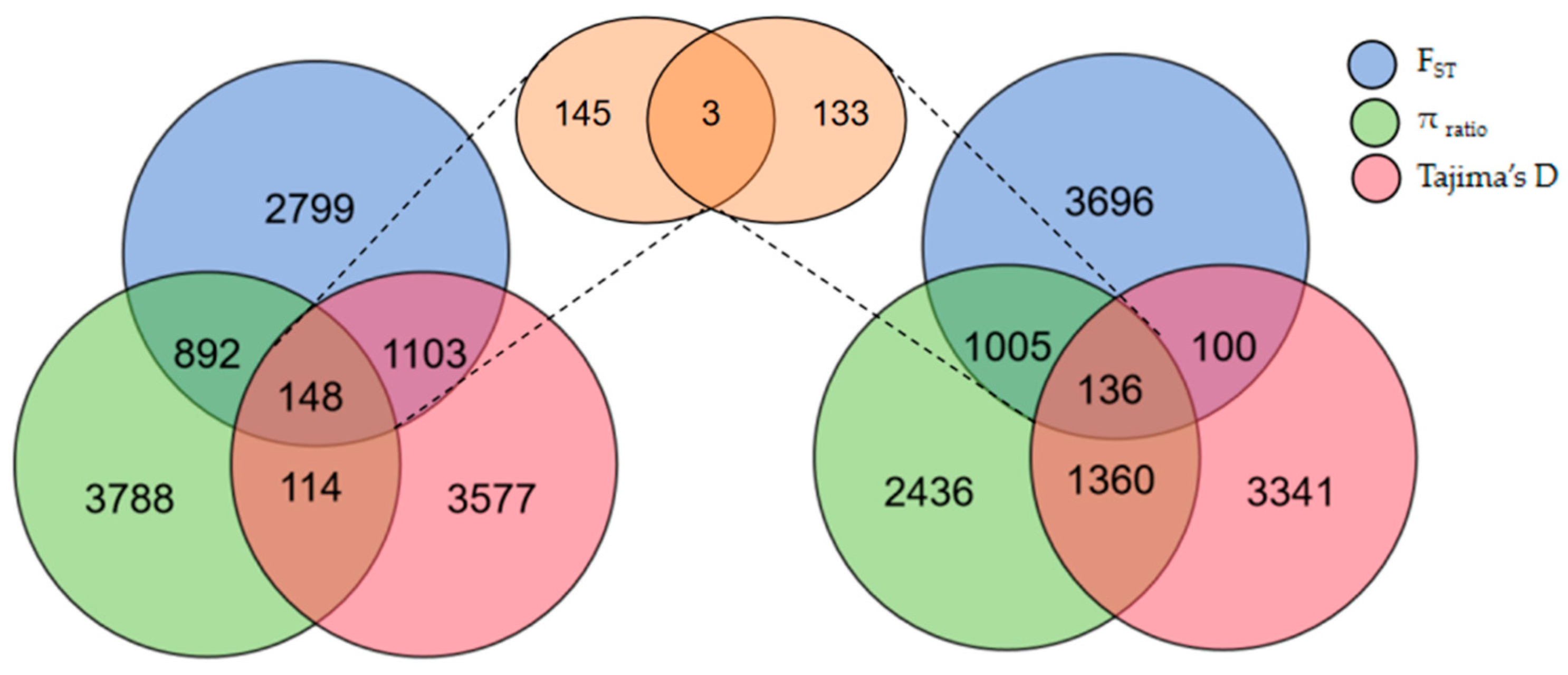
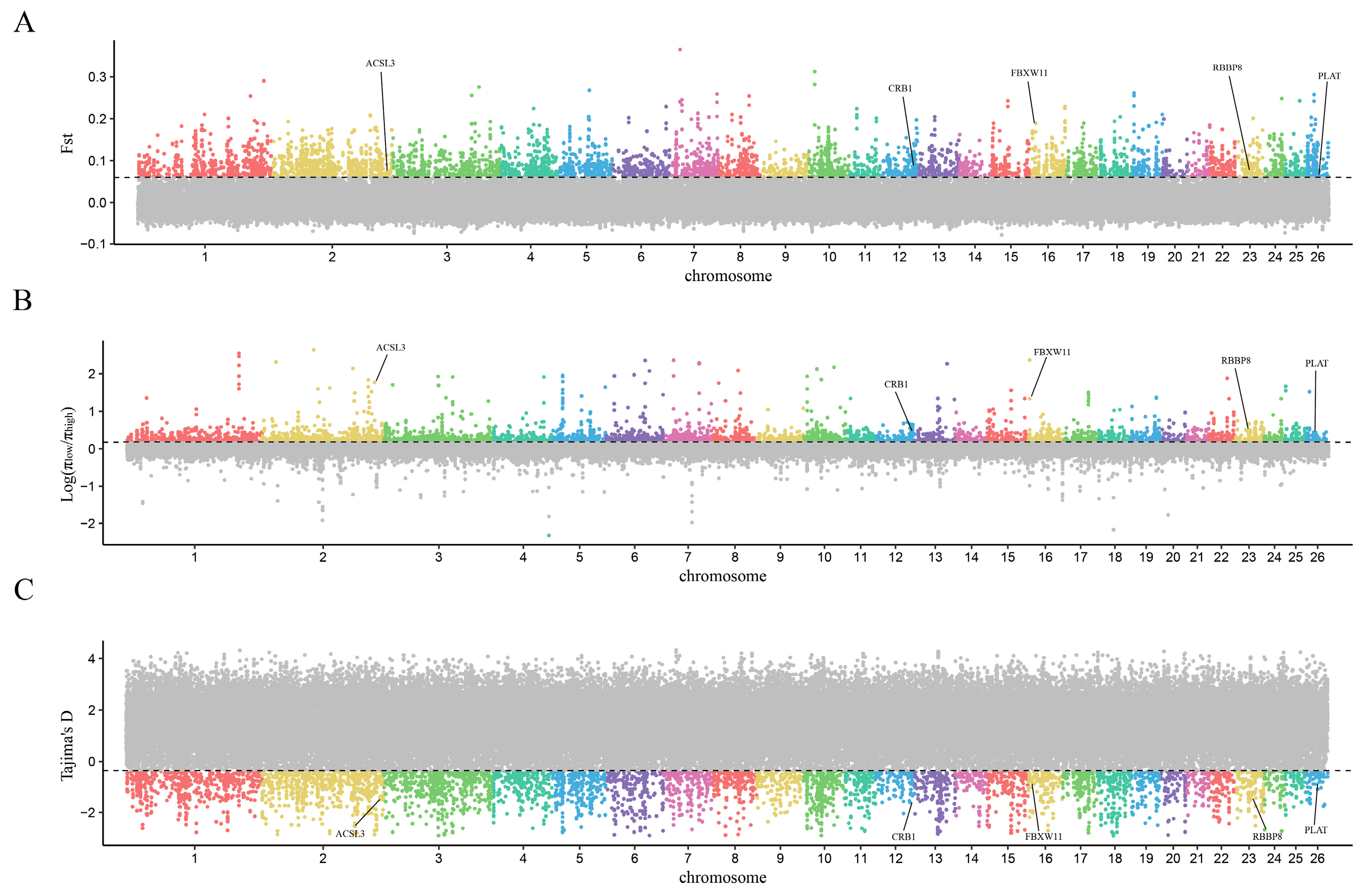
3.3. Select Signal Analysis
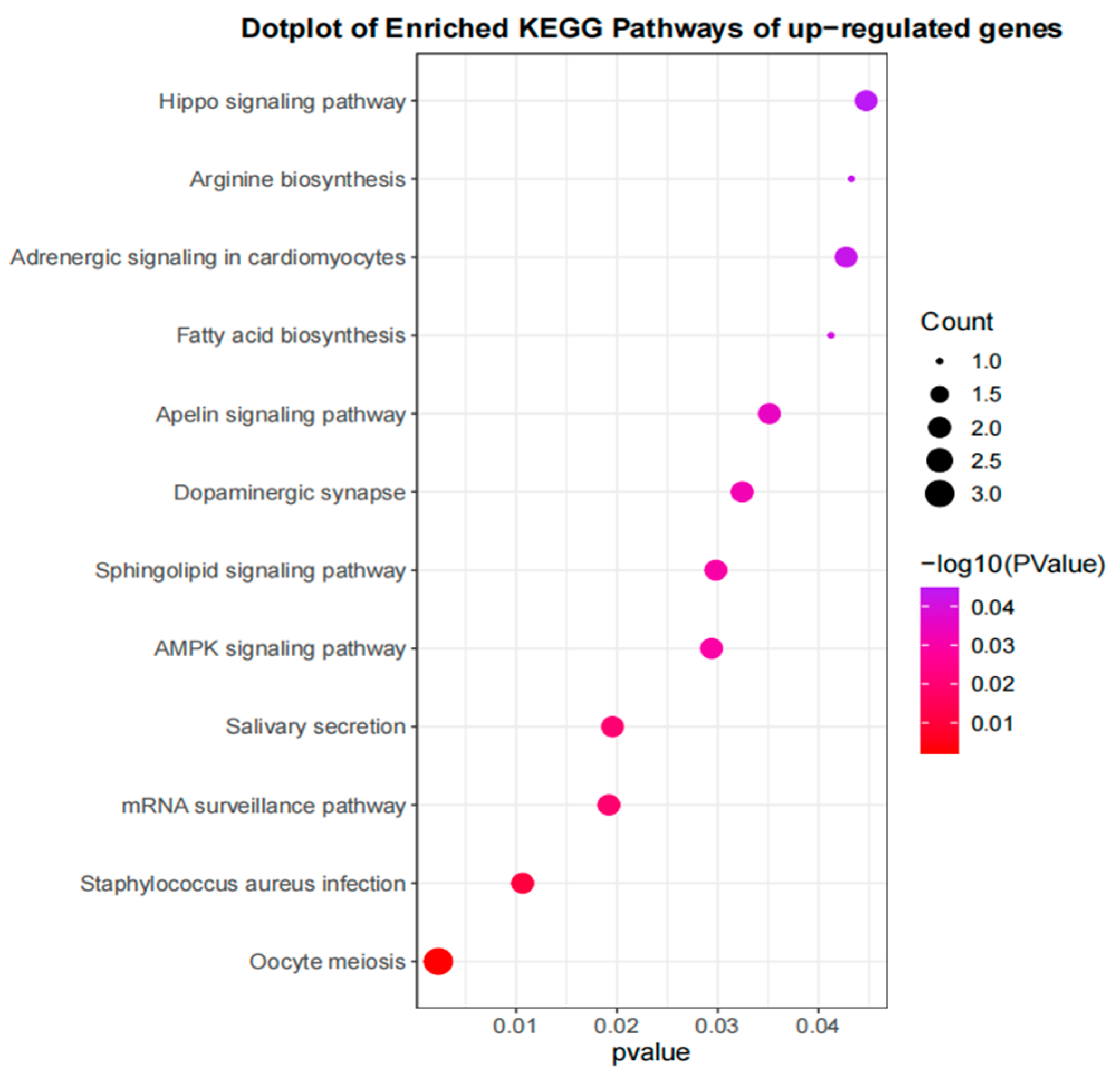
3.4. Gene Enrichment and Analysis
4. Discussion
4.1. Phenotypic Value Data and the Sequencing Data
4.2. Genes Associated with the Growth
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diamond, J. Evolution, consequences and future of plant and animal domestication. Nature 2002, 418, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Zeder, M.A. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc. Natl. Acad. Sci. USA 2008, 105, 11597–11604. [Google Scholar] [CrossRef] [PubMed]
- Chessa, B.; Pereira, F.; Arnaud, F.; Amorim, A.; Goyache, F.; Mainland, I.; Kao, R.R.; Pemberton, J.M.; Beraldi, D.; Stear, M.J.; et al. Revealing the history of sheep domestication using retrovirus integrations. Science 2009, 324, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Kim, T.-H.; Lee, K.-T.; Kwak, W.; Lee, T.; Lee, S.-W.; Kim, M.-J.; Cho, K.; Kim, N.; Chung, W.-H.; et al. A genome-wide scan for signatures of directional selection in domesticated pigs. BMC Genom. 2015, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Sun, L.; Pan, H.; Zhang, S.; Xu, J.; He, M.; Zhang, K.; Zhou, J.; Zhang, D.; Wu, C.; et al. Genomic Dissection through Whole-Genome Resequencing of Five Local Pig Breeds from Shanghai, China. Animals 2023, 13, 3727. [Google Scholar] [CrossRef] [PubMed]
- Rafiepour, M.; Ebrahimie, E.; Vahidi, M.F.; Salekdeh, G.H.; Niazi, A.; Dadpasand, M.; Liang, D.; Si, J.; Ding, X.; Han, J.; et al. Whole-Genome Resequencing Reveals Adaptation Prior to the Divergence of Buffalo Subspecies. Genome Biol. Evol. 2021, 13, evaa231. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.S.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. McCombie, Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Rangel, P.X.M.; Cross, E.; Liu, C.; Pedigo, C.E.; Tian, X.; Gutiérrez-Calabrés, E.; Nagata, S.; Priyadarshini, A.; Lerner, G.; Bunda, P.; et al. Cell Cycle and Senescence Regulation by Podocyte Histone Deacetylase 1 and 2. J. Am. Soc. Nephrol. 2023, 34, 433–450. [Google Scholar] [CrossRef]
- Zhou, X.; He, Y.; Quan, H.; Pan, X.; Zhou, Y.; Zhang, Z.; Yuan, X.; Li, J. HDAC1-Mediated lncRNA Stimulatory Factor of Follicular Development to Inhibit the Apoptosis of Granulosa Cells and Regulate Sexual Maturity through miR-202-3p-COX1 Axis. Cells 2023, 12, 2734. [Google Scholar] [CrossRef]
- Xu, Y.; Fan, S.; Liu, Y.; Shi, J.; Xie, X.; Wang, X.; Wang, C.; Liu, X.; Xia, G. HDAC1 in the Ovarian Granulosa Cells of Tan Sheep Improves Cumulus Cell Expansion and Oocyte Maturation Independently of the EGF-like Growth Factors. Biology 2022, 11, 1464. [Google Scholar] [CrossRef] [PubMed]
- Boon, H.; Sjögren, R.J.O.; Massart, J.; Egan, B.; Kostovski, E.; Iversen, P.O.; Hjeltnes, N.; Chibalin, A.V.; Widegren, U.; Zierath, J.R. MicroRNA-208b progressively declines after spinal cord injury in humans and is inversely related to myostatin expression. Physiol. Rep. 2015, 3, e12622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, Z.; Hu, G.; Zhang, Y.; Ao, Z. Exploring characteristics of placental transcriptome and cord serum metabolome associated with low birth weight in Kele pigs. Trop. Anim. Health Prod. 2023, 55, 340. [Google Scholar] [CrossRef]
- Rigueur, D.; Brugger, S.; Anbarchian, T.; Kim, J.K.; Lee, Y.; Lyons, K.M. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res. 2015, 30, 733–741. [Google Scholar] [CrossRef]
- Zhao, X.; Mo, D.; Li, A.; Gong, W.; Xiao, S.; Zhang, Y.; Qin, L.; Niu, Y.; Guo, Y.; Liu, X.; et al. Comparative analyses by sequencing of transcriptomes during skeletal muscle development between pig breeds differing in muscle growth rate and fatness. PLoS ONE 2011, 6, e19774. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gu, Y.; Cao, X.; Ge, L.; He, M.; Zhang, W.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Quan, K.; et al. The identification and validation of target genes of IGFBP3 protein in sheep skeletal muscle cells. Anim. Biotechnol. 2023, 34, 4580–4587. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Charbeneau, R.A.; Fu, Y.; Kaur, K.; Gerin, I.; MacDougald, O.A.; Neubig, R.R. Resistance to diet-induced obesity and improved insulin sensitivity in mice with a regulator of G protein signaling-insensitive G184S Gnai2 allele. Diabetes 2008, 57, 77–85. [Google Scholar] [CrossRef]
- Li, F.; Han, H.; Lei, Q.; Gao, J.; Liu, J.; Liu, W.; Zhou, Y.; Li, H.; Cao, D. Genome-wide association study of body weight in Wenshang Barred chicken based on the SLAF-seq technology. J. Appl. Genet. 2018, 59, 305–312. [Google Scholar] [CrossRef]
- Lai, Z.; Li, S.; Wu, F.; Zhou, Z.; Gao, Y.; Yu, J.; Lei, C.; Dang, R. Genotypes and haplotype combination of ACSL3 gene sequence variants is associated with growth traits in Dezhou donkey. Gene 2020, 743, 144600. [Google Scholar] [CrossRef]
- Yuan, Y.; Ida, J.M.; Paczkowski, M.; Krisher, R.L. Identification of developmental competence-related genes in mature porcine oocytes. Mol. Reprod. Dev. 2011, 78, 565–575. [Google Scholar] [CrossRef]
- Dalle Carbonare, L.; Gomez Lira, M.; Minoia, A.; Bertacco, J.; Orsi, S.; Lauriola, A.; Li Vigni, V.; Gandini, A.; Antoniazzi, F.; Zipeto, D.; et al. Expression of FBXW11 in normal and disease-associated osteogenic cells. J. Cell. Mol. Med. 2023, 27, 1580–1591. [Google Scholar] [CrossRef]
- Schierding, W.; Antony, J.; Cutfield, W.S.; Horsfield, J.A.; O’sullivan, J.M. Intergenic GWAS SNPs are key components of the spatial and regulatory network for human growth. Hum. Mol. Genet. 2016, 25, 3372–3382. [Google Scholar] [CrossRef] [PubMed]
- Salek Ardestani, S.; Aminafshar, M.; Zandi Baghche Maryam, M.B.; Banabazi, M.H.; Sargolzaei, M.; Miar, Y. Signatures of selection analysis using whole-genome sequence data reveals novel candidate genes for pony and light horse types. Genome 2020, 63, 387–396. [Google Scholar] [CrossRef]
- Yu, B.Y.; Subudeng, G.; Du, C.G.; Liu, Z.H.; Zhao, Y.F.; Namei, E.; Bai, Y.; Yang, B.X.; Li, H.J. Plasminogen activator, tissue type regulates germinal vesicle breakdown and cumulus expansion of bovine cumulus-oocyte complex in vitrodagger. Biol. Reprod. 2019, 100, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lou, G.; Li, C.; Wang, X.; Cederbaum, A.I.; Gan, L.; Xie, B. HBx inhibits CYP2E1 gene expression via downregulating HNF4alpha in human hepatoma cells. PLoS ONE 2014, 9, e107913. [Google Scholar]
- Son, M.H.; Kang, K.W.; Kim, E.J.; Ryu, J.H.; Cho, H.; Kim, S.H.; Kim, W.B.; Kim, S.G. Role of glucose utilization in the restoration of hypophysectomy-induced hepatic cytochrome P450 2E1 by growth hormone in rats. Chem. Biol. Interact. 2000, 127, 13–28. [Google Scholar] [CrossRef]
- Wang, T.; Chen, M.; Yan, Y.E.; Xiao, F.Q.; Pan, X.L.; Wang, H. Growth retardation of fetal rats exposed to nicotine in utero: Possible involvement of CYP1A1, CYP2E1, and P-glycoprotein. Environ. Toxicol. 2009, 24, 33–42. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, P.; Shang, M.; Bao, J.; Liu, T.; Xiong, J.; Huang, J.; Sun, J.; Zhang, L. Whole-Genome Resequencing Revealed Selective Signatures for Growth Traits in Hu and Gangba Sheep. Genes 2024, 15, 551. https://doi.org/10.3390/genes15050551
Yang P, Shang M, Bao J, Liu T, Xiong J, Huang J, Sun J, Zhang L. Whole-Genome Resequencing Revealed Selective Signatures for Growth Traits in Hu and Gangba Sheep. Genes. 2024; 15(5):551. https://doi.org/10.3390/genes15050551
Chicago/Turabian StyleYang, Peifu, Mingyu Shang, Jingjing Bao, Tianyi Liu, Jinke Xiong, Jupeng Huang, Jinghua Sun, and Li Zhang. 2024. "Whole-Genome Resequencing Revealed Selective Signatures for Growth Traits in Hu and Gangba Sheep" Genes 15, no. 5: 551. https://doi.org/10.3390/genes15050551
APA StyleYang, P., Shang, M., Bao, J., Liu, T., Xiong, J., Huang, J., Sun, J., & Zhang, L. (2024). Whole-Genome Resequencing Revealed Selective Signatures for Growth Traits in Hu and Gangba Sheep. Genes, 15(5), 551. https://doi.org/10.3390/genes15050551