One NF1 Mutation may Conceal Another

 , , , , add
Show full author list
, , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Samples
2.2. DNA and RNA Extractions
2.3. Microsatellite Typing
2.4. NF1 and SPRED1 Next-Generation Sequencing (NGS)
2.5. DNA and RNA Sequencing with PCR
2.6. Variant Classification
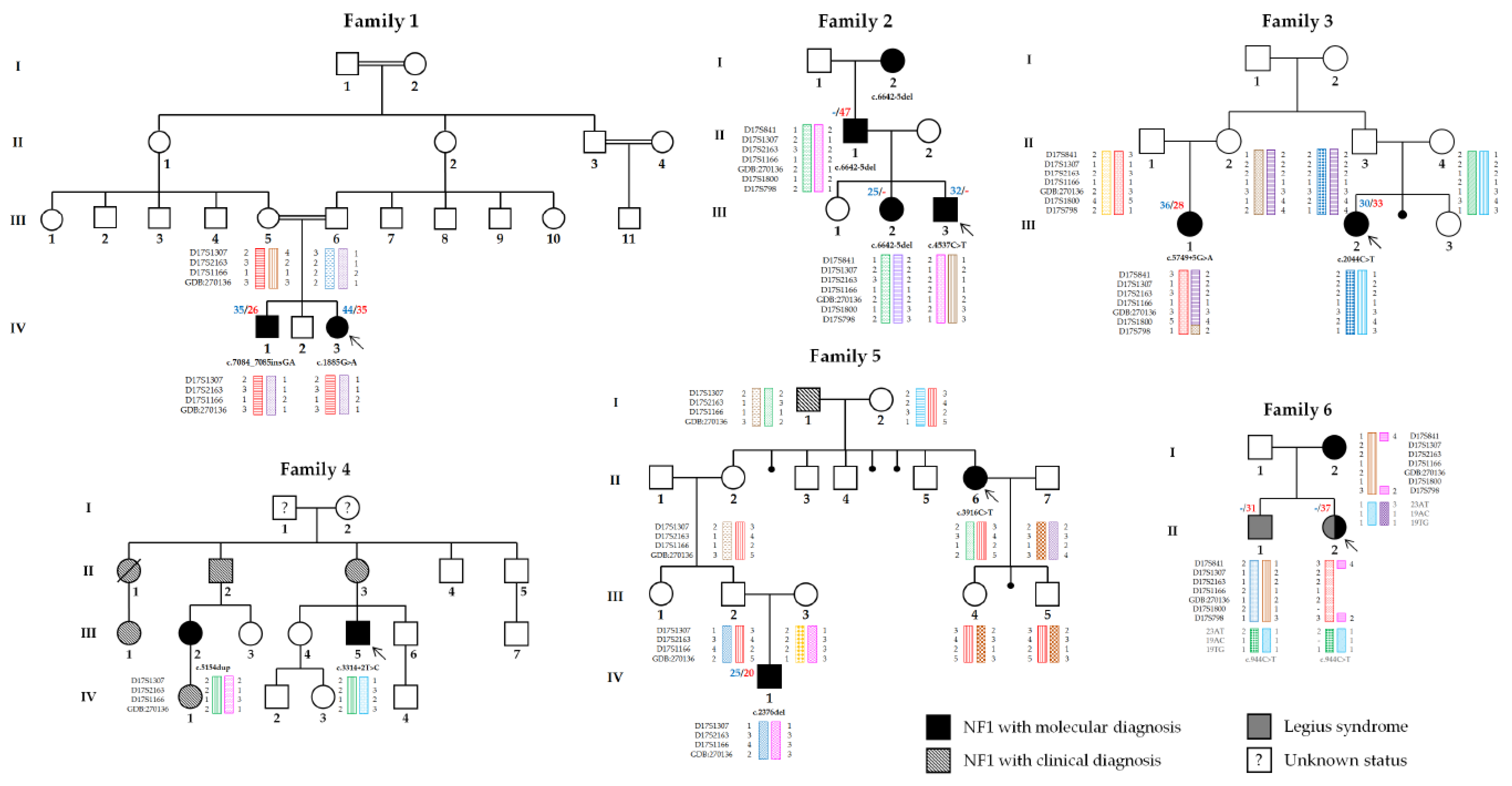
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, V.C.; Lucas, J.; Babcock, M.A.; Gutmann, D.H.; Korf, B.; Maria, B.L. Neurofibromatosis type 1 revisited. Pediatrics 2009, 123, 124–133. [Google Scholar] [CrossRef]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef]
- Tucker, T.; Wolkenstein, P.; Revuz, J.; Zeller, J.; Friedman, J.M. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology 2005, 65, 205–211. [Google Scholar] [CrossRef]
- Stumpf, D.A. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Friedman, J.M. Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 1–6. [Google Scholar] [CrossRef]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef]
- Von Deimling, A.; Krone, W.; Menon, A.G. Neurofibromatosis type 1: Pathology, clinical features and molecular genetics. Brain Pathol. 1995, 5, 153–162. [Google Scholar] [CrossRef]
- Xu, G.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R.; et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- Clementi, M.; Barbujani, G.; Turolla, L.; Tenconi, R. Neurofibromatosis-1: A maximum likelihood estimation of mutation rate. Hum. Genet. 1990, 84, 116–118. [Google Scholar] [CrossRef]
- Kluwe, L.; Siebert, R.; Gesk, S.; Friedrich, R.E.; Tinschert, S.; Kehrer-Sawatzki, H.; Mautner, V.F. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum. Mutat. 2004, 23, 111–116. [Google Scholar] [CrossRef]
- Mautner, V.F.; Kluwe, L.; Friedrich, R.E.; Roehl, A.C.; Bammert, S.; Högel, J.; Spöri, H.; Cooper, D.N.; Kehrer-Sawatzki, H. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J. Med. Genet. 2010, 47, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Hanna, N.; Masliah-Planchon, J.; Jolly, E.; Goussard, P.; Ballerini, P.; Cartault, F.; Barbarot, S.; Landman-Parker, J.; et al. SPRED1 germline mutations caused a neurofibromatosis type 1 overlapping phenotype. J. Med. Genet. 2009, 46, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Brems, H.; Pasmant, E.; Van Minkelen, R.; Wimmer, K.; Upadhyaya, M.; Legius, E.; Messiaen, L. Review and update of SPRED1 mutations causing Legius syndrome. Hum. Mutat. 2012, 33, 1538–1546. [Google Scholar] [CrossRef]
- Wakioka, T.; Sasaki, A.; Kato, R.; Shouda, T.; Matsumoto, A.; Miyoshi, K.; Tsuneoka, M.; Komiya, S.; Baron, R.; Yoshimura, A. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001, 412, 647–651. [Google Scholar] [CrossRef]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ars, E.; Kruyer, H.; Morell, M.; Pros, E.; Serra, E.; Ravella, A.; Estivill, X.; Lázaro, C. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J. Med. Genet. 2003, 40, e82. [Google Scholar] [CrossRef]
- Side, L.; Taylor, B.; Cayouette, M.; Conner, E.; Thompson, P.; Luce, M.; Shannon, K. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N. Engl. J. Med. 1997, 336, 1713–1720. [Google Scholar] [CrossRef]
- Pros, E.; Gómez, C.; Martín, T.; Fábregas, P.; Serra, E.; Lázaro, C. Nature and mRNA effect of 282 different NF1 point mutations: Focus on splicing alterations. Hum. Mutat. 2008, 29, E173–E193. [Google Scholar] [CrossRef]
- Park, V.M.; Pivnick, E.K. Neurofibromatosis type 1 (NF1): A protein truncation assay yielding identification of mutations in 73% of patients. J. Med. Genet. 1998, 35, 813–820. [Google Scholar] [CrossRef]
- Klose, A.; Peters, H.; Hoffmeyer, S.; Buske, A.; Lüder, A.; Hess, D.; Lehmann, R.; Nürnberg, P.; Tinschert, S. Two independent mutations in a family with neurofibromatosis type 1 (NF1). Am. J. Med. Genet. 1999, 83, 6–12. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Majounie, E.; Thompson, P.; Han, S.; Consoli, C.; Krawczak, M.; Cordeiro, I.; Cooper, D.N. Three different pathological lesions in the NF1 gene originating de novo in a family with neurofibromatosis type 1. Hum. Genet. 2003, 112, 12–17. [Google Scholar] [CrossRef]
- Messiaen, L.; Yao, S.; Brems, H.; Callens, T.; Sathienkijkanchai, A.; Denayer, E.; Spencer, E.; Arn, P.; Babovic-Vuksanovic, D.; Bay, C.; et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA 2009, 302, 2111–2118. [Google Scholar] [CrossRef]
- Baralle, D.; Mattocks, C.; Kalidas, K.; Elmslie, F.; Whittaker, J.; Lees, M.; Ragge, N.; Patton, M.A.; Winter, R.M.; ffrench-Constant, C. Different mutations in the NF1 gene are associated with Neurofibromatosis-Noonan syndrome (NFNS). Am. J. Med. Genet. Part A 2003, 119, 1–8. [Google Scholar] [CrossRef]
- De Luca, A.; Bottillo, I.; Sarkozy, A.; Carta, C.; Neri, C.; Bellacchio, E.; Schirinzi, A.; Conti, E.; Zampino, G.; Battaglia, A.; et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am. J. Hum. Genet. 2005, 77, 1092–1101. [Google Scholar] [CrossRef]
- Stevenson, D.A.; Viskochil, D.H.; Rope, A.F.; Carey, J.C. Clinical and molecular aspects of an informative family with neurofibromatosis type 1 and Noonan phenotype. Clin. Genet. 2006, 69, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Hüffmeier, U.; Zenker, M.; Hoyer, J.; Fahsold, R.; Rauch, A. A variable combination of features of Noonan syndrome and neurofibromatosis type I are caused by mutations in the NF1 gene. Am. J. Med. Genet. Part A 2006, 140, 2749–2756. [Google Scholar] [CrossRef]
- Nyström, A.M.; Ekvall, S.; Allanson, J.; Edeby, C.; Elinder, M.; Holmström, G.; Bondeson, M.L.; Annerén, G. Noonan syndrome and neurofibromatosis type I in a family with a novel mutation in NF1. Clin. Genet. 2009, 76, 524–534. [Google Scholar] [CrossRef]
- Ekvall, S.; Sjörs, K.; Jonzon, A.; Vihinen, M.; Annerén, G.; Bondeson, M.L. Novel association of neurofibromatosis type 1-causing mutations in families with neurofibromatosis-Noonan syndrome. Am. J. Med. Genet. Part A 2014, 164, 579–587. [Google Scholar] [CrossRef]
- Bahuau, M.; Houdayer, C.; Assouline, B.; Blanchet-Bardon, C.; Le Merrer, M.; Lyonnet, S.; Giraud, S.; Récan, D.; Lakhdar, H.; Vidaud, M.; et al. Novel recurrent nonsense mutation causing neurofibromatosis type 1 (NF1) in a family segregating both NF1 and Noonan syndrome. Am. J. Med. Genet. 1998, 75, 265–272. [Google Scholar] [CrossRef]
- Pasmant, E.; Amiel, J.; Rodriguez, D.; Vidaud, M.; Vidaud, D.; Parfait, B. Two independent de novo mutations as a cause for neurofibromatosis type 1 and Noonan syndrome in a single family. Am. J. Med. Genet. Part A 2012, 158, 2290–2291. [Google Scholar] [CrossRef]
- Bertola, D.R.; Pereira, A.C.; Passetti, F.; de Oliveira, P.S.L.; Messiaen, L.; Gelb, B.D.; Kim, C.A.; Krieger, J.E. Neurofibromatosis-Noonan syndrome: Molecular evidence of the concurrence of both disorders in a patient. Am. J. Med. Genet. Part A 2005, 136, 242–245. [Google Scholar] [CrossRef]
- Thiel, C.; Wilken, M.; Zenker, M.; Sticht, H.; Fahsold, R.; Gusek-Schneider, G.-C.; Rauch, A. Independent NF1 and PTPN11 mutations in a family with neurofibromatosis-Noonan syndrome. Am. J. Med. Genet. Part A 2009, 149, 1263–1267. [Google Scholar] [CrossRef]
- Prada, C.E.; Zarate, Y.A.; Hagenbuch, S.; Lovell, A.; Schorry, E.K.; Hopkin, R.J. Lethal presentation of neurofibromatosis and Noonan syndrome. Am. J. Med. Genet. Part A 2011, 155, 1360–1366. [Google Scholar] [CrossRef]
- Goriely, A. Decoding germline de novo point mutations. Nat. Genet. 2016, 48, 823–824. [Google Scholar] [CrossRef]
- Lázaro, C.; Gaona, A.; Ainsworth, P.; Tenconi, R.; Vidaud, D.; Kruyer, H.; Ars, E.; Volpini, V.; Estivill, X. Sex differences in mutational rate and mutational mechanism in the NF1 gene in neurofibromatosis type 1 patients. Hum. Genet. 1996, 98, 696–699. [Google Scholar] [CrossRef]
- Goriely, A.; Wilkie, A.O.M. Paternal age effect mutations and selfish spermatogonial selection: Causes and consequences for human disease. Am. J. Hum. Genet. 2012, 90, 175–200. [Google Scholar] [CrossRef]
- Maher, G.J.; Ralph, H.K.; Ding, Z.; Koelling, N.; Mlcochova, H.; Giannoulatou, E.; Dhami, P.; Paul, D.S.; Stricker, S.H.; Beck, S.; et al. Selfish mutations dysregulating RAS-MAPK signaling are pervasive in aged human testes. Genome Res. 2018, 28, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Snajderova, M.; Riccardi, V.M.; Petrak, B.; Zemkova, D.; Zapletalova, J.; Mardesic, T.; Petrakova, A.; Lanska, V.; Marikova, T.; Bendova, S.; et al. The importance of advanced parental age in the origin of neurofibromatosis type 1. Am. J. Med. Genet. Part A 2012, 158, 519–523. [Google Scholar] [CrossRef]
- Dubov, T.; Toledano-Alhadef, H.; Bokstein, F.; Constantini, S.; Ben-Shachar, S. The effect of parental age on the presence of de novo mutations—Lessons from neurofibromatosis type I. Mol. Genet. Genomic Med. 2016, 4, 480–486. [Google Scholar] [CrossRef]


{kind=link}
| Family | Patient | Sex | Age | CALS * | Freckling | Subcutaneous/Cutaneous NFs | Plexiform NFs | Lisch Nodules | OPG | Skeletal Abnormalities | Other | NF1 Variant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IV-1 | M | 21 | >6 | No | Yes | No | Yes, bilateral | No | Mild scoliosis, cubitus valgus | Anemic nevus, learning disabilities, headaches, myopia | c.7084_7085insGA |
| IV-3 # | F | 18 | >6 | Yes, Ax & In bilateral | Yes | No | Yes, bilateral | No | Mild scoliosis | Facial nevus, left hemiparesis, hyperopia, mild intellectual disability | c.1885G>A | |
| 2 | II-1 | M | 41 | >6 | No | No | No | No | ND | None | c.6642-5del | |
| III-2 | F | 14 | >6 | No | No | No | No | ND | None | c.6642-5del | ||
| III-3 # | M | 2 | >6 | Yes, Ax bilateral | Yes | Yes | Yes, bilateral | Yes | Scoliosis | Spinal NFs, xanthogranulomas, headaches, attention deficit disorder | c.4537C>T | |
| 3 | III-1 | F | 20 | >6 | Yes, SM bilateral | Yes | Yes | No | No | Scoliosis | c.5749+5G>A | |
| III-2 # | F | 15 | >6 | Yes, Ax bilateral | No | No | Yes | Yes | None | c.2044C>T | ||
| 4 | III-2 | F | 26 | >6 | Yes, Ax bilateral | Yes | No | Yes, left | No | None | Short stature, low-set ears | c.5154dup |
| III-5 # | M | 39 | >6 | Yes, Ax bilateral | Yes | No | Yes, bilateral | No | None | Brugada syndrome (paternal origin) | c.3314+2T>C | |
| 5 | II-6 # | F | 23 | >6 | Yes, Ax | Yes | Yes | Yes, bilateral | No | Scoliosis, short stature | Breast cancer, headaches | c.3916C>T |
| IV-1 | M | 18 | >6 | Yes | Yes | Yes | No | Yes | Pseudarthrosis, sphenoid wing dysplasia | Learning disabilities | c.2376del | |
| 6 | I-2 | F | 45 | >6 | No | Yes | No | ND | ND | Scoliosis | Complete deletion | |
| II-1 | M | 14 | >6 | Yes | No | No | No | No | None | |||
| II-2 # | F | 10 | >6 | Yes | Yes | No | ND | ND | None | Hypertelorism, congenital coronarocardiac fistula | Complete deletion |
| Gene | Variant | Exon | Protein | Consequence | Studied Sample | Techniques | ACMG-AMP classification | Evidence of Pathogenicity | Reference |
|---|---|---|---|---|---|---|---|---|---|
| NF1 | c.7084_7085insGA | 48 | p.Asn2362Argfs*14 | Frameshift | DNA + RNA | RT-PCR + Sanger | Pathogenic | PVS1+ PS2+PM2+PP4+PP5 | Sabbagh et al., 2013 [6] |
| c.1885G>A | 17 | p.Gln616Glyfs*4 | Creation of a new 3′ splice site | DNA + RNA | RT-PCR + Sanger | Pathogenic | PVS1+PS2+PP3+PP4+PP5 | Ars et al., 2003 [19] | |
| c.6642-5del | 45 | p.Phe2215Hisfs*6 | Exon 45 skip | DNA + RNA | NGS + RT-PCR + Sanger | Pathogenic | PVS1+PM2+PP4 | ND | |
| c.4537C>T | 35 | p.Arg1513* | Premature stop codon | DNA + RNA | NGS + RT-PCR + Sanger | Pathogenic | PVS1+PP4+PP5 | Side et al., 1997 [20] | |
| c.5749+5G>A | 39 | p.Ser1850fs*2 | Exon 39 skip | DNA + RNA | NGS + RT-PCR + Sanger | Pathogenic | PVS1+PS2+PM2+PP3+PP4+PP5 | Pros et al., 2008 [21] | |
| c.2044C>T | 18 | p.(Gln682*) | Premature stop codon (predicted) | DNA | NGS + Sanger | Pathogenic | PVS1+PS2+PM2+PP4 | ND | |
| c.5154dup | 37 | p.(Phe1719Ilefs*17) | Frameshift (predicted) | DNA | NGS + Sanger | Pathogenic | PVS1+PM2+PP4 | ND | |
| c.3314+2T>C | 25 | p.? | Unknown | DNA | NGS + Sanger | Pathogenic | PVS1+PM2+PP4 | ND | |
| c.3916C>T | 29 | p.Arg1306* | Premature stop codon | DNA + RNA | RT-PCR + Sanger | Pathogenic | PVS1+PS2+PP4+PP5 | Park & Pivnick, 1998 [22] | |
| c.2376del | 20 | p.Asn793Thrfs*28 | Frameshift | DNA + RNA | RT-PCR + Sanger | Pathogenic | PVS1+PS2+PM2+PP4+PP5 | Sabbagh et al., 2013 [6] | |
| SPRED1 | c.944C>T | 7 | p.(Pro315Leu) | Missense (predicted) | DNA | NGS + Sanger | Uncertain significance | PP3 | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacot, L.; Burin des Roziers, C.; Laurendeau, I.; Briand-Suleau, A.; Coustier, A.; Mayard, T.; Tlemsani, C.; Faivre, L.; Thomas, Q.; Rodriguez, D.; et al. One NF1 Mutation may Conceal Another. Genes 2019, 10, 633. https://doi.org/10.3390/genes10090633
Pacot L, Burin des Roziers C, Laurendeau I, Briand-Suleau A, Coustier A, Mayard T, Tlemsani C, Faivre L, Thomas Q, Rodriguez D, et al. One NF1 Mutation may Conceal Another. Genes. 2019; 10(9):633. https://doi.org/10.3390/genes10090633
Chicago/Turabian StylePacot, Laurence, Cyril Burin des Roziers, Ingrid Laurendeau, Audrey Briand-Suleau, Audrey Coustier, Théodora Mayard, Camille Tlemsani, Laurence Faivre, Quentin Thomas, Diana Rodriguez, and et al. 2019. "One NF1 Mutation may Conceal Another" Genes 10, no. 9: 633. https://doi.org/10.3390/genes10090633