Affinity Purification of NF1 Protein–Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners

 ,
,
Abstract
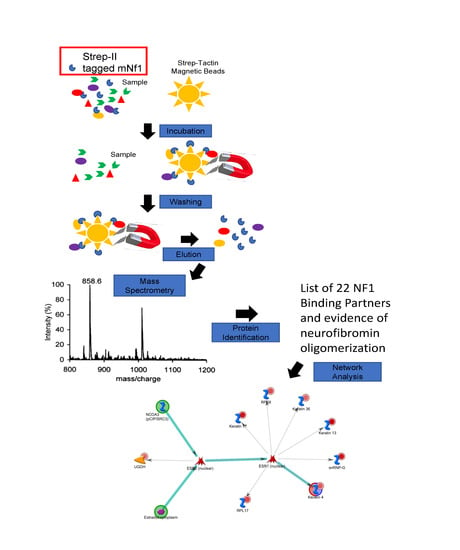
:
1. Introduction
2. Materials and Methods
2.1. Tagged NF1 cDNAs and Empty Vector Controls
2.2. Cell Lines and cDNA Transient Transfection
2.3. Affinity Purification (AP)
2.4. Reciprocal Immunoprecipitation
2.5. Mass Spectrometry
2.6. MS Data Conversion and Searches
2.7. Peptide Filtering, Grouping, and Quantification
2.8. Statistical Analysis
2.9. Systems Analysis
3. Results
3.1. Neurofibromin Oligomerization
3.2. Neurofibromin PPIs
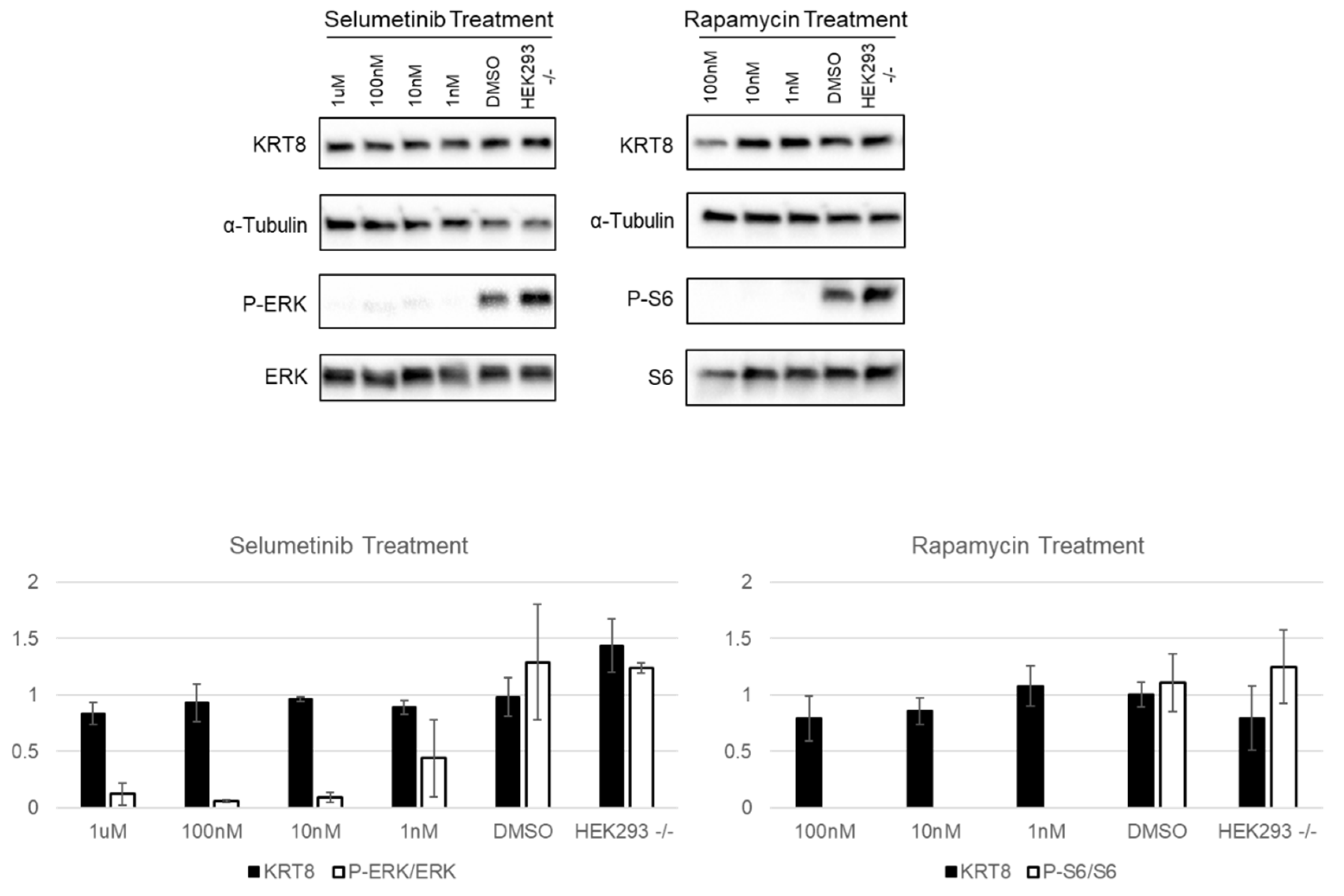
3.3. Neurofibromin and Keratins
3.4. Correlation with the Cancer Genome Atlas (TCGA)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharafi, P.; Ayter, S. Possible modifier genes in the variation of neurofibromatosis type 1 clinical phenotypes. J. Neurogenet. 2018, 32, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Welti, S. Neurofibromin: Protein domains and functional characteristics. In Neurofibromatosis Type 1: Molecular and Cellular Biology; Upadhyaya, M., Cooper, D.N., Eds.; Springer: New York, NY, USA, 2012; pp. 305–325. [Google Scholar]
- Phan, V.T.; Ding, V.W.; Li, F.; Chalkley, R.J.; Burlingame, A.; McCormick, F. The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol. Cell. Biol. 2010, 30, 2264–2279. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meffre, J.; Chaumont-Dubel, S.; Mannoury la Cour, C.; Loiseau, F.; Watson, D.J.; Dekeyne, A.; Seveno, M.; Rivet, J.M.; Gaven, F.; Deleris, P.; et al. 5-HT(6) receptor recruitment of mTOR as a mechanism for perturbed cognition in schizophrenia. EMBO Mol. Med. 2012, 4, 1043–1056. [Google Scholar] [CrossRef]
- Deraredj Nadim, W.; Chaumont-Dubel, S.; Madouri, F.; Cobret, L.; De Tauzia, M.L.; Zajdel, P.; Benedetti, H.; Marin, P.; Morisset-Lopez, S. Physical interaction between neurofibromin and serotonin 5-HT6 receptor promotes receptor constitutive activity. Proc. Natl. Acad. Sci. USA 2016, 113, 12310–12315. [Google Scholar] [CrossRef]
- Hirata, Y.; Brems, H.; Suzuki, M.; Kanamori, M.; Okada, M.; Morita, R.; Llano-Rivas, I.; Ose, T.; Messiaen, L.; Legius, E.; et al. Interaction between a Domain of the Negative Regulator of the Ras-ERK Pathway, SPRED1 Protein, and the GTPase-activating Protein-related Domain of Neurofibromin Is Implicated in Legius Syndrome and Neurofibromatosis Type 1. J. Biol. Chem. 2016, 291, 3124–3134. [Google Scholar] [CrossRef] [Green Version]
- Dunzendorfer-Matt, T.; Mercado, E.L.; Maly, K.; McCormick, F.; Scheffzek, K. The neurofibromin recruitment factor Spred1 binds to the GAP related domain without affecting Ras inactivation. Proc. Natl. Acad. Sci. USA 2016, 113, 7497–7502. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Yan, L.H.; Huang, B.; Liu, M.; Liu, X.; Huang, C. Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum-associated protein degradation. J. Neurochem. 2014, 129, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, P.E.; Cichowski, K. Identifying the Ubiquitin Ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov. 2013, 3, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Cichowski, K.; Santiago, S.; Jardim, M.; Johnson, B.W.; Jacks, T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 2003, 17, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandru, G.; Graumann, J.; Smith, G.T.; Kolawa, N.J.; Fang, R.; Deshaies, R.J. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 2008, 134, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Nonami, A.; Taketomi, T.; Kimura, A.; Saeki, K.; Takaki, H.; Sanada, T.; Taniguchi, K.; Harada, M.; Kato, R.; Yoshimura, A. The Sprouty-related protein, Spred-1, localizes in a lipid raft/caveola and inhibits ERK activation in collaboration with caveolin-1. Genes Cells 2005, 10, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Yunoue, S.; Tokuo, H.; Ozawa, T.; Zhang, D.; Patrakitkomjorn, S.; Ichimura, T.; Saya, H.; Araki, N. PKA phosphorylation and 14-3-3 interaction regulate the function of neurofibromatosis type I tumor suppressor, neurofibromin. FEBS Lett. 2004, 557, 275–282. [Google Scholar] [CrossRef]
- Carnes, R.M.; Mobley, J.A.; Crossman, D.K.; Liu, H.; Korf, B.R.; Kesterson, R.A.; Wallis, D. Multi-Omics Profiling for NF1 Target Discovery in Neurofibromin (NF1) Deficient Cells. Proteomics 2019, 19, e1800334. [Google Scholar] [CrossRef]
- Popov, I.K.; Hiatt, S.M.; Whalen, S.; Keren, B.; Ruivenkamp, C.; van Haeringen, A.; Chen, M.J.; Cooper, G.M.; Korf, B.R.; Chang, C. A YWHAZ Variant Associated with Cardiofaciocutaneous Syndrome Activates the RAF-ERK Pathway. Front. Physiol. 2019, 10, 388. [Google Scholar] [CrossRef]
- Molzan, M.; Schumacher, B.; Ottmann, C.; Baljuls, A.; Polzien, L.; Weyand, M.; Thiel, P.; Rose, R.; Rose, M.; Kuhenne, P.; et al. Impaired binding of 14-3-3 to C-RAF in Noonan syndrome suggests new approaches in diseases with increased Ras signaling. Mol. Cell. Biol. 2010, 30, 4698–4711. [Google Scholar] [CrossRef]
- Wallis, D.; Li, K.; Lui, H.; Hu, K.; Chen, M.J.; Li, J.; Kang, J.; Das, S.; Korf, B.R.; Kesterson, R.A. Neurofibromin (NF1) genetic variant structure-function analyses using a full-length mouse cDNA. Hum. Mutat. 2018, 39, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Woo, A.S.; Messiaen, L.M.; Gutmann, D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015, 24, 3518–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Weatherly, D.B.; Atwood, J.A., 3rd; Minning, T.A.; Cavola, C.; Tarleton, R.L.; Orlando, R. A Heuristic method for assigning a false-discovery rate for protein identifications from Mascot database search results. Mol. Cell. Proteom. 2005, 4, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sadygov, R.G.; Yates, J.R., 3rd. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Old, W.M.; Meyer-Arendt, K.; Aveline-Wolf, L.; Pierce, K.G.; Mendoza, A.; Sevinsky, J.R.; Resing, K.A.; Ahn, N.G. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteom. 2005, 4, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Beissbarth, T.; Hyde, L.; Smyth, G.K.; Job, C.; Boon, W.M.; Tan, S.S.; Scott, H.S.; Speed, T.P. Statistical modeling of sequencing errors in SAGE libraries. Bioinformatics 2004, 20, i31–i39. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.N.; Perlman, D.H.; Costello, C.E.; McComb, M.E. Software tool for researching annotations of proteins: Open-source protein annotation software with data visualization. Anal. Chem. 2009, 81, 9819–9823. [Google Scholar] [CrossRef]
- Ekins, S.; Bugrim, A.; Brovold, L.; Kirillov, E.; Nikolsky, Y.; Rakhmatulin, E.; Sorokina, S.; Ryabov, A.; Serebryiskaya, T.; Melnikov, A.; et al. Algorithms for network analysis in systems-ADME/Tox using the MetaCore and MetaDrug platforms. Xenobiotica 2006, 36, 877–901. [Google Scholar] [CrossRef]
- Mellert, K.; Lechner, S.; Ludeke, M.; Lamla, M.; Moller, P.; Kemkemer, R.; Scheffzek, K.; Kaufmann, D. Restoring functional neurofibromin by protein transduction. Sci. Rep. 2018, 8, 6171. [Google Scholar] [CrossRef] [PubMed]
- Toonen, J.A.; Anastasaki, C.; Smithson, L.J.; Gianino, S.M.; Li, K.; Kesterson, R.A.; Gutmann, D.H. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum. Mol. Genet. 2016, 25, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Frayling, I.M.; Mautner, V.F.; van Minkelen, R.; Kallionpaa, R.A.; Aktas, S.; Baralle, D.; Ben-Shachar, S.; Callaway, A.; Cox, H.; Eccles, D.M.; et al. Breast cancer risk in neurofibromatosis type 1 is a function of the type of NF1 gene mutation: A new genotype-phenotype correlation. J. Med. Genet. 2019, 56, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Coover, R.A.; Healy, T.E.; Guo, L.; Chaney, K.E.; Hennigan, R.F.; Thomson, C.S.; Aschbacher-Smith, L.E.; Jankowski, M.P.; Ratner, N. Tonic ATP-mediated growth suppression in peripheral nerve glia requires arrestin-PP2 and is evaded in NF1. Acta Neuropathol. Commun. 2018, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef]
- Yang, H.M.; Hsiao, S.J.; Schaeffer, D.F.; Lai, C.; Remotti, H.E.; Horst, D.; Mansukhani, M.M.; Horst, B.A. Identification of recurrent mutational events in anorectal melanoma. Mod. Pathol. 2017, 30, 286–296. [Google Scholar] [CrossRef]
- McClure, R.F.; Ewalt, M.D.; Crow, J.; Temple-Smolkin, R.L.; Pullambhatla, M.; Sargent, R.; Kim, A.S. Clinical Significance of DNA Variants in Chronic Myeloid Neoplasms: A Report of the Association for Molecular Pathology. J. Mol. Diagn. 2018, 20, 717–737. [Google Scholar] [CrossRef]
- Ganguly, B.B.; Kadam, N.N. Mutations of myelodysplastic syndromes (MDS): An update. Mutat. Res. Rev. Mutat. Res. 2016, 769, 47–62. [Google Scholar] [CrossRef]
- Gill, H.; Leung, A.Y.; Kwong, Y.L. Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy. Int. J. Mol. Sci. 2016, 17, 440. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar, S.C. Genetic and epigenetic pathways in myelodysplastic syndromes: A brief overview. Adv. Biol. Regul. 2015, 58, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Dmello, C.; Srivastava, S.S.; Tiwari, R.; Chaudhari, P.R.; Sawant, S.; Vaidya, M.M. Multifaceted role of keratins in epithelial cell differentiation and transformation. J. Biosci. 2019, 44, 33. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Wong, P.; Coulombe, P.A. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006, 441, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, L.; Cao, L.; Wen, Q. Keratin 17 Promotes Lung Adenocarcinoma Progression by Enhancing Cell Proliferation and Invasion. Med. Sci. Monit. 2018, 24, 4782–4790. [Google Scholar] [CrossRef]
- Casanova, M.L.; Bravo, A.; Martinez-Palacio, J.; Fernandez-Acenero, M.J.; Villanueva, C.; Larcher, F.; Conti, C.J.; Jorcano, J.L. Epidermal abnormalities and increased malignancy of skin tumors in human epidermal keratin 8-expressing transgenic mice. FASEB J. 2004, 18, 1556–1558. [Google Scholar] [CrossRef]
- Malminen, M.; Peltonen, S.; Koivunen, J.; Peltonen, J. Functional expression of NF1 tumor suppressor protein: Association with keratin intermediate filaments during the early development of human epidermis. BMC Dermatol. 2002, 2, 10. [Google Scholar] [CrossRef]
- Li, X.; Gao, M.; Choi, J.M.; Kim, B.J.; Zhou, M.T.; Chen, Z.; Jain, A.N.; Jung, S.Y.; Yuan, J.; Wang, W.; et al. Clustered, Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-coupled Affinity Purification/Mass Spectrometry Analysis Revealed a Novel Role of Neurofibromin in mTOR Signaling. Mol. Cell. Proteom. 2017, 16, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Giurato, G.; Nassa, G.; Salvati, A.; Alexandrova, E.; Rizzo, F.; Nyman, T.A.; Weisz, A.; Tarallo, R. Quantitative mapping of RNA-mediated nuclear estrogen receptor β interactome in human breast cancer cells. Sci. Data 2018, 5, 180031. [Google Scholar] [CrossRef]
- Theos, A.; Korf, B.R.; American College of, P.; American Physiological, S. Pathophysiology of neurofibromatosis type 1. Ann. Intern. Med. 2006, 144, 842–849. [Google Scholar] [CrossRef]
- Dischinger, P.S.; Tovar, E.A.; Essenburg, C.J.; Madaj, Z.B.; Gardner, E.E.; Callaghan, M.E.; Turner, A.N.; Challa, A.K.; Kempston, T.; Eagleson, B.; et al. NF1 deficiency correlates with estrogen receptor signaling and diminished survival in breast cancer. NPJ Breast Cancer 2018, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Wu, Z.; Yin, X.; Zhao, Y.; Chen, M.; Si, Y.; Yang, J.; Fu, X.; Han, W. Keratin 18 attenuates estrogen receptor α-mediated signaling by sequestering LRP16 in cytoplasm. BMC Cell Biol. 2009, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Boldt, K.; van Reeuwijk, J.; Lu, Q.; Koutroumpas, K.; Nguyen, T.M.; Texier, Y.; van Beersum, S.E.; Horn, N.; Willer, J.R.; Mans, D.A.; et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun. 2016, 7, 11491. [Google Scholar] [CrossRef]
- De Schepper, S.; Boucneau, J.M.; Westbroek, W.; Mommaas, M.; Onderwater, J.; Messiaen, L.; Naeyaert, J.M.; Lambert, J.L. Neurofibromatosis type 1 protein and amyloid precursor protein interact in normal human melanocytes and colocalize with melanosomes. J. Investig. Dermatol. 2006, 126, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Godin, F.; Villette, S.; Vallee, B.; Doudeau, M.; Morisset-Lopez, S.; Ardourel, M.; Hevor, T.; Pichon, C.; Benedetti, H. A fraction of neurofibromin interacts with PML bodies in the nucleus of the CCF astrocytoma cell line. Biochem. Biophys. Res. Commun. 2012, 418, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.P.; Roberts, A.M.; Volta, M.; Sheng, M.; Roberts, R.G. Bipartite interaction between neurofibromatosis type I protein (neurofibromin) and syndecan transmembrane heparan sulfate proteoglycans. J. Neurosci. 2001, 21, 3764–3770. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Gonzalez-Pecchi, V.; Khuri, L.F.; Niu, Q.; Wang, Y.; Xu, Y.; Bai, Y.; Mo, X.; Prochownik, E.V.; Johns, M.A.; et al. OncoPPi-informed discovery of mitogen-activated protein kinase kinase 3 as a novel binding partner of c-Myc. Oncogene 2017, 36, 5852–5860. [Google Scholar] [CrossRef] [Green Version]
- Vallee, B.; Doudeau, M.; Godin, F.; Gombault, A.; Tchalikian, A.; de Tauzia, M.L.; Benedetti, H. Nf1 RasGAP inhibition of LIMK2 mediates a new cross-talk between Ras and Rho pathways. PLoS ONE 2012, 7, e47283. [Google Scholar] [CrossRef]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef]
- Wang, H.F.; Shih, Y.T.; Chen, C.Y.; Chao, H.W.; Lee, M.J.; Hsueh, Y.P. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Investig. 2011, 121, 4820–4837. [Google Scholar] [CrossRef]
- Zhao, L.H.; Ba, X.Q.; Wang, X.G.; Zhu, X.J.; Wang, L.; Zeng, X.L. BAF complex is closely related to and interacts with NF1/CTF and RNA polymerase II in gene transcriptional activation. Acta Biochim. Biophys. Sin. (Shanghai) 2005, 37, 440–446. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProtKB ID | UniProt | Function | TCGA Overlap with NF1/Total Cases |
|---|---|---|---|
| 40S ribosomal protein S8 | P62241 | Ribosomal protein that is a part of the 40S subunit. Increased gene expression seen in colorectal tumors and colon polyps. | 177/755 |
| 60S ribosomal protein L17 | P18621 | Ribosomal protein that is a part of the L22P family and is a component of the 60S subunit. | 136/527 |
| βETA-arrestin-2 | P32121 | Regulates agonist-mediated G-protein coupled receptor (GPCR) signaling through desensitization and resensitization. | 189/593 |
| Dermcidin | P81605 | The N-terminal peptide promotes neural cell survival under oxidative stress. Has oncogenic effects in breast tumors. | 141/579 |
| Eukaryotic translation initiation factor 3 subunit K | Q9UBQ5 | Component of the eIF3 complex which associates with the 40S ribosome to help initiate protein synthesis. | 248/954 |
| Insulin-like growth factor 2 mRNA-binding protein 2 | Q9Y6M1 | RNA-binding factor that recruits target transcripts to cytoplasmic protein-RNA complexes (mRNPs). | 440/1729 |
| Interferon-induced protein with tetratricopeptide repeats 3 | O14879 | An antiviral protein which acts as an inhibitor of cellular and viral processes, such as cell migration, proliferation, signaling, and viral replication. | 192/965 |
| Keratin 13 | A1A4E9 | Member of the type I cytokeratin family. Works in tandem with keratin 4 | 465/846 |
| Keratin, type I cuticular Ha1 | Q15323 | Member of the type I cytokeratin family. | |
| Keratin, type I cuticular Ha6 | O76013 | Member of the type I cytokeratin family. | 458/825 |
| Keratin, type I cytoskeletal 17 | Q04695 | Member of the type I cytokeratin family involved in determing the shape and direction of hair development. | 471/862 |
| Keratin, type II cytoskeletal 4 | P19013 | Member of the type II cytokeratin family. Heterodimerizes with keratin 13. | 188/757 |
| Large proline-rich protein BAG6 | P46379 | An ATP-independent molecular chaperone preventing the aggregation of misfolded and hydrophobic patches-containing proteins | 238/933 |
| Low-density lipoprotein receptor-related protein 2 | P98164 | Acts together with CUBN to mediate endocytosis of high-density lipoproteins | 395/1525 |
| Nuclear protein localization protein 4 homolog | Q8TAT6 | Forms a ternary complex with UFD1 and VCP which transports misfolded proteins from the ER to the cytoplasm for proteasomal degradation. | 425/1399 |
| Proteasome inhibitor PI31 subunit | Q92530 | Inhibits the hydrolysis of peptides by the 20S proteasome | 208/804 |
| Protein disulfide-isomerase A4 | P13667 | Catalyzes the rearrangement of disulfide bonds within proteins. | 274/1211 |
| Regulator of G-protein signaling 10 | O43665 | Regulates GPCR signaling cascades by driving the G-protein α subunit into its GDP bound form. | 201/854 |
| Small nuclear ribonucleoprotein G | P62308 | Major component of the SMN-Sm complex which takes part in splicing pre-mRNAs. | 107/446 |
| Splicing factor 3B subunit 1 | O75533 | Component of the SF3B complex which takes part in splicing pre-mRNAs. | 249/958 |
| UDP-glucose 6-dehydrogenase | O60701 | Catalyzes the formation of UDP-α-D-glucuronate. Required for embryonic development via its role in the biosynthesis of glycosaminoglycans. | 143/481 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carnes, R.M.; Kesterson, R.A.; Korf, B.R.; Mobley, J.A.; Wallis, D. Affinity Purification of NF1 Protein–Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners. Genes 2019, 10, 650. https://doi.org/10.3390/genes10090650
Carnes RM, Kesterson RA, Korf BR, Mobley JA, Wallis D. Affinity Purification of NF1 Protein–Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners. Genes. 2019; 10(9):650. https://doi.org/10.3390/genes10090650
Chicago/Turabian StyleCarnes, Rachel M., Robert A. Kesterson, Bruce R. Korf, James A. Mobley, and Deeann Wallis. 2019. "Affinity Purification of NF1 Protein–Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners" Genes 10, no. 9: 650. https://doi.org/10.3390/genes10090650