Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders

 , , ,
, , ,  ,
,  , and
, and 
Abstract
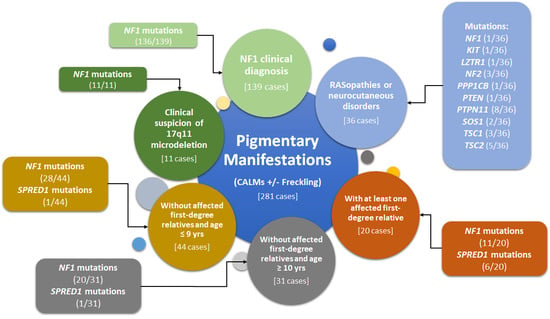
:
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Clinical Classification
2.2. Primer Design for NF1 and SPRED1 Mutation Screening
2.3. Mutation Screening by RT-PCR
2.4. Targeted NGS-Based Mutational Screening
2.5. Multiplex Ligation-Dependent Probe Amplification
2.6. Real-Time PCR
2.7. Validation of Variants by Sanger Sequencing
3. Results
3.1. Molecular Diagnosis
3.2. NF1 Mutation Screening
3.3. SPRED1 Mutation Screening
3.4. Phenotype-Genotype Overview
3.5. Mutation Screening in Non-NF1 or NF1-Like Conditions and Unsolved Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
| Family ID 1 | Group | Exon | Type 2 | Genomic | cDNA | Effect | Protein | ClinVar/HGMD/LOVD ID3 |
|---|---|---|---|---|---|---|---|---|
| 216 (3) | 5 | 01(01) | SNV | 3G>A | RNA decay | RNA decay | ? | LOVD: NF1_001130 |
| 220 (1) | 1 | 01(01) | SNV | 59A>C (splicing) | RNA decay | RNA decay | ? | New |
| 212 (1) | 1 | 01(01) | SNV | 60G>C (splicing) | RNA decay | RNA decay | ? | New |
| 35 (2) | 1 | 02(02) | SNV | 128T>C | 128T>C | Missense | Leu43Pro | LOVD: NF1_000049 |
| 67 (1) | 1 | 03(03) | SNV | 277T>C | 277T>C | Missense | Cys93Arg | LOVD: NF1_002264 |
| 294 (1) | 3 | 03(03) | SNV | 288+1delG | 288del | Splicing | Glu97Asnfs*6 | LOVD: NF1_001615 |
| 81 (1) | 3 | 03(03) | SNV | 288+1G>A (Splicing) | 205_288del | Splicing | Arg69_Gly96del | LOVD: NF1_001484 |
| 262 (1) | 5 | 03(03) | SNV | 288+4A>G | 205_288del | Splicing | Arg69_Gly96del | LOVD: NF1_000270 |
| 93 (3) | 1 | 03(03) | DIM | 289-2956C>T (cryptic splice site) | 288_289insAGTCTCACTCTGCGGCACAGGCTGAAGTGCAGTGGCACCCTCTCGGCTCATTGCAACCTCCACTTCCCGGGTTCAAGCTATTCTCATGCCTCAGCCTCCCAAGTAGCTGGGATTACAG | Intronic cryptic splice site | Gln97Valfs*8 | New |
| 72 (1) | 1 | 04(04a) | DEL | 363del | 363del | Frame-shift | His122Thrfs*43 | New |
| 31 (1) | 1 | 05(04b) | DEL | 499_502del | 499_502del | Frame-shift | Cys167Glnfs*10 | LOVD: NF1_000605 |
| 251 (1) | 1 | |||||||
| 2 (1) | 1 | 05(04b) | SNV | 574C>T | 574C>T | Nonsense | Arg192* | LOVD: NF1_000702 |
| 68 (1) | 1 | |||||||
| 121 (1) | 3 | 07(05) | SNV | 667T>A | 667T>A | Missense | Trp223Arg | LOVD: NF1_000800 |
| 128 (1) | 3 | 08(06) | SNV | 818T>C | 818T>C | Missense | Leu273Pro | New |
| 221 (1) | 1 | 09(07) | DEL | 1019_1020del | 1019_1020del | Frame-shift | Ser340Cysfs*12 | LOVD: NF1_000005 |
| 172 (1) | 1 | 10(08) | DEL | 1110del | 1110del | Frame-shift | Ala371Glnfs*5 | New |
| 49 (1) | 3 | 10(08) | DEL | 1123del | 1123del | Frame-shift | Leu375* | New |
| 296 (1) | 4 | 10(08) | SNV | 1144T>C | 1144T>C | Missense | Ser382Pro | New |
| 46 (1) | 1 | 10(08) | SNV | 1185+1G>A (splicing) | 1063_1185del | Splicing | Asn355_Lys395del | LOVD: NF1_000019 |
| 65 (1) | 1 | |||||||
| 218 (1) | 1 | 10(08) | SNV | 1185G>T (splicing) | 1063_1185del | Splicing | Asn355_Lys395del | HGMD: CS147216 |
| 125 (1) | 3 | 11(09) | SNV | 1186-3T>G (splicing) | 1185_1186dup | Splicing | Ile396Glufs*17 | New |
| 292 (1) | 1 | 11(09) | SNV | 1198C>T | 1198C>T | Nonsense | Gln400* | LOVD: NF1_000024 |
| 110 (1) | 3 | 11(09) | INS | 1243dup | 1243dup | Frame-shift | His415Profs*14 | ClinVar: 426651 |
| 23 (2) | 1 | 11(09) | SNV | 1246C>T | 1246C>T | Nonsense | Arg416* | LOVD: NF1_000034 |
| 152 (1) | 1 | |||||||
| 45 (1) | 3 | 11(09) | DIM | 1260+1604A>G (cryptic splice site) | 1260_1261insCTGACTACATAGAGCACTTTCAAGCATGGACTTGGCACTGCT | Intronic cryptic splice site | Ser421Leufs*4 | LOVD: NF1_000035 |
| 165 (1) | 1 | 11(09) | SNV | 1260+1G>A | 1260_1261insATAAGTCCAAAAG | Splicing | Ser421Ilefs*12 | LOVD: NF1_000036 |
| 289 (1) | 1 | 12(10a) | SNV | 1261-2A>C (cryptic splice site) | 1261_1284del | Splicing | Ser421_Lys428del | LOVD: NF1_000045 |
| 82 (1) | 1 | 12(10a) | SNV | 1318C>T | 1318C>T | Nonsense | Arg440* | LOVD: NF1_000052 |
| 153 (1) | 1 | |||||||
| 54 (1) | 4 | 12(10a) | DEL | 1329del | 1329del | Frame-shift | Phe443Leufs*30 | LOVD: NF1_001174 |
| 52 (1) | 1 | 12(10a) | INS | 1378dup | 1378dup | Frame-shift | Ile460Asnfs*10 | New |
| 21 (1) | 1 | 12(10a) | SNV | 1381C>T | 1381C>T | Nonsense | Arg461* | LOVD: NF1_000056 |
| 57 (1) | 3 | |||||||
| 144 (3) | 1 | |||||||
| 160 (1) | 1 | 12(10a) | DIM | 1393-1554C>G (cryptic splice site) | 1392_1393insTGAAGATTTGTTTACACCAGCATCACTACAAACAATAACGCATTGTGCTTGGACATCACGATGGCTATGATA | Intronic cryptic splice site | Ser465* | New |
| 17 (3) | 1 | 13(10b) | DEL | 1393-2del | 1393_1527del | Splicing | Ser465_Cys509del | LOVD: NF1_000061 |
| 116 (1) | 1 | 13(10b) | INS | 1399dup | 1399dup | Frame-shift | Thr467Asnfs*3 | LOVD: NF1_002283 |
| 288 (1) | 1 | 13(10b) | DEL | 1423del | 1423del | Frame-shift | Lys476Asnfs*22 | New |
| 13 (1) | 4 | 13(10b) | SNV | 1466A>G (cryptic splice site) | 1466_1527del | Splicing | Tyr489* | ClinVar: 354 |
| 222 (1) | 1 | |||||||
| 159 (1) | 1 | 13(10b) | INS | 1470_1471insATACG | 1470_1471insATACG | Frame-shift | Tyr491Ilefs*9 | New |
| 132 (2) | 1 | 13(10b) | SNV | 1487T>G | 1487T>G | Missense | Met496Arg | New |
| 174 (1) | 1 | 13(10b) | INDEL | 1499_1501delinsAAA | 1499_1501delinsAAA | DelIns | Ile500_His501delinsLysAsn | New |
| 107 (1) | 2 | 13(10b) | DEL | 1500del | 1500del | Frame-shift | His501Metfs*25 | New |
| 138 (1) | 3 | 13(10b) | DIM | 1527+1165T>A (cryptic splice site) | 1527_1528insGATGACATGTTTAACCTTTGTTGAGCTTCTTCAGTCCCTGGAGAGCAGCATCAAGCAAG | Intronic cryptic splice site | Asn510Aspfs*7 | New |
| 76 (1) | 1 | 14(10c) | DEL | 1541_1542del | 1541_1542del | Frame-shift | Gln514Argfs*43 | LOVD: NF1_000074 |
| 44 (3) | 1 | 14(10c) | SNV | 1595T>G | 1595T>G | Missense | Leu532Arg | LOVD: NF1_002498 |
| 5 (1) | 4 | 15(11) | SNV | 1642-1G>A (splicing) | 1642_1721del | Splicing | Ala548Leufs*13 | LOVD: NF1_000084 |
| 147 (3) | 5 | 15(11) | SNV | 1642G>T | 1642_1721del | Splicing | Ala548Leufs*13 | New |
| 59 (1) | 4 | 15(11) | SNV | 1658A>G | 1658A>G | Missense | His553Arg | LOVD: NF1_000091 |
| 186 (1) | 1 | 15(11) | SNV | 1721+3A>G (Splicing) | 1642_1721del | Splicing | Ala548Leufs*13 | LOVD: NF1_000101 |
| 85 (1) | 1 | 15(11) | SNV | 1721G>C (Splicing) | 1642_1721del | Splicing | Ala548Leufs*13 | New |
| 150 (1) | 1 | 15-36 | DEL | 1642-?_4772+?del (intragenic deletion ex. 15-36) | 1642_4772del | Intragenic deletion | Ala548Valfs*9 | New |
| 15 (1) | 1 | 16(12a) | SNV | 1748A>G (cryptic splice site) | 1722_1748del | Splicing | Ser574_Lys583delinsArg | LOVD: NF1_000110 |
| 102 (2) | 1 | 17(12b) | SNV | 1846C>T | 1846C>T | Nonsense | Gln616* | LOVD: NF1_000125 |
| 133 (2) | 1 | 17(12b) | DEL | 1863del | 1863del | Frame-shift | Cys622Valfs*9 | LOVD: NF1_001427 |
| 168 (1) | 2 | |||||||
| 149 (1) | 2 | |||||||
| 214 (1) | 1 | 17(12b) | SNV | 1942G>T | 1942G>T | Nonsense | Glu648* | LOVD: NF1_002848 |
| 55 (2) | 1 | 17(12b) | INS | 1995dup | 1995dup | Frame-shift | Ser666Leufs*4 | New |
| 217 (1) | 1 | 18(13) | SNV | 2002-10T>A (cryptic splice site) | 2001_2002insACTCTCAG | Splicing | Asp668Thrfs*23 | New |
| 8 (1) | 1 | 18(13) | SNV | 2002-1G>A (cryptic splice site) | 2002_2011del | Splicing | Asp668Glnfs*17 | LOVD: NF1_000143 |
| 192 (1) | 1 | 18(13) | INDEL | 2027_2028delinsA | 2027_2028delinsA | DelIns | Thr676Asnfs*12 | New |
| 29 (2) | 1 | 18(13) | INS | 2033dup | 2033dup | Frame-shift | Ile679Aspfs*21 | LOVD: NF1_000148 |
| 63 (1) | 1 | 18(13) | SNV | 2041C>T | 2041C>T | Nonsense | Arg681* | LOVD: NF1_000153 |
| 39 (2) | 1 | 18(13) | INS | 2167dup | 2167dup | Frame-shift | Val723Glyfs*3 | New |
| 27 (2) | 1 | 18(13) | SNV | 2251G>C (splicing) | 2002_2251del | Splicing | Asp668Glufs*9 | ClinVar: 584927 |
| 209 (1) | 1 | 19(14) | SNV | 2266C>T | 2252_2325del | Splicing | Arg752Leufs*17 | LOVD: NF1_000174 |
| 75 (1) | 1 | 19(14) | SNV | 2288T>C | 2288T>C | Missense | Leu763Pro | LOVD: NF1_000177 |
| 7 (2) | 1 | 19(14) | INS | 2307dup | 2307dup | Frame-shift | Thr770Hisfs*6 | New |
| 53 (1) | 3 | 20(15) | SNV | 2326G>A | 2326_2409del | Splicing | Trp777_Ala804del | New |
| 126 (1) | 1 | |||||||
| 184 (2) | 1 | 20(15) | SNV | 2339C>A | 2339C>A | Missense | Thr780Lys | LOVD: NF1_000190 |
| 234 (2) | 1 | 20(15) | SNV | 2339C>G | 2339C>G | Missense | Thr780Arg | LOVD: NF1_001397 |
| 6 (3) | 1 | 20(15) | SNV | 2351G>C | 2351G>C | Missense | Trp784Ser | LOVD: NF1_000196 |
| 33 (2) | 1 | 20(15) | SNV | 2352G>C | 2352G>C | Missense | Trp784Cys | LOVD: NF1_001853 |
| 106 (1) | 4 | 21(16) | SNV | 2540T>G | 2540T>G | Missense | Leu847Arg | LOVD: NF1_000229 |
| 188 (1) | 1 | |||||||
| 300 (1) | 1 | 21(16) | SNV | 2557C>T | 2557C>T | Nonsense | Gln853* | LOVD: NF1_001222 |
| 241 (1) | 1 | |||||||
| 38 (1) | 1 | 21(16) | SNV | 2693T>C | 2693T>C | Missense | Leu898Pro | LOVD: NF1_000241 |
| 48 (1) | 1 | 21(16) | SNV | 2850+1G>A (cryptic plice site) | 2707_2850del | Splicing | Cys904_Val951del | LOVD: NF1_000259 |
| 18 (1) | 1 | 22(17) | SNV | 2851G>T (splicing) | 2851_2990del | Splicing | Leu952Cysfs*22 | LOVD: NF1_001526 |
| 190 (2) | 1 | |||||||
| 14 (1) | 1 | 22(17) | SNV | 2887C>T | 2887C>T | Nonsense | Gln963* | ClinVar: 233495 |
| 109 (1) | 1 | 22(17) | DEL | 2948del | 2948del | Frame-shift | Leu983Glnfs*9 | New |
| 19 (2) | 1 | 22(17) | DEL | 2970_2972del | 2970_2972del | In-frame deletion | Met992del | LOVD: NF1_000277 |
| 134 (1) | 1 | 23(18) | SNV | 3040A>T | 3040A>T | Nonsense | Lys1014* | ClinVar: 431616 |
| 140 (1) | 4 | 23(18) | SNV | 3104T>A | 3104T>A | Missense | Met1035Lys | New |
| 245 (1) | 4 | 23(18) | SNV | 3106A>G | 3106A>G | Missense | Lys1036Glu | New |
| 182 (1) | 1 | 23(18) | SNV | 3113+1G>A (splicing) | 2991_3113del | Splicing | Tyr998_Arg1038del | LOVD: NF1_000306 |
| 230 (1) | 3 | 25(19b) | SNV | 3277G>A (cryptic splice site) | 3275_3314del | Splicing | Gly1092Aspfs*7 | LOVD: NF1_000340 |
| 74 (1) | 1 | 26(20) | SNV | 3326T>G | 3326T>G | Nonsense | Leu1109* | New |
| 41 (1) | 4 | 26(20) | DEL | 3347_3350del | 3347_3350del | Frame-shift | Asp1116Alafs*25 | LOVD: NF1_000351 |
| 255 (1) | 1 | |||||||
| 115 (1) | 3 | 26(20) | SNV | 3445A>G | 3445A>G | Missense | Met1149Val | LOVD: NF1_000356 |
| 215 (1) | 3 | |||||||
| 20 (2) | 1 | 26(20) | SNV | 3496+1G>A | 3315_3496del | Splicing | Tyr1106Leufs*28 | HGMD: CS072245 |
| 40 (2) | 5 | 27(21) | DEL | 3502_3519del | 3502_3519del | In-frame deletion | Gly1168_Leu1173del | New |
| 284 (2) | 6 | 27(21) | SNV | 3592G>A | 3592G>A | Missense | Glu1198Lys | New |
| 256 (1) | 1 | 27(21) | SNV | 3610C>G | 3610C>G | Missense | Arg1204Gly | LOVD: NF1_000372 |
| 104 (1) | 1 | 28(22) | SNV | 3826C>T | 3826C>T | Nonsense | Arg1276* | LOVD: NF1_000403 |
| 108 (1) | 1 | |||||||
| 166 (1) | 1 | |||||||
| 225 (1) | 1 | 28-29 | DEL | (3708+1_3709-1)_(3973+1_3974-1)del (intragenic deletion ex. 28-29) | not determined | Intragenic deletion | ? | New |
| 275 (1) | 3 | 29(23) | DEL | 3899del | 3899del | Frame-shift | Leu1300Profs*9 | New |
| 180 (1) | 1 | 29(23) | SNV | 3916C>T | 3916C>T | Nonsense | Arg1306* | LOVD: NF1_000416 |
| 58 (1) | 1 | 29(23) | DEL | 3972del | 3972del | Frame-shift | Arg1325Glyfs*2 | New |
| 247 (1) | 1 | 29(23) | SNV | 3974G>A | 3873_3976del | Splicing | Tyr1292Argfs*7 | LOVD: NF1_001992 |
| 129 (1) | 1 | 30(23-1) | INS | 4100_4103dup | 4100_4103dup | Frame-shift | Tyr1369Phefs*6 | New |
| 36 (1) | 2 | 32(24) | DEL | 4168del | 4168del | Frame-shift | Leu1390Serfs*17 | LOVD: NF1_000458 |
| 162 (1) | 1 | 32(24) | SNV | 4172G>C | 4172G>C | Missense | Arg1391Thr | LOVD: NF1_000461 |
| 285 (1) | 1 | 32(24) | SNV | 4269+2T>C | not determined | Splicing | ? | New |
| 261 (1) | 5 | 33(25) | SNV | 4276C>G | 4276C>G | Missense | Gln1426Glu | LOVD: NF1_001275 |
| 154 (1) | 1 | 33(25) | SNV | 4278G>C | 4278G>C | Missense | Gln1426His | LOVD: NF1_000483 |
| 32 (4) | 5 | 35(27a) | SNV | 4515-21T>G (splicing) | 4514_4515insTTTGCTGTATCTAG | Splicing | Arg1505Serfs*53 | New |
| 16 (13) | 1 | 35(27a) | SNV | 4515-2A>G | 4514_4515insTTTGCTGTATCTGG | Splicing | Arg1505Serfs*53 | LOVD: NF1_000518 |
| 28 (1) | 1 | 35(27a) | SNV | 4537C>T | 4537C>T | Nonsense | Arg1513* | LOVD: NF1_000521 |
| 9 (2) | 1 | 35(27a) | SNV | 4637C>A | 4637C>A | Nonsense | Ser1546* | LOVD: NF1_000534 |
| 181 (1) | 1 | 35(27a) | DEL | 4644del | 4644del | Frame-shift | Phe1548Leufs*5 | New |
| 66 (1) | 1 | 36(27b) | DEL | 4680_4683del | 4680_4683del | Frame-shift | Glu1561Asnfs*5 | New |
| 173 (1) | 4 | 36(27b) | DEL | 4691del | 4691del | Frame-shift | Lys1564Argfs*3 | New |
| 70 (1) | 4 | 36(27b) | SNV | 4768C>T | 4768C>T | Missense | Arg1590Trp | HGMD: CM971051 |
| 4 (1) | 1 | 37(28) | SNV | 4780del | 4780del | Frame-shift | Thr1594Leufs*9 | New |
| 119 (1) | 2 | 37(28) | DEL | 4840_4854del | 4840_4854del | In-frame deletion | Tyr1614_Tyr1618del | LOVD: NF1_001657 |
| 89 (1) | 1 | 37(28) | DEL | 4914_4917del | 4914_4917del | Frame-shift | Lys1640Glyfs*36 | LOVD: NF1_000586 |
| 193 (1) | 1 | 37(28) | SNV | 4922G>A | 4922G>A | Nonsense | Trp1641* | LOVD: NF1_001303 |
| 80 (2) | 1 | 37(28) | DEL | 4973_4978del | 4973_4978del | In-frame deletion | Ile1658_Tyr1659del | LOVD: NF1_000597 |
| 95 (2) | 4 | 37-51 | DUP | 5035-?_7426-?dup (intragenic duplication ex. 37-51) | not determined | Intragenic duplication | ? | New |
| 10 (2) | 1 | 38(29) | SNV | 5264C>G | 5264C>G | Nonsense | Ser1755* | HGMD: CM001260 |
| 1 (2) | 4 | 38(29) | SNV | 5401C>T | 5401C>T | Nonsense | Gln1801* | LOVD: NF1_001390 |
| 101 (2) | 5 | 38(29) | SNV | 5425C>T | 5425C>T | Missense | Arg1809Cys | LOVD: NF1_000653 |
| 112 (5) | 5 | |||||||
| 178 (2) | 5 | |||||||
| 302 (1) | 3 | |||||||
| 155 (1) | 4 | 38(29) | SNV | 5426G>C | 5426G>C | Missense | Arg1809Pro | ClinVar: 208855 |
| 124 (3) | 5 | 38(29) | SNV | 5426G>T | 5426G>T | Missense | Arg1809Leu | LOVD: NF1_000654 |
| 156 (1) | 4 | |||||||
| 229 (1) | 5 | |||||||
| 175 (1) | 3 | 38(29) | SNV | 5437T>C | 5437T>C | Missense | Ser1813Pro | New |
| 164 (2) | 1 | 38(29) | SNV | 5483A>T | 5483A>T | Missense | Asp1828Val | LOVD: NF1_000666 |
| 259 (1) | 1 | 38(29) | SNV | 5543T>A | 5543T>A | Nonsense | Leu1848* | LOVD: NF1_000670 |
| 163 (1) | 1 | 39(30) | DEL | 5592_5596del | 5592_5596del | Frame-shift | Asn1864Lysfs*26 | New |
| 231 (1) | 3 | 39(30) | SNV | 5608C>T | 5608C>T | Nonsense | Gln1870* | ClinVar: 237577 |
| 22 (1) | 1 | 39(30) | SNV | 5676G>T | 5676G>T | Missense | Lys1892Asn | New |
| 177 (3) | 1 | 39(30) | SNV | 5719G>T | 5719G>T | Nonsense | Glu1907* | ClinVar: 187652 |
| 26 (2) | 1 | 39(30) | DEL | 5739del | 5739del | Frame-shift | Phe1913Leufs*8 | New |
| 24 (2) | 1 | 40(31) | SNV | 5839C>T | 5839C>T | Nonsense | Arg1947* | LOVD: NF1_000711 |
| 171 (1) | 1 | 40(31) | SNV | 5842C>T | 5842C>T | Nonsense | Gln1948* | LOVD: NF1_001913 |
| 151 (1) | 1 | 40(31) | SNV | 5928G>A | 5928G>A | Nonsense | Trp1976* | LOVD: NF1_002495 |
| 71 (1) | 4 | 40(31) | SNV | 5938G>C | 5938G>C | Missense | Gly1980Arg | ClinVar: 457773 |
| 282 (1) | 3 | 41(32) | SNV | 5944-1G>C (cryptic splice site) | 5946_5952del | Splicing | Thr1983Cysfs*6 | ClinVar: 431977 |
| 135 (1) | 3 | 41(32) | SNV | 5944-5A>G (cryptic splice site) | 5943_5944insCTAG | Splicing | Ile1982Leufs*7 | LOVD: NF1_001321 |
| 34 (2) | 1 | 42(33) | SNV | 6085-2A>T (splicing) | 6085_6364del | Splicing | Val2029Lysfs*7 | LOVD: NF1_001919 |
| 83 (1) | 1 | 42(33) | SNV | 6243C>A | 6243C>A | Nonsense | Y2081* | New |
| 223 (1) | 1 | 42(33) | SNV | 6335T>C | 6335T>C | Missense | Leu2112Pro | LOVD: NF1_000756 |
| 233 (1) | 1 | 42(33) | SNV | 6364+4A>G | 6085_6364del | Splicing | Val2029Lysfs*7 | HGMD: CS941517 |
| 97 (1) | 3 | 43(34) | SNV | 6579+1G>T (splicing) | 6365_6579del | Splicing | Glu2122Glyfs*27 | LOVD: NF1_000784 |
| 253 (1) | 3 | 43(34) | SNV | 6579+2T>C | not determined | Splicing | ? | New |
| 42 (1) | 3 | 44(35) | SNV | 6606C>A | 6606C>A | Nonsense | Cys2202* | LOVD: NF1_001338 |
| 137 (1) | 1 | 44(35) | SNV | 6611G>A | 6611G>A | Nonsense | Trp2204* | LOVD: NF1_001584 |
| 170 (1) | 3 | 44(35) | SNV | 6641+1G>C | 6580_6641del | Splicing | Ala2194Ilefs*6 | LOVD: NF1_000796 |
| 139 (1) | 1 | 45(36) | DEL | 6688del | 6688del | Frame-shift | Val2230Serfs*14 | LOVD: NF1_001670 |
| 50 (1) | 1 | 45(36) | SNV | 6709C>T | 6709C>T | Nonsense | Arg2237* | LOVD: NF1_000802 |
| 73 (1) | 1 | 46(37) | INS | 6791_6792insAA | 6791_6792insAA | Frame-shift | Tyr2264* | LOVD: NF1_001349 |
| 51 (2) | 1 | 46(37) | INS | 6791dup | 6791dup | Frame-shift | Tyr2264* | LOVD: NF1_000815 |
| 118 (1) | 3 | 46(37) | SNV | 6792C>A (STOP determining splicing) | 6757_6858del | Splicing | Ala2253_Lys2286del | LOVD: NF1_000816 |
| 143 (1) | 4 | |||||||
| 176 (1) | 1 | |||||||
| 299 (1) | 3 | 46(37) | SNV | 6792C>G (STOP determining splicing) | 6757_6858del | Splicing | Ala2253_Lys2286del | LOVD: NF1_000817 |
| 30 (1) | 1 | 46(37) | SNV | 6858+1G>T (splicing) | 6757_6858del | Splicing | Ala2253_Lys2286del | LOVD: NF1_000824 |
| 268 (1) | 1 | 46(37) | SNV | 6858+2T>C | 6757_6858del | Splicing | Ala2253_Lys2286del | HGMD: CS073509 |
| 47 (1) | 3 | 47(38) | DEL | 6881del | 6881del | Frame-shift | Leu2294Profs*4 | LOVD: NF1_001726 |
| 84 (1) | 1 | 47(38) | DEL | 6898_6903del | 6898_6903del | In-frame deletion | Ala2300_Val2301del | New |
| 88 (2) | 1 | 47(38) | SNV | 6955C>T | 6955C>T | Nonsense | Gln2319* | New |
| 276 (1) | 1 | 47(38) | DEL | 6974_6977del | 6974_6977del | Frame-shift | Asp2325Valfs*49 | LOVD: NF1_001352 |
| 94 (1) | 1 | 48(39) | INS | 7089dup | 7089dup | Frame-shift | Asn2364* | LOVD: NF1_001359 |
| 183 (1) | 1 | |||||||
| 43 (3) | 1 | 48(39) | DEL | 7125del | 7125del | Frame-shift | Tyr2377Thrfs*20 | LOVD: NF1_000849 |
| 158 (3) | 1 | |||||||
| 169 (1) | 1 | 49(40) | DEL | 7169_7170del | 7169_7170del | Frame-shift | Arg2390Asnfs*10 | New |
| 79 (2) | 1 | 49(40) | SNV | 7184T>C | 7184T>C | Missense | Leu2395Pro | LOVD: NF1_000857 |
| 227 (1) | 1 | 49(40) | INS | 7232dup | 7232dup | Frame-shift | Asn2411Lysfs*16 | New |
| 185 (1) | 1 | 50(41) | SNV | 7259C>A | 7259C>A | Missense | Ala2420Asp | LOVD: NF1_000867 |
| 3 (1) | 1 | 50(41) | SNV | 7285C>T | 7285C>T | Nonsense | Arg2429* | LOVD: NF1_000871 |
| 87 (1) | 1 | |||||||
| 37 (1) | 3 | 51(42) | DEL | 7518del | 7518del | Frame-shift | Gln2507Asnfs*20 | ClinVar: 237598 |
| 130 (1) | 4 | 52(43) | SNV | not determined | 7553_7675del | In-frame deletion | Gly2518_Met2558del | |
| 12 (2) | 1 | 53(44) | DEL | 7686del | 7686del | Frame-shift | Ile2563Phefs*40 | LOVD: NF1_002529 |
| 25 (2) | 1 | 54(45) | INS | 7874_7875dup | 7874_7875dup | Frame-shift | Ser2626Profs*33 | New |
| 103 (1) | 1 | 56(47) | SNV | 8051-1G>C (splicing) | 8051_8097del | Splicing | Ser2684Thrfs*9 | New |
| 260 (1) | 4 | 57(48) | INS | 8207_8231dup | 8207_8231dup | Frame-shift | Leu2745Serfs*14 | New |
| 61 (1) | 2 | all | DEL | -718-?_8375+?del (Microdeletion 17q11.2) | Microdeletion 17q11.2 | Microdeletion 17q11.2 | ? | LOVD: NF1_000001 |
| 64 (1) | 2 | |||||||
| 69 (1) | 2 | |||||||
| 77 (1) | 2 | |||||||
| 78 (1) | 1 | |||||||
| 99 (1) | 2 | |||||||
| 127 (1) | 2 | |||||||
| 136 (1) | 1 | |||||||
| 211 (1) | 1 | |||||||
| 277 (1) | 1 |
| Family ID 1 | Group | Exon | Type 2 | Genomic | cDNA | Effect | Protein | ClinVar/HGMD/LOVD ID 3 |
|---|---|---|---|---|---|---|---|---|
| 11(10) | 5 | 2 | DEL | 49_53del | 49_53del | Frame-shift | Val17Serfs*8 | New |
| 224(1) | 3 | 2 | SNV | 52C>T | 52C>T | Nonsense | Arg18* | LOVD: SPRED1_000177 |
| 92(3) | 5 | 2 | SNV | 70C>T | 70C>T | Nonsense | Arg24* | LOVD: SPRED1_000014 |
| 179(3) | 5 | 2 | SNV | 74A>G | 74A>G | Missense | Asp25Gly | ClinVar: 391600 |
| 161(2) | 5 | 3 | SNV | 229A>T | 229A>T | Nonsense | Lys77* | LOVD: SPRED1_000121 |
| 157(1) | 4 | 6–7 | DEL | 618-?_*91+?del (intragenic deletion ex. 6-7) | ? | ? | ? | New |
| 167(4) | 5 | 7 | SNV | 973C>T | 973C>T | Nonsense | Arg325* | LOVD: SPRED1_000077 |
| 286(2) | 5 | 7 | DUP | 993dup | 993dup | Frame-shift | Arg332Thrfs*12 | New |
| Group | Family ID | Patient ID | Sex | Molecularly Characterized Affected Relatives | Sporadic (Y/N) | Paternal/Maternal Inheritance | Age (yy:mm) | CALMs (≥6) | Freckling | Lisch Nodules | OPG | Bone Dysplasia | Neurofibromas (Cutaneous or Plexiform) | Other Clinical Features 1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T0 | T1 | T0 | T1 | T0 | T1 | T0 | T1 | T0 | T1 | T0 | T1 | T0 | T1 | ||||||||
| 1 | 2 | 4 | F | 0 | N | Maternal | 2 | 12:06 | + | + | - | + | + | + | - | - | - | - | + | + | LGG, DLL |
| 1 | 3 | 5 | M | 0 | N | Maternal | 6:08 | 14 | + | + | + | + | - | - | - | - | - | - | - | + | Thor |
| 1 | 4 | 6 | M | 0 | N | Paternal | 14:02 | 18:05 | + | + | + | + | - | - | n.a. | n.a. | - | - | - | - | |
| 1 | 6 | 8 | M | 2 | N | Maternal | 6 | 13 | + | + | + | + | + | + | - | - | - | - | + | + | DLL, DS, ID |
| 1 | 7 | 10 | M | 1 | N | Paternal | 20 | 22 | + | + | + | + | + | + | - | - | - | - | - | - | NBOs, Epy |
| 1 | 8 | 12 | M | 0 | N | Paternal | 10 | 19:08 | + | + | + | + | + | + | - | - | - | - | + | + | DLL |
| 1 | 9 | 13 | F | 1 | N | Paternal | 14:09 | 17 | + | + | + | + | + | + | - | - | - | - | + | + | UH |
| 1 | 10 | 16 | F | 1 | N | Maternal | 10 | 15:04 | + | + | - | + | - | - | + | + | - | - | + | + | NBOs |
| 1 | 12 | 116 | M | 3 (2 twin) | N | Paternal | 10 | 14:08 | + | + | - | + | - | - | - | - | - | - | - | - | LD, SP |
| 1 | 14 | 23 | M | 0 | Y | 9:09 | 11:08 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 15 | 24 | M | 0 | N | Maternal | 10 | 11 | + | + | + | + | + | + | - | - | - | - | - | - | DS |
| 1 | 16 | 26 | M | 12 | N | Maternal | 10:01 | 12:04 | + | + | + | + | + | + | - | - | - | - | + | + | UH, MMS, Thor, NBOs, DLL, PmD, LD |
| 1 | 17 | 37 | F | 1 | N | Paternal | 3:01 | 9:01 | + | + | - | + | - | - | - | - | - | - | - | + | LD, NBOs |
| 1 | 18 | 38 | F | 0 | N | Paternal | 5:09 | 8:11 | + | + | + | + | - | - | - | - | - | - | + | + | Noon, MacroC, SP, Thor |
| 1 | 19 | 40 | F | 1 | N | Maternal | 12:01 | 15:08 | + | + | + | + | + | + | - | - | - | - | - | - | Noon, MacroC, SP, Thor, SS, BvP, BP, |
| 1 | 20 | 41 | M | 1 | N | Maternal | 11 | 19 | + | + | n.a. | n.a. | + | + | - | - | - | - | - | - | Thor |
| 1 | 21 | 43 | M | 0 | N | Paternal | 21:11 | 22:08 | + | + | + | + | + | + | - | - | - | - | + | + | |
| 1 | 22 | 45 | M | 0 | N | Maternal | 14:08 | 15:06 | + | + | + | + | + | + | - | - | - | - | + | + | |
| 1 | 23 | 48 | F | 1 (twin) | Y | 15 | 17:06 | + | + | + | + | + | + | - | - | - | - | + | + | DS, DE, VS, LD, SP | |
| 1 | 24 | 50 | M | 1 | N | Maternal | 13:09 | 17:06 | + | + | + | + | n.a. | n.a. | + | + | - | - | + | + | SS, DE, DS, NBOs |
| 1 | 25 | 51 | F | 1 | N | Maternal | 11:02 | 11:07 | + | + | + | + | + | + | - | - | - | - | - | - | |
| 1 | 26 | 53 | F | 1 | N | Maternal | 6:01 | 10 | + | + | - | + | - | - | - | - | n.a. | n.a. | - | - | LD |
| 1 | 27 | 54 | M | 1 (twin) | Y | 15:03 | 18:11 | + | + | + | + | + | + | - | - | - | - | - | + | Thor | |
| 1 | 28 | 56 | F | 0 | N | Maternal | 7 | 8:11 | + | + | + | + | n.a. | n.a. | - | - | - | - | + | + | |
| 1 | 29 | 58 | F | 1 | N | Paternal | 19:04 | 22:11 | + | + | + | + | + | + | - | - | - | - | + | + | DS, NBOs |
| 1 | 30 | 61 | M | 0 | N | Paternal | 11:09 | 13:01 | + | + | + | + | - | n.a. | - | - | - | - | - | - | Thor |
| 1 | 31 | 62 | M | 0 | N | Paternal | 9:07 | 10:03 | + | + | + | + | + | + | + | + | - | - | - | - | DLL, NBOs |
| 1 | 33 | 65 | F | 1 | N | Paternal | 14:02 | 14:08 | + | + | + | + | + | + | n.a. | n.a. | - | - | - | - | SS, DLL, MacroC, DS, Noon |
| 1 | 34 | 66 | M | 1 | N | Maternal | 14:05 | 16 | + | + | + | + | + | + | - | - | - | - | - | - | MMS |
| 1 | 35 | 67 | F | 1 | N | Paternal | 22 | 27 | + | + | + | + | - | - | - | - | - | - | + | + | |
| 1 | 38 | 72 | F | 0 | N | Paternal | 6:04 | 7:05 | + | + | - | + | - | - | - | - | - | - | - | - | NA, MacroC |
| 1 | 39 | 76 | M | 1 | N | Maternal | 8:06 | 9:05 | + | + | + | + | n.a. | n.a. | n.a. | n.a. | - | - | - | - | BP |
| 1 | 43 | 88 | F | 2 | N | Maternal | 7 | 8:01 | + | + | + | + | - | - | + | + | - | - | + | + | Leuk, DLL, MacroC, NBOs |
| 1 | 44 | 91 | M | 2 | N | n.a. | 35 | 36 | + | + | + | + | - | - | n.a. | n.a. | - | - | + | + | |
| 1 | 46 | 93 | M | 0 | Y | 14 | 18 | + | + | + | + | - | + | - | - | - | + | + | + | Thor, DS | |
| 1 | 48 | 99 | F | 0 | Y | 20:01 | 21:09 | + | + | + | + | + | + | - | - | - | - | + | + | ||
| 1 | 50 | 101 | M | 0 | Y | 4 | 6:06 | + | + | - | + | + | + | - | + | - | - | + | + | SS, NBOs | |
| 1 | 51 | 102 | F | 1 | N | Maternal | 5 | 7:08 | + | + | + | + | + | + | - | - | - | - | + | + | |
| 1 | 52 | 104 | F | 0 | Y | 13 | 14:02 | + | + | + | + | + | + | - | - | - | - | - | - | BvP, NA, LD, SP, CD | |
| 1 | 55 | 107 | M | 1 (twin) | Y | 9:02 | 14:04 | + | + | + | + | + | + | - | + | - | - | + | + | DLL, NBOs | |
| 1 | 58 | 112 | F | 0 | N | Maternal | 7 | 10 | + | + | + | + | + | + | n.a. | n.a. | - | - | + | + | mild ID |
| 1 | 63 | 125 | F | 0 | Y | 16:04 | 16:07 | + | + | + | + | + | + | - | - | - | - | + | + | DLL, NBOs, Thor, MacroC | |
| 1 | 65 | 128 | F | 0 | Y | 12 | 19:02 | + | + | + | + | + | + | - | - | - | - | + | + | LD, DS, NBOs, Thor | |
| 1 | 66 | 129 | M | 0 | Y | 7 | 13:03 | + | + | + | + | - | - | - | - | - | - | + | + | ||
| 1 | 67 | 130 | M | 0 | Y | 12 | 13:03 | + | + | + | + | n.a. | n.a. | - | - | - | - | + | + | ||
| 1 | 68 | 132 | M | 0 | Y | 7 | 13:06 | + | + | - | - | + | + | - | - | - | - | + | + | DLL, NBOs | |
| 1 | 72 | 137 | M | 0 | Y | 21 | 22:02 | + | + | + | + | + | + | - | - | + | + | - | - | ||
| 1 | 73 | 138 | F | 0 | Y | 17:03 | 19:05 | + | + | + | + | - | - | - | - | - | - | + | + | MacroC | |
| 1 | 74 | 139 | F | 0 | Y | 12:04 | 13:04 | + | + | + | + | + | + | - | - | - | - | + | + | NBOs, LD, SP | |
| 1 | 75 | 140 | M | 0 | N | Maternal | 6:05 | 8:02 | + | + | + | + | - | - | - | - | - | - | - | - | Thor, LD, NBOs |
| 1 | 76 | 141 | F | 0 | Y | 9:09 | 12:03 | + | + | + | + | + | + | - | - | - | - | + | + | DLL | |
| 1 | 78 | 149 | F | 0 | Y | 16 | 18 | + | + | n.a. | + | - | + | - | - | - | - | + | + | ||
| 1 | 79 | 151 | M | 1 | N | Maternal | 5:06 | 8:05 | + | + | + | + | - | + | - | + | - | - | + | + | NBOs, CIm, SP, DS, Cry, Noon |
| 1 | 80 | 155 | M | 1 | N | Paternal | 4:03 | 5:05 | + | + | - | + | - | - | - | - | - | - | - | - | NBOs, ID |
| 1 | 82 | 158 | M | 0 | N | Maternal | 4:11 | 5:11 | + | + | - | - | - | + | - | - | - | - | - | - | DLL |
| 1 | 83 | 160 | F | 0 | Y | 8 | 9:07 | + | + | + | + | - | + | - | - | - | - | + | + | NBOs, BvP | |
| 1 | 84 | 161 | F | 0 | Y | 23:01 | 25:11 | + | + | + | + | + | + | - | - | - | -- | + | + | NBOs | |
| 1 | 85 | 162 | F | 0 | N | Paternal | 1:11 | 3:01 | + | + | + | + | - | - | - | - | - | - | - | - | MacroC, UH |
| 1 | 87 | 171 | F | 0 | N | Paternal | 7 | 8:01 | + | + | + | + | - | + | - | - | - | - | + | + | DS, DE, VS, LD, SP, DLL, |
| 1 | 88 | 253 | F | 1 | N | Maternal | 7:06 | 8:06 | + | + | + | + | + | + | - | - | - | - | - | - | NBOs |
| 1 | 89 | 175 | F | 0 | Y | 20 | 21 | + | + | + | + | + | + | - | - | - | - | - | - | LD, NBOs | |
| 1 | 93 | 185 | M | 2 | N | Maternal | 7:06 | 11 | + | + | + | + | + | + | - | - | - | - | + | + | PmD, SP |
| 1 | 94 | 187 | M | 0 | N | Maternal | 15 | 18:04 | + | + | + | + | - | - | - | - | - | - | - | - | BvP |
| 1 | 98 | 204 | F | 0 | N | Paternal | 13:06 | 16:06 | + | + | + | + | + | + | - | + | - | - | + | + | DS |
| 1 | 102 | 218 | F | 1 | N | Maternal | 25 | 27 | + | + | + | + | - | - | - | - | - | - | - | - | Myo, MLyn |
| 1 | 103 | 220 | F | 0 | Y | 21 | 23 | + | + | - | - | + | + | - | - | - | - | + | + | SF | |
| 1 | 104 | 223 | F | 0 | Y | 15 | 16:05 | + | + | + | + | n.a. | n.a. | - | - | - | - | + | + | NBOs, BvP, MacroC | |
| 1 | 108 | 228 | M | 0 | Y | 0:05 | 3:05 | - | + | - | + | - | - | - | - | - | - | + | + | NA, MacroC, Thor, PmD | |
| 1 | 109 | 236 | M | 1 | N | Maternal | 12:07 | 13:01 | + | + | + | + | + | + | - | - | - | - | + | + | |
| 1 | 114 | 247 | M | 0 | Y | 18 | 21:08 | + | + | - | - | + | + | - | - | - | - | - | - | ||
| 1 | 116 | 250 | M | 0 | Y | 12:03 | 13:03 | + | + | + | + | - | - | - | - | - | - | + | + | ||
| 1 | 126 | 271 | M | 0 | N | Maternal | 4:05 | 5:03 | + | + | - | - | + | + | + | + | - | - | + | + | UH, NBOs, BvP, LD |
| 1 | 129 | 276 | M | 0 | Y | 6:04 | 7 | + | + | + | + | - | - | - | - | - | - | + | + | PmD, DLL, Epy | |
| 1 | 132 | 286 | M | 1 | N | Paternal | 11:09 | 12:01 | + | + | + | + | + | + | + | + | - | - | - | - | NBOs, SP, PmD |
| 1 | 133 | 287 | F | 1 | N | n.a. | 1 | 1:06 | + | + | + | + | + | + | - | - | - | - | - | - | Noon |
| 1 | 134 | 289 | M | 0 | Y | 10 | 10:08 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 136 | 291 | M | 0 | Y | 15:05 | 15:09 | + | + | + | + | n.a. | n.a. | - | - | - | - | + | + | LD, PmD, SD, NBOs, BvP, SF | |
| 1 | 137 | 309 | F | 0 | Y | 0:08 | 1:01 | + | + | - | - | - | - | - | - | - | - | + | + | ||
| 1 | 139 | 301 | M | 1 | N | n.a. | 30:06 | 31:02 | + | + | - | - | + | + | - | - | - | - | + | + | |
| 1 | 144 | 317 | F | 2 | N | Maternal | 30 | 30:03 | + | + | + | + | + | + | - | - | - | - | + | + | MacroC, Noon |
| 1 | 150 | 325 | M | 0 | Y | 11:11 | 13 | + | + | + | + | + | + | - | - | - | - | + | + | BSL, LD | |
| 1 | 151 | 326 | F | 0 | Y | 14:01 | 14:08 | + | + | + | + | + | + | + | + | - | - | - | - | MMS | |
| 1 | 152 | 328 | M | 0 | Y | 13 | 14:06 | + | + | + | + | + | + | + | + | - | - | - | - | ||
| 1 | 153 | 329 | M | 0 | Y | 19 | 19:6 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 154 | 331 | M | 0 | Y | 20 | 20:06 | + | + | + | + | + | + | - | - | - | - | + | + | ||
| 1 | 158 | 341 | F | 2 | N | Maternal | 15:03 | 15:09 | + | + | + | + | + | + | - | - | - | - | - | - | |
| 1 | 159 | 343 | M | 0 | Y | 14:11 | 15:05 | + | + | + | + | + | + | + | + | - | - | - | - | ||
| 1 | 160 | 385 | M | 0 | Y | 29:01 | 30 | + | + | - | - | + | + | - | - | - | - | - | - | PS, CD, MacroC, Noon | |
| 1 | 162 | 354 | F | 0 | Y | 42:06 | 43 | + | + | + | + | n.a. | n.a. | - | - | - | - | + | + | PS, SS, BvP | |
| 1 | 163 | 350 | F | 0 | Y | 49:01 | 50:08 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 164 | 384 | F | 1 | N | Paternal | 12:06 | 13:04 | + | + | + | + | + | + | - | - | - | - | - | - | NBOs, MMS |
| 1 | 165 | 52 | F | 0 | N | Paternal | 20 | 26 | + | + | + | + | + | + | - | - | - | - | + | + | |
| 1 | 166 | 28 | F | 0 | N | Maternal | 17:01 | 19 | + | + | + | + | + | + | - | - | - | - | + | + | HY |
| 1 | 169 | 334 | M | 0 | Y | 18 | 19 | + | + | + | + | + | + | - | - | - | - | + | + | Thor, MacroC | |
| 1 | 171 | 405 | M | 0 | Y | 5:05 | 6:01 | + | + | - | - | + | + | + | + | - | - | + | + | ||
| 1 | 172 | 395 | M | 0 | Y | 37:02 | 37:08 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 174 | 435 | F | 0 | Y | 60:02 | 60:09 | + | + | + | + | - | - | - | - | - | - | + | + | ||
| 1 | 176 | 463 | F | 0 | Y | 37 | 37:03 | + | + | + | + | + | + | - | - | - | - | + | + | MacroC | |
| 1 | 177 | 199 | M | 2 | N | Paternal | 10:03 | 13:09 | + | + | + | + | + | + | n.a. | n.a. | - | - | - | - | PmD, SP, Cry |
| 1 | 180 | 340 | M | 0 | Y | 13 | 15 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 181 | 450 | F | 0 | Y | 15:01 | 16:03 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 182 | 95 | M | 0 | Y | 20 | 21 | + | + | n.a. | n.a. | n.a. | n.a. | - | - | - | - | + | + | NBOs | |
| 1 | 183 | 146 | M | 0 | Y | Paternal | 15 | 17 | + | + | + | + | - | - | n.a. | n.a. | - | - | - | - | HY |
| 1 | 184 | 166 | M | 1 | N | Maternal | 24 | 31 | + | + | n.a. | n.a. | + | + | - | - | - | - | + | + | NBOs, LD, DLL, DS, PmD, ID, VS |
| 1 | 185 | 146 | M | 0 | Y | 69 | 76 | + | + | n.a. | n.a. | + | + | - | - | - | - | + | + | ||
| 1 | 186 | 189 | F | 0 | Y | 28 | 34 | + | + | + | + | + | + | - | - | - | - | + | + | NBOs, LD, SP, DS, PmD, ID, BvP, SS, Epy, AD | |
| 1 | 188 | 255 | M | 0 | Y | 41 | 42 | + | + | + | + | n.a. | n.a. | + | - | - | - | + | + | LGG, Malignancies, SS | |
| 1 | 190 | 367 | F | 1 | N | Maternal | 68 | 70 | + | + | + | + | n.a. | n.a. | - | - | - | - | + | + | DS |
| 1 | 192 | 425 | M | 0 | Y | 20 | 22 | + | + | + | + | + | + | + | + | - | - | + | + | NBOs, LD, DLL, DS, MacroC, HY | |
| 1 | 193 | 431 | M | 0 | Y | 31 | 36 | + | + | + | + | + | + | - | - | - | - | + | + | NBOs, LD, SP, DLL, PmD, Malignancies, HY | |
| 1 | 195 | 86 | F | 0 | Y | 3 | 20 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 1 | 209 | 345 | M | 0 | Y | 4 | 6 | + | + | + | + | + | + | - | - | - | - | - | - | ||
| 1 | 211 | 432 | M | 0 | Y | 33 | 33 | + | + | + | + | + | + | - | - | - | - | + | + | ||
| 1 | 212 | 442 | M | 0 | Y | 34 | 36:8 | + | + | + | + | n.a. | n.a. | n.a. | n.a. | - | - | - | - | Dlip | |
| 1 | 214 | 453 | F | 0 | Y | 13 | 14:5 | + | + | + | + | + | + | n.a. | n.a. | - | - | + | + | BvP | |
| 1 | 217 | 470 | F | 0 | Y | 20 | 21 | + | + | + | + | + | + | + | + | - | - | + | + | NBOs | |
| 1 | 218 | 471 | M | 0 | Y | 7 | 7:09 | + | + | - | - | - | - | + | + | - | - | + | + | NBOs, ID, SP, Noon | |
| 1 | 220 | 480 | F | 0 | Y | 19 | 19 | + | + | + | + | - | - | - | - | - | - | + | + | NBOS, DLL, DS, SS | |
| 1 | 221 | 481 | M | 0 | Y | 9 | 10:02 | + | + | + | + | - | - | n.a. | - | + | + | - | - | NBOs, ID, Bover | |
| 1 | 222 | 483 | M | 0 | N | 0:08 | 1:06 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | CD | |
| 1 | 223 | 491 | M | 0 | Y | 37 | 40 | + | + | + | + | - | - | - | - | - | - | + | + | DS, SF | |
| 1 | 225 | 495 | F | 0 | Y | 8 | 10:01 | + | + | + | + | - | - | - | - | - | - | - | - | DE, VS, Noon, SS, NBOs | |
| 1 | 227 | 500 | M | 0 | Y | 13 | 13 | + | + | + | + | + | + | + | + | - | - | + | + | ||
| 1 | 233 | 523 | F | 0 | Y | 21 | 21:05 | + | + | - | - | - | - | - | - | - | - | + | + | ||
| 1 | 234 | 524 | M | 1 | N | Paternal | 17 | 18:2 | + | + | + | + | - | - | - | - | - | - | + | + | Hyd, NA, Noon, Chem, SS |
| 1 | 241 | 548 | M | 0 | Y | 30 | 33 | + | + | + | + | - | - | - | - | - | - | + | + | NBOs, LD, DS, BP | |
| 1 | 247 | 567 | M | 0 | N | Maternal | 5 | 7 | + | + | + | + | - | - | + | + | - | - | + | + | NBOs |
| 1 | 251 | 575 | M | 0 | Y | 10 | 11 | + | + | + | + | - | - | + | + | - | - | + | + | NBOs | |
| 1 | 255 | 585 | M | 0 | Y | 12 | 13:03 | + | + | - | - | - | - | - | - | - | - | - | - | ID, NBOs | |
| 1 | 256 | 592 | M | 0 | Y | 19 | 20 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 1 | 259 | 602 | M | 0 | Y | 13 | 14:25 | + | + | + | + | - | - | - | - | + | + | + | + | AD | |
| 1 | 268 | 632 | M | 0 | Y | 15 | 17 | + | + | - | - | - | - | - | - | - | - | + | + | ||
| 1 | 276 | 646 | M | 0 | Y | 1 | 30:05 | + | + | - | - | - | - | + | + | - | - | - | - | ||
| 1 | 277 | 647 | M | 0 | Y | 46 | 48 | + | + | + | + | - | - | - | - | - | - | + | + | DLL, DS | |
| 1 | 285 | 657 | M | 0 | Y | 51 | 54 | + | + | - | - | - | - | - | - | - | - | + | + | HY, SF | |
| 1 | 288 | 665 | M | 0 | Y | 5 | 6 | + | + | + | + | - | - | n.a. | n.a. | - | - | + | + | DD, SP | |
| 1 | 289 | 667 | F | 0 | Y | 55 | 58 | + | + | + | + | + | + | - | - | - | - | + | + | HY, Malignancies | |
| 1 | 292 | 670 | M | 0 | Y | 11 | 11:04 | + | + | + | + | n.a. | n.a. | - | - | - | - | + | + | NBOs, SP, LD | |
| 1 | 300 | 681 | M | 0 | Y | 45 | 47 | + | + | + | + | + | + | - | - | - | - | + | + | NBOs, DS | |
| 2 | 36 | 70 | M | 0 | N | Maternal | 10 | 12:11 | + | + | + | + | + | + | + | + | - | - | + | + | RMS, NBOs, MacroC, DS, VS |
| 2 | 61 | 118 | M | 0 | Y | 2:01 | 3:11 | + | + | - | + | - | - | - | - | - | - | + | + | ID, BP, MacroC, UH, Noon, Thor, | |
| 2 | 64 | 127 | M | 0 | Y | 14:06 | 16:02 | + | + | + | + | + | + | - | - | - | - | + | + | ID, Thor | |
| 2 | 69 | 134 | M | 0 | Y | 16 | 17:02 | + | + | + | + | + | + | - | - | - | - | + | + | Thor, NBOs, DLL, ID | |
| 2 | 77 | 147 | M | 0 | Y | 4:09 | 6:06 | + | + | - | - | - | - | - | - | - | - | + | + | ID, BvP, NBOs, Thor, | |
| 2 | 99 | 205 | F | 0 | Y | 29 | 29:08 | + | + | + | + | + | + | - | - | - | - | - | - | ID, NBOs, MPNST | |
| 2 | 107 | 227 | M | 0 | Y | 5:03 | 7 | + | + | + | + | + | + | - | - | - | - | - | - | ID, SP, Noon, NA | |
| 2 | 119 | 259 | M | 0 | Y | 11:02 | 13 | + | + | + | + | + | + | - | - | - | - | + | + | DE, DS, VS, ID, SP, LD, Noon, macroC, NBOs, DLL, BSL | |
| 2 | 127 | 272 | M | 0 | Y | 15 | 15:09 | + | + | + | + | - | - | - | - | - | - | + | + | ID, PS, Epy | |
| 2 | 149 | 324 | M | 0 | Y | 14 | 16 | + | + | + | + | - | - | + | + | - | - | + | + | ID | |
| 2 | 168 | 97 | M | 0 | N | Paternal | 20 | 23 | + | + | + | + | + | + | + | + | - | - | + | + | ID |
| 3 | 37 | 71 | M | 0 | Y | 9 | 12:11 | + | + | + | + | - | - | - | - | - | - | - | - | Leuk | |
| 3 | 42 | 83 | M | 0 | Y | 6 | 8:09 | + | + | - | - | - | + | - | - | - | - | - | - | ||
| 3 | 45 | 92 | F | 0 | Y | 9:02 | 14:08 | + | + | - | + | - | - | - | - | - | - | - | + | Thor, DS, DLL | |
| 3 | 47 | 96 | F | 0 | Y | 4:11 | 5:11 | + | + | + | + | - | + | - | - | - | - | - | - | ||
| 3 | 49 | 100 | M | 0 | Y | 1:04 | 3:01 | + | + | - | + | - | - | - | - | - | - | - | + | BP, XG | |
| 3 | 53 | 105 | M | 0 | Y | 8 | 10 | + | + | - | + | - | - | - | - | - | - | - | - | LD | |
| 3 | 57 | 110 | F | 0 | Y | 3:09 | 4:03 | + | + | - | + | - | - | - | - | - | - | - | - | ||
| 3 | 81 | 157 | M | 0 | Y | 2:01 | 4:11 | + | + | - | + | - | - | - | - | - | - | - | - | SP, Cry, NA, Thor, Noon | |
| 3 | 91 | 180 | M | 0 | Y | 3:05 | 4:07 | + | + | - | - | - | - | - | - | - | - | - | - | DD, MacroC, SP | |
| 3 | 97 | 203 | F | 0 | Y | 3 | 7:2 | + | + | - | + | - | - | - | - | - | - | - | + | ||
| 3 | 110 | 238 | F | 0 | Y | 1:01 | 4:02 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 3 | 113 | 245 | F | 0 | Y | 9:01 | 12:02 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 3 | 115 | 249 | M | 0 | Y | 9 | 9:02 | + | + | + | + | - | - | - | - | - | - | - | - | DLL | |
| 3 | 118 | 257 | M | 0 | Y | 5:08 | 8 | + | + | - | + | - | - | - | - | - | - | - | - | XG | |
| 3 | 120 | 260 | F | 0 | Y | 8:08 | 12 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 3 | 121 | 261 | F | 0 | Y | 1:09 | 3:05 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 3 | 125 | 268 | F | 0 | Y | 3:05 | 3:06 | + | + | + | + | - | - | - | - | - | - | - | - | UH, XG | |
| 3 | 128 | 273 | M | 0 | Y | 8:08 | 10:04 | + | + | - | - | - | - | - | - | - | - | - | - | ID, UH, Thor | |
| 3 | 131 | 282 | F | 0 | Y | 4 | 6 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | NA | |
| 3 | 135 | 290 | F | 0 | Y | 9:03 | 10:01 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 3 | 138 | 293 | M | 0 | Y | 5:08 | 5:01 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 3 | 142 | 314 | M | 0 | Y | 8:06 | 10 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 3 | 145 | 321 | F | 0 | Y | 9:01 | 10:01 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 3 | 146 | 322 | F | 0 | Y | 6 | 7 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 3 | 170 | 145 | M | 0 | Y | 3:05 | 6:02 | + | + | + | + | - | - | - | - | - | - | - | - | MacroC, NBOs | |
| 3 | 175 | 358 | F | 0 | Y | 2:08 | 4:04 | + | + | - | + | - | - | - | - | - | - | - | - | SS, SP | |
| 3 | 202 | 374 | M | 0 | Y | 8 | 9 | + | + | - | - | - | - | - | - | - | - | - | - | Thor | |
| 3 | 215 | 464 | F | 0 | Y | 2 | 3:03 | + | + | - | - | - | - | - | - | - | - | - | - | SP, CD | |
| 3 | 224 | 494 | M | 0 | Y | 9 | 12 | + | + | - | - | - | - | - | - | - | - | - | - | LD, BvP | |
| 3 | 228 | 503 | M | 0 | Y | 2 | 3:08 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 3 | 230 | 507 | M | 0 | Y | 7 | 8:03 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 3 | 231 | 508 | F | 0 | Y | 1 | 1:03 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 3 | 239 | 536 | M | 0 | Y | 5 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | MacroC | ||
| 3 | 244 | 555 | M | 0 | Y | 7 | 8:01 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 3 | 252 | 576 | M | 0 | Y | 3 | 5 | + | + | + | + | - | - | - | + | - | - | - | + | ID, SP, NBOs, CIm, MBG, secondary Epy, DE, VS | |
| 3 | 253 | 579 | M | 0 | Y | 7 | 9 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | SP | |
| 3 | 275 | 644 | M | 0 | Y | 6 | 7:02 | + | + | + | + | - | - | - | - | - | - | - | + | SP, DD | |
| 3 | 278 | 649 | M | 0 | Y | 3 | 3:08 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | NA | |
| 3 | 279 | 650 | M | 0 | Y | 2 | 3:25 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | AtC | |
| 3 | 282 | 653 | M | 0 | Y | 8 | 8:06 | + | + | + | + | - | - | n.a. | n.a. | - | - | - | - | ||
| 3 | 294 | 672 | F | 0 | Y | 2 | 2:10 | + | + | - | + | - | - | n.a. | n.a. | - | - | - | - | CD | |
| 3 | 297 | 677 | F | 0 | Y | 7 | 8 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | AtC | |
| 3 | 299 | 680 | M | 0 | Y | 6 | 8 | + | + | - | - | - | + | - | - | - | - | - | - | ID | |
| 3 | 302 | 683 | M | 0 | Y | 1 | 2 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 4 | 1 | 3 | M | 1 (twin) | Y | 12 | 14 | + | + | + | + | n.a. | n.a. | - | - | - | - | - | - | LD | |
| 4 | 5 | 7 | M | 0 | Y | 13:03 | 17 | + | + | - | - | - | + | - | - | - | - | - | - | Thor | |
| 4 | 13 | 20 | M | 0 | Y | 10:07 | 12:08 | + | + | - | - | - | + | - | - | - | - | - | - | ArC, NBOs, BP, Noon, Thor, SP, MMS | |
| 4 | 41 | 82 | F | 0 | Y | 16 | 18 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 4 | 54 | 106 | M | 0 | Y | 12:03 | 13:01 | + | + | + | + | - | - | - | - | - | - | - | - | UH, NBOs | |
| 4 | 56 | 109 | M | 0 | Y | 15:04 | 16:02 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 4 | 59 | 113 | F | 0 | Y | 11:03 | 13:05 | + | + | - | + | - | - | - | - | - | - | - | - | DS, SS | |
| 4 | 70 | 135 | M | 0 | Y | 17:06 | 18 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | SS, BvP | |
| 4 | 71 | 136 | M | 0 | Y | 15:07 | 17:05 | + | + | - | - | - | - | - | - | - | - | - | - | ID, SP, DLL | |
| 4 | 86 | 163 | F | 0 | Y | 14:02 | 17:01 | + | + | - | - | - | - | - | - | - | - | - | - | LD | |
| 4 | 95 | 198 | F | 1 | Y | 13:05 | 15:03 | + | + | - | + | - | - | - | - | - | - | - | + | ||
| 4 | 96 | 202 | F | 0 | Y | 14:01 | 15 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 4 | 106 | 225 | M | 0 | Y | 10:06 | 12:09 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 4 | 117 | 251 | M | 0 | Y | 13:04 | 14:04 | + | + | - | - | - | - | - | - | - | - | - | - | Myo, AtIN | |
| 4 | 123 | 264 | M | 0 | Y | 11 | 14:07 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 4 | 130 | 278 | M | 0 | Y | 16:03 | 18:01 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 4 | 140 | 306 | F | 0 | Y | 16:04 | 17 | + | + | + | + | - | - | - | - | - | - | - | - | DS, BvP, DLL | |
| 4 | 143 | 315 | F | 0 | Y | 14:01 | 14:08 | + | + | + | + | - | + | - | - | - | - | - | - | ||
| 4 | 155 | 339 | F | 0 | Y | 11:09 | 12:04 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | MacroC, LD | |
| 4 | 156 | 195 | F | 0 | Y | 11:03 | 13:09 | + | + | - | - | - | - | - | - | - | - | - | - | ||
| 4 | 157 | 196 | F | 0 | Y | 11:09 | 14:08 | + | + | - | + | - | - | - | - | - | - | - | - | ||
| 4 | 173 | 404 | F | 0 | Y | 15:01 | 15:07 | + | + | + | + | - | - | - | - | - | - | - | - | SS, MacroC | |
| 4 | 196 | 87 | F | 0 | Y | 14 | 11 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | OD | |
| 4 | 201 | 265 | M | 0 | Y | 13 | 14 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 4 | 237 | 532 | M | 0 | Y | 13 | 14 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 4 | 245 | 557 | F | 0 | Y | 10 | 11:05 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | NA | |
| 4 | 260 | 608 | F | 0 | Y | 34 | 36 | + | + | + | + | - | - | - | - | - | - | - | - | ||
| 4 | 269 | 635 | M | 0 | Y | 14 | 15 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 4 | 280 | 651 | F | 0 | Y | 14 | 15 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 4 | 281 | 652 | M | 0 | Y | 10 | 12 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | ||
| 4 | 296 | 675 | F | 0 | Y | 29 | 29 | + | + | - | - | n.a. | n.a. | - | - | - | - | - | - | ||
| 5 | 11 | 17 | M | 9 | N | Paternal | 12 | 15:08 | + | + | - | - | - | - | - | - | - | - | - | - | Cry |
| 5 | 32 | 170 | M | 3 | N | Maternal | 12:09 | 12:11 | + | + | + | + | - | - | - | - | - | - | - | - | |
| 5 | 40 | 79 | F | 1 | N | Maternal | 18 | 19:01 | + | + | + | + | - | - | - | - | - | - | - | - | |
| 5 | 62 | 121 | F | 0 | N | Paternal | 8:05 | 12 | + | + | + | + | - | - | - | - | - | - | - | - | |
| 5 | 92 | 183 | F | 2 | N | Paternal | 5 | 6:02 | + | + | - | - | - | - | - | - | - | - | - | - | NA |
| 5 | 100 | 212 | M | 0 | N | Paternal | 15:08 | 17 | + | + | - | - | - | - | - | - | - | - | - | - | Thor |
| 5 | 101 | 216 | F | 1 | N | Maternal | 7:06 | 8:02 | + | + | + | + | - | - | - | - | - | - | - | - | LD |
| 5 | 112 | 243 | F | 4 | N | Paternal | 10:01 | 10:06 | + | + | - | + | - | - | - | - | - | - | - | - | Thor |
| 5 | 124 | 269 | M | 2 | N | Maternal | 3:08 | 5:06 | + | + | - | - | - | - | - | - | - | - | - | - | UH |
| 5 | 147 | 323 | F | 2 | N | Paternal | 3:05 | 4:03 | + | + | - | - | - | - | - | - | - | - | - | - | PS, Noon, IH |
| 5 | 161 | 391 | F | 1 | N | Paternal | 10:01 | 10:04 | + | + | - | - | - | - | - | - | - | - | - | - | |
| 5 | 167 | 416 | F | 3 | N | Paternal | 0:07 | 1:01 | + | + | - | - | - | - | - | - | - | - | - | - | |
| 5 | 178 | 492 | M | 1 | N | Paternal | 5 | 5:03 | + | + | - | - | - | - | - | - | - | - | - | - | MacroC, Noon |
| 5 | 179 | 362 | F | 2 | N | Paternal | 8:01 | 9:01 | + | + | + | + | - | - | - | - | - | - | - | - | IPP |
| 5 | 207 | 335 | F | 0 | N | Paternal | 9 | 10 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | |
| 5 | 216 | 638 | M | 2 | N | Paternal | 1 | 4 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | Chem |
| 5 | 229 | 504 | F | 3 | N | Paternal | 9 | 11:09 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | SS |
| 5 | 261 | 609 | F | 0 | N | Maternal | 32 | 34 | + | + | + | + | - | - | n.a. | n.a. | - | - | - | - | Noon |
| 5 | 262 | F | 0 | N | Maternal | 28 | 29 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | Noon, MacroC | |
| 5 | 286 | 658 | M | 1 | N | Maternal | 7 | 7:05 | + | + | - | - | - | - | n.a. | n.a. | - | - | - | - | AD |
| Family ID 1 | Gene | Exon | Type 2 | Genomic | cDNA | Effect | Protein | ClinVar/HGMD/LOVD ID 3 |
|---|---|---|---|---|---|---|---|---|
| 240 (1) | KIT | 14 | DEL | 2027del | 2027del | Frame-shift | Gly676Valfs*4 | New |
| 298 (1) | LZTR1 | 4 | SNV | 353G>A | 353G>A | Missense | Arg118His | LOVD: LZTR1_000051 |
| 232 (1) | NF2 | 5 | DEL | 465del | 465del | Frame-shift | Ser156Valfs*18 | ClinVar: 547705 |
| 238 (1) | 13 | SNV | 1396C>T | 1396C>T | Nonsense | Arg466* | ClinVar: 3295 | |
| 226 (1) | 2–6 | DUP | (114+1_115-1)_(599+1_600-1)dup (intragenic duplication ex. 2-6) | n.a. | Intragenic duplication | ? | New | |
| 263 (1) | PPP1CB | 3 | SNV | 146C>G | 146C>G | Missense | Pro49Arg | ClinVar: 254648 |
| 258 (2) | PTEN | 7 | INS | 778_779insG | 778_779insG | Frame-shift | Lys260Argfs*38 | New |
| 287 (1) | PTPN11 | 3 | SNV | 235C>A | 235C>A | Missense | Gln79Lys | ClinVar: 44605 |
| 242 (1) | 8 | SNV | 923A>G | 923A>G | Missense | Asn308Ser | ClinVar: 13327 | |
| 191 (1) | 12 | SNV | 1403C>T | 1403C>T | Missense | Thr468Met | ClinVar: 13331 | |
| 204 (1) | ||||||||
| 257 (1) | ||||||||
| 272 (1) | ||||||||
| 236 (1) | 13 | SNV | 1492C>T | 1492C>T | Missense | Arg498Trp | ClinVar: 40553 | |
| 301 (1) | SNV | 1528C>G | 1528C>G | Missense | Gln510Glu | ClinVar: 40566 | ||
| 270 (1) | SOS1 | 4 | SNV | 429G>T | 429G>T | Missense | Lys143Asn | New |
| 187 (2) | 10 | SNV | 1642A>C | 1642A>C | Missense | Ser548Arg | LOVD: SOS1_000142 | |
| 189 (2) | TSC1 | 13 | DEL | 1326_1327del | 1326_1327del | Frame-shift | Gly443Ilefs*15 | ClinVar: 421669 |
| 265 (1) | 18 | SNV | 2293C>T | 2293C>T | Nonsense | Gln765* | ClinVar: 48934 | |
| 274 (1) | 19 | INS | 2421dup | 2421dup | Frame-shift | Ala808Cysfs*18 | LOVD: TSC1_00419 | |
| 305 (1) | TSC2 | 1–16 | DEL | (-30+1_-29-1)_(1716+1_1717-1)del (intragenic deletion ex. 1-16) | n.a. | Intragenic deletion | ? | New |
| 267 (1) | 1–22 | DEL | c.(-30+1_-29-1)_(2545+1_2546-1)del (intragenic deletion ex. 1-22) | n.a. | Intragenic deletion | ? | New | |
| 295 (1) | 7 | SNV | 648+1G>T | n.a. | Splicing | ? | LOVD: TSC2_03572 | |
| 303 (1) | 37 | DEL | c.(4662+1_4663-1)_(4849+1_4850-1)del (intragenic deletion ex. 37) | n.a. | Intragenic deletion | ? | LOVD: TSC2_03465 | |
| 248 (1) | 41 | DEL | 5238_5255del | 5238_5255del | In-frame deletion | His1746_Arg1751del | LOVD: TSC2_00149 |
References
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.-P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Denayer, E.; de Ravel, T.; Legius, E. Clinical and molecular aspects of RAS related disorders. J. Med. Genet. 2008, 45, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Gelb, B.D. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: Phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 2010, 1214, 99–121. [Google Scholar] [CrossRef] [PubMed]
- Tidyman, W.E.; Rauen, K.A. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, V.C.; Lucas, J.; Babcock, M.A.; Gutmann, D.H.; Korf, B.; Maria, B.L. Neurofibromatosis Type 1 Revisited. Pediatrics 2009, 123, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferner, R.E. Neurofibromatosis 1. Eur. J. Hum. Genet. 2006, 15, 131–138. [Google Scholar] [CrossRef]
- Shen, M.H.; Harper, P.S.; Upadhyaya, M. Molecular genetics of neurofibromatosis type 1 (NF1). J. Med. Genet. 1996, 33, 2–17. [Google Scholar] [CrossRef]
- Li, Y.; O’Connell, P.; Breidenbach, H.H.; Cawthon, R.; Stevens, J.; Xu, G.; Neil, S.; Robertson, M.; White, R.; Viskochil, D. Genomic organization of the neurofibromatosis 1 gene (NF1). Genomics 1995, 25, 9–18. [Google Scholar] [CrossRef]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Ferner, R.E. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol. 2007, 6, 340–351. [Google Scholar] [CrossRef]
- National Institutes of Health Consensus Development, C. Neurofibromatosis: Conference Statement. Arch. Neurol. 1988, 45, 575–578. [Google Scholar] [CrossRef]
- Santoro, C.; Pacileo, G.; Limongelli, G.; Scianguetta, S.; Giugliano, T.; Piluso, G.; Ragione, F.D.; Cirillo, M.; Mirone, G.; Perrotta, S. LEOPARD syndrome: Clinical dilemmas in differential diagnosis of RASopathies. BMC Med. Genet. 2014, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970–2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Maietta, A.; Giugliano, T.; Melis, D.; Perrotta, S.; Nigro, V.; Piluso, G. Arg(1809) substitution in neurofibromin: Further evidence of a genotype-phenotype correlation in neurofibromatosis type 1. Eur. J. Hum. Genet. 2015, 23, 1460–1461. [Google Scholar] [CrossRef] [PubMed]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef] [PubMed]
- Wakioka, T.; Sasaki, A.; Kato, R.; Shouda, T.; Matsumoto, A.; Miyoshi, K.; Tsuneoka, M.; Komiya, S.; Baron, R.; Yoshimura, A. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001, 412, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Stowe, I.B.; Mercado, E.L.; Stowe, T.R.; Bell, E.L.; Oses-Prieto, J.A.; Hernández, H.; Burlingame, A.L.; McCormick, F. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012, 26, 1421–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, Y.; Brems, H.; Suzuki, M.; Kanamori, M.; Okada, M.; Morita, R.; Llano-Rivas, I.; Ose, T.; Messiaen, L.; Legius, E.; et al. Interaction between a domain of a negative regulator of the RAS-ERK pathway, SPRED1, and the GTPase-Activating Protein-Related Domain of neurofibromin is implicated in Legius Syndrome and Neurofibromatosis Type 1. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, L.; Yao, S.; Brems, H.; Callens, T.; Sathienkijkanchai, A.; Denayer, E.; Spencer, E.; Arn, P.; Babovic-Vuksanovic, D.; Bay, C.; et al. Clinical and Mutational Spectrum of Neurofibromatosis Type 1-like Syndrome. JAMA 2009, 302, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, G.; Bennett, E.; Chuzhanova, N.; Thomas, N.; Jim, H.P.; Side, L.; Davies, S.; Haan, E.; Kerr, B.; Huson, S.M.; et al. SPRED1 mutations (Legius syndrome): Another clinically useful genotype for dissecting the neurofibromatosis type 1 phenotype. J. Med. Genet. 2009, 46, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Suerink, M.; Ripperger, T.; Messiaen, L.; Menko, F.H.; Bourdeaut, F.; Colas, C.; Jongmans, M.; Goldberg, Y.; Nielsen, M.; Muleris, M.; et al. Constitutional mismatch repair deficiency as a differential diagnosis of neurofibromatosis type 1: Consensus guidelines for testing a child without malignancy. J. Med. Genet. 2019, 56, 53–62. [Google Scholar] [CrossRef]
- Ars, E.; Serra, E.; GarcÃ-a, J.; Kruyer, H.; Gaona, A.; Lázaro, C.; Estivill, X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum. Mol. Genet. 2000, 9, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Eckart, M.; Rehder, H.; Fonatsch, C. Illegitimate splicing of the NF1 gene in healthy individuals mimics mutation-induced splicing alterations in NF1 patients. Hum. Genet. 2000, 106, 311–313. [Google Scholar]
- Luijten, M.; Wang, Y.; Smith, B.T.; Westerveld, A.; Smink, L.J.; Dunham, I.; Roe, B.A.; Hulsebos, T.J. Mechanism of spreading of the highly related neurofibromatosis type 1 (NF1) pseudogenes on chromosomes 2, 14 and 22. Eur. J. Hum. Genet. 2000, 8, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Mutarelli, M.; Marwah, V.; Rispoli, R.; Carrella, D.; Dharmalingam, G.; Oliva, G.; di Bernardo, D. A community-based resource for automatic exome variant-calling and annotation in Mendelian disorders. BMC Genom. 2014, 15 (Suppl. S3), S5. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Klimke, W.; Maglott, D.R. NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res. 2009, 37, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protocols 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluwe, L.; Siebert, R.; Gesk, S.; Friedrich, R.E.; Tinschert, S.; Kehrer-Sawatzki, H.; Mautner, V.F. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum. Mutat. 2004, 23, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Yao, S.; Claes, K.; Kehrer-Sawatzki, H.; Tinschert, S.; De Raedt, T.; Legius, E.; Callens, T.; Beiglböck, H.; Maertens, O.; et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer 2006, 45, 265–276. [Google Scholar] [CrossRef]
- Bandipalliam, P. Syndrome of early onset colon cancers, hematologic malignancies & features of neurofibromatosis in HNPCC families with homozygous mismatch repair gene mutations. Fam. Cancer 2005, 4, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Charest, D.L.; Mordret, G.; Harder, K.W.; Jirik, F.; Pelech, S.L. Molecular cloning, expression, and characterization of the human mitogen-activated protein kinase p44erk1. Mol. Cell. Biol. 1993, 13, 4679–4690. [Google Scholar] [CrossRef] [PubMed]
- Tsipi, M.; Poulou, M.; Fylaktou, I.; Kosma, K.; Tsoutsou, E.; Pons, M.R.; Kokkinou, E.; Kitsiou-Tzeli, S.; Fryssira, H.; Tzetis, M. Phenotypic expression of a spectrum of Neurofibromatosis Type 1 (NF1) mutations identified through NGS and MLPA. J. Neurol. Sci. 2018, 395, 95–105. [Google Scholar] [CrossRef]
- Balla, B.; Arvai, K.; Horvath, P.; Tobias, B.; Takacs, I.; Nagy, Z.; Dank, M.; Fekete, G.; Kosa, J.P.; Lakatos, P. Fast and robust next-generation sequencing technique using ion torrent personal genome machine for the screening of neurofibromatosis type 1 (NF1) gene. J. Mol. Neurosci.: MN 2014, 53, 204–210. [Google Scholar] [CrossRef]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef]
- Griffiths, S.; Thompson, P.; Frayling, I.; Upadhyaya, M. Molecular diagnosis of neurofibromatosis type 1: 2 years experience. Fam. Cancer 2007, 6, 21–34. [Google Scholar] [CrossRef]
- Valero, M.C.; Martin, Y.; Hernandez-Imaz, E.; Marina Hernandez, A.; Melean, G.; Valero, A.M.; Javier Rodriguez-Alvarez, F.; Telleria, D.; Hernandez-Chico, C. A highly sensitive genetic protocol to detect NF1 mutations. J. Mol. Diagn.: JMD 2011, 13, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xiong, H.; Han, Y.; Li, C.; Mai, S.; Huang, Z.; Ai, X.; Guo, Z.; Zeng, F.; Guo, Q. Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2018, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Bottillo, I.; Sarkozy, A.; Carta, C.; Neri, C.; Bellacchio, E.; Schirinzi, A.; Conti, E.; Zampino, G.; Battaglia, A.; et al. NF1 Gene Mutations Represent the Major Molecular Event Underlying Neurofibromatosis-Noonan Syndrome. Am. J. Hum. Genet. 2005, 77, 1092–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, C.; Di Rocco, F.; Kossorotoff, M.; Zerah, M.; Boddaert, N.; Calmon, R.; Vidaud, D.; Cirillo, M.; Cinalli, G.; Mirone, G.; et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am. J. Med. Genet. Part A 2017, 173, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Giugliano, T.; Kraemer, M.; Torella, A.; Schwitalla, J.C.; Cirillo, M.; Melis, D.; Berlit, P.; Nigro, V.; Perrotta, S.; et al. Whole exome sequencing identifies MRVI1 as a susceptibility gene for moyamoya syndrome in neurofibromatosis type 1. PLoS ONE 2018, 13, e0200446. [Google Scholar] [CrossRef] [PubMed]
- De Raedt, T.; Brems, H.; Wolkenstein, P.; Vidaud, D.; Pilotti, S.; Perrone, F.; Mautner, V.; Frahm, S.; Sciot, R.; Legius, E. Elevated risk for MPNST in NF1 microdeletion patients. Am. J. Hum. Genet. 2003, 72, 1288–1292. [Google Scholar] [CrossRef]
- D’Amico, A.; Mazio, F.; Ugga, L.; Cuocolo, R.; Cirillo, M.; Santoro, C.; Perrotta, S.; Melis, D.; Brunetti, A. Medullary unidentified bright objects in Neurofibromatosis type 1: A case series. BMC Pediatr. 2018, 18, 91. [Google Scholar] [CrossRef]
- Wang, H.F.; Shih, Y.T.; Chen, C.Y.; Chao, H.W.; Lee, M.J.; Hsueh, Y.P. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Investig. 2011, 121, 4820–4837. [Google Scholar] [CrossRef]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Opitz, J.M.; Weaver, D.D. The neurofibromatosis-Noonan syndrome. Am. J. Med. Genet. 1985, 21, 477–490. [Google Scholar] [CrossRef]
- Allanson, J.E.; Hall, J.G.; Van Allen, M.I. Noonan phenotype associated with neurofibromatosis. Am. J. Med. 1985, 21, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Bertola, D.R.; Pereira, A.C.; Passetti, F.; de Oliveira, P.S.; Messiaen, L.; Gelb, B.D.; Kim, C.A.; Krieger, J.E. Neurofibromatosis-Noonan syndrome: Molecular evidence of the concurrence of both disorders in a patient. Am. J. Med. Genet. Part A 2005, 136, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Wilken, M.; Zenker, M.; Sticht, H.; Fahsold, R.; Gusek-Schneider, G.C.; Rauch, A. Independent NF1 and PTPN11 mutations in a family with neurofibromatosis-Noonan syndrome. Am. J. Med. Genet. Part A 2009, 149A, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Giugliano, T.; Melone, M.A.B.; Cirillo, M.; Schettino, C.; Bernardo, P.; Cirillo, G.; Perrotta, S.; Piluso, G. Multiple spinal nerve enlargement and SOS1 mutation: Further evidence of overlap between neurofibromatosis type 1 and Noonan phenotype. Clin. Genet. 2018, 93, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Trevisson, E.; Morbidoni, V.; Forzan, M.; Daolio, C.; Fumini, V.; Parrozzani, R.; Cassina, M.; Midena, E.; Salviati, L.; Clementi, M. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol. Genet. Genom. Med. 2019, 7, e616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet. Med.: Off. J. Am. Coll. Med. Genet. 2019, 21, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [CrossRef]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef]
| Group | Criteria for Molecular Testing | Number of Selected Patients | Number of Mutated Patients | Mutation Detection Rate (%) | Number of Patients without Molecular Diagnosis (%) |
|---|---|---|---|---|---|
| 1 | Clinical diagnosis of NF1 (test requested by parents or milder phenotype) | 139 | 136 (NF1 = 136; SPRED1 = 0; OTHER = 0) | 97.8% (NF1 = 97.8%; SPRED1 = 0.0%; OTHER = 0.0%) | 3 (2.2%) |
| 2 | Severe NF1 phenotype with suspicion of 17q11.2 microdeletion | 11 | 11 (NF1 = 11; SPRED1 = 0; OTHER = 0) | 100.0% (NF1 = 100.0%; SPRED1 = 0.0%; OTHER = 0.0%) | 0 (0.0%) |
| 3 | Isolated CALMs in patients without affected first-degree relatives and age ≤ 9 y | 44 | 29 (NF1 = 28; SPRED1 = 1; OTHER = 0) | 65.9% (NF1 = 63.6%; SPRED1 = 2.3%; OTHER = 0.0%) | 15 (34.1%) |
| 4 | Isolated CALMs in patients without affected first-degree relatives and age ≥ 10 y | 31 | 20 (NF1 = 19; SPRED1 = 1; OTHER = 0) | 64.5% (NF1 = 61.3%; SPRED1 = 3.2%; OTHER = 0.0%) | 11 (35.5%) |
| 5 | Isolated CALMs in patients and at least one affected first-degree relative | 20 | 17 (NF1 = 11; SPRED1 = 6; OTHER = 0) | 85.0% (NF1 = 55.0%; SPRED1 = 30.0%; OTHER = 0.0%) | 3 (15.0%) |
| 6 | Other RASopathies or neurocutaneous disorders | 36 | 26 (NF1 = 1; SPRED1 = 0; OTHER = 25) | 72.2% (NF1 = 2.7%; SPRED1 = 0.0%; OTHER = 69.4%) | 10 (27.8%) |
| Total | 281 | 239 (NF1 = 206; SPRED1 = 8; OTHER = 25) | 85.0% (NF1 = 73.3%; SPRED1 = 2.8%; OTHER = 8.9%) | 42 (15.0%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giugliano, T.; Santoro, C.; Torella, A.; Del Vecchio Blanco, F.; Grandone, A.; Onore, M.E.; Melone, M.A.B.; Straccia, G.; Melis, D.; Piccolo, V.; et al. Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders. Genes 2019, 10, 580. https://doi.org/10.3390/genes10080580
Giugliano T, Santoro C, Torella A, Del Vecchio Blanco F, Grandone A, Onore ME, Melone MAB, Straccia G, Melis D, Piccolo V, et al. Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders. Genes. 2019; 10(8):580. https://doi.org/10.3390/genes10080580
Chicago/Turabian StyleGiugliano, Teresa, Claudia Santoro, Annalaura Torella, Francesca Del Vecchio Blanco, Anna Grandone, Maria Elena Onore, Mariarosa Anna Beatrice Melone, Giulia Straccia, Daniela Melis, Vincenzo Piccolo, and et al. 2019. "Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders" Genes 10, no. 8: 580. https://doi.org/10.3390/genes10080580