
Regulation of the Adaptive Immune Response by the IκB Family Protein Bcl-3

{kind=link}
Abstract
:1. NF-κB
2. Bcl-3
3. The Adaptive Immune Response
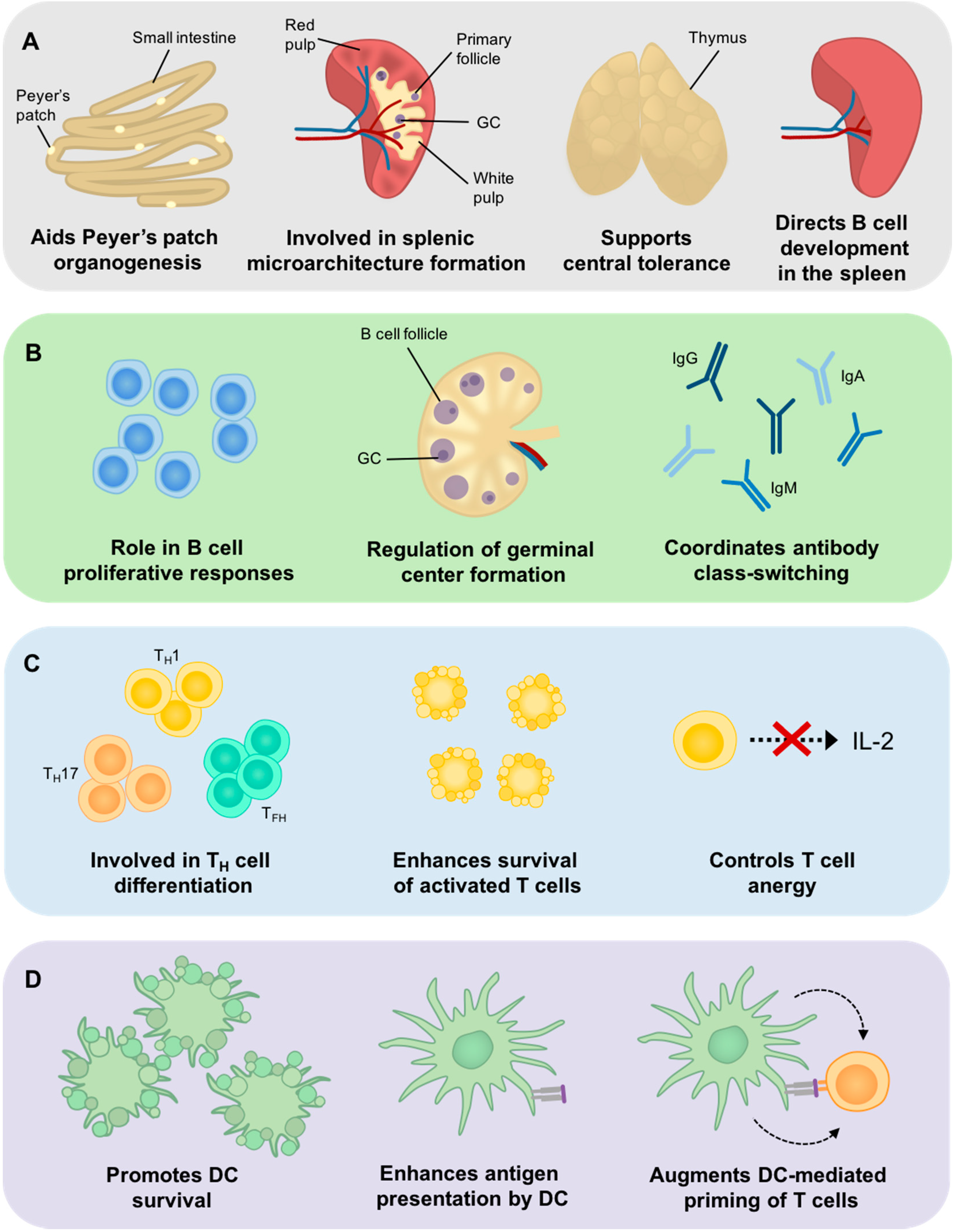
4. Bcl-3 Involvement in Central Tolerance
5. The Role of Bcl-3 in SLO Development
6. The Role of Bcl-3 in B Cell Development and Function
7. The Role of Bcl-3 in T Cells
8. The Role of Bcl-3 in DC Development and Function
9. Bcl-3 and Immunopathology
10. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| NF-κB | Nuclear factor κB |
| BAFF | B-cell activating factor |
| IκB | inhibitor of κB |
| Ig | immunoglobulin |
| TNF | tumor necrosis factor |
| PTM | post-translational modification |
| Ags | antigens |
| TCR | T cell receptor |
| BCR | B cell receptor |
| Tregs | regulatory T cells |
| FO | follicular |
| MZ | marginal zone |
| SLO | secondary lymphoid organ |
| LN | lymph node |
| MHC | major histocompatibility complex |
| DCs | dendritic cells |
| Ab | antibody |
| ASCs | antibody-secreting cells |
| TH cell | helper T cell |
| TFH cell | follicular T helper cell |
| GC | germinal centre |
| mTECs | medullary thymic epithelial cells |
| SED | sub-epithelial dome |
| FDCs | follicular dendritic cells |
| RA | rheumatoid arthritis |
| IFNγ | interferon-γ |
| DSS | dextran-sodium sulfate |
References
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Foxwell, B.; Browne, K.; Bondeson, J.; Clarke, C.; de Martin, R.; Brennan, F.; Feldmann, M. Efficient adenoviral infection with IκBα reveals that macrophage tumor necrosis factor α production in rheumatoid arthritis is NF-κB dependent. Proc. Natl. Acad. Sci. USA 1998, 95, 8211–8215. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G.; et al. Characterization of a Functional NF-κB Site in the Human Interleukin 1β Promoter: Evidence for a Positive Autoregulatory Loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Aono, H.; Hasunuma, T.; Yamamoto, K.; Mita, S.; Nishioka, K. Activation of transcription factor NF-κB in human synovial cells in response to tumor necrosis factor α. Arthritis Rheum. 1996, 39, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A. NF-κB, chemokine gene transcription and tumour growth. Nat. Rev. Immunol. 2002, 2, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Ley, S. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.A.; Baldwin, A.S. NF-κB pathways in the immune system: Control of the germinal center reaction. Immunol. Res. 2008, 41, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Ghosh, S. NF-κB: Roles and regulation in different CD4+ T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J. Return to homeostasis: Downregulation of NF-κB responses. Nat. Immunol. 2011, 12, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Arslan, S.Ç.; Scheidereit, C. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol. Rev. 2012, 246, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.N.; Nieters, A.; Wulczyn, F.G.; Naumann, M.; Meyer, R.; Nucifora, G.; McKeithan, T.W.; Scheidereit, C. The ankyrin repeat domains of the NF-κB precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-κB DNA binding. Proc. Natl. Acad. Sci. USA 1992, 89, 2489–2493. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, E.; Schmitz, I.; Marissen, W.E.; Osborn, S.L.; Touma, M.; Sasada, T.; Reche, P.A.; Tibaldi, E.V.; Hussey, R.E.; Kruisbeek, A.M.; et al. Peptide-Induced Negative Selection of Thymocytes Activates Transcription of an NF-κB Inhibitor. Mol. Cell 2002, 9, 637–648. [Google Scholar] [CrossRef]
- Yamazaki, S.; Muta, T.; Takeshige, K. A Novel IκB Protein, IκB-ζ, Induced by Proinflammatory Stimuli, Negatively Regulates Nuclear Factor-κB in the Nuclei. J. Biol. Chem. 2001, 276, 27657–27662. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Ito, H.; Miyajima, A. IκBη, a nuclear IκB protein, positively regulates the NF-κB–mediated expression of proinflammatory cytokines. Proc. Natl. Acad. Sci. USA 2010, 107, 11924–11929. [Google Scholar] [CrossRef] [PubMed]
- Ueshima, Y.; Bird, M.L.; Vardiman, J.W.; Rowley, J.D. A 14;19 translocation in B-cell chronic lymphocytic leukemia: A new recurring chromosome aberration. Int. J. Cancer 1985, 36, 287–290. [Google Scholar] [PubMed]
- Crossen, P.E.; Kennedy, M.A.; Heaton, D.C.; Morrison, M.J. Cloning and sequencing of a t(14;19) breakpoint that involves the Cμ switch region. Genes Chromosom. Cancer 1993, 8, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Asou, H.; Takechi, M.; Tanaka, K.; Tashiro, S.; Dohy, H.; Ohno, R.; Kamada, N. Japanese B cell chronic lymphocytic leukaemia: A cytogenetic and molecular biological study. Br. J. Haematol. 1993, 85, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Jöhrens, K.; Joos, S.; Lietz, A.; Hummel, F.; Janz, M.; Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Lichter, P.; et al. Elevated NF-κB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005, 106, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- McKeithan, T.W.; Takimoto, G.S.; Ohno, H.; Bjorling, V.S.; Morgan, R.; Hecht, B.K.; Dubé, I.; Sandberg, A.A.; Rowley, J.D. BCL3 rearrangements and t(14;19) in chronic lymphocytic leukemia and other B-cell malignancies: A molecular and cytogenetic study. Genes Chromosom. Cancer 1997, 20, 64–72. [Google Scholar] [CrossRef]
- Gao, C.; Wang, X.; Chen, L.; Wang, J.-H.; Gao, Z.-T.; Wang, H. Knockdown of Bcl-3 Inhibits Cell Growth and Induces DNA Damage in HTLV-1-infected Cells. Asian Pac. J. Cancer Prev. 2013, 14, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, P.C.; Guttridge, D.C.; Funkhouser, W.K.; Baldwin, A.B., Jr. Selective activation of NF-κB subunits in human breast cancer: Potential roles for NF-κB2/p52 and for Bcl-3. Oncogene 2000, 19, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Urban, B.C.; Collard, T.J.; Eagle, C.J.; Southern, S.L.; Greenhough, A.; Hamdollah-Zadeh, M.; Ghosh, A.; Poulsom, R.; Paraskeva, C.; Silver, A.; et al. BCL-3 expression promotes colorectal tumorigenesis through activation of AKT signalling. Gut 2015. gutjnl-2014. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Nolan, G.P.; Liou, H.C.; Scott, M.L.; Baltimore, D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers. Genes Dev. 1993, 7, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Kerr, L.D.; Duckett, C.S.; Wamsley, P.; Zhang, Q.; Chiao, P.; Nabel, G.; McKeithan, T.W.; Baeuerle, P.A.; Verma, I.M. The proto-oncogene BCL-3 encodes an IκB protein. Genes Dev. 1992, 6, 2352–2363. [Google Scholar] [CrossRef] [PubMed]
- Wulczyn, F.G.; Naumann, M.; Scheidereit, C. Candidate proto-oncogene bcl-3 encodes a subunit-specific inhibitor of transcription factor NF-κB. Nature 1992, 358, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Takahara, T.; Akizawa, T.; Hino, O. Bcl-3, a member of the I kappa B proteins, has distinct specificity towards the Rel family of proteins. Oncogene 1993, 8, 2067–2073. [Google Scholar] [PubMed]
- Franzoso, G.; Bours, V.; Park, S.; Tomita-Yamaguchi, M.; Kelly, K.; Siebenlist, U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB-mediated inhibition. Nature 1992, 359, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Nolan, G.P.; Fujita, T.; Bhatia, K.; Huppi, C.; Liou, H.C.; Scott, M.L.; Baltimore, D. The bcl-3 proto-oncogene encodes a nuclear IκB-like molecule that preferentially interacts with NF-κB p50 and p52 in a phosphorylation-dependent manner. Mol. Cell. Biol. 1993, 13, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Wulczyn, F.G.; Scheidereit, C. The NF-κB precursor p105 and the proto-oncogene product Bcl-3 are IκB molecules and control nuclear translocation of NF-κB. EMBO J. 1993, 12, 213–222. [Google Scholar] [PubMed]
- Watanabe, N.; Iwamura, T.; Shinoda, T.; Fujita, T. Regulation of NFKB1 proteins by the candidate oncoprotein BCL-3: Generation of NF-κB homodimers from the cytoplasmic pool of p50–p105 and nuclear translocation. EMBO J. 1997, 16, 3609–3620. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G.; Bours, V.; Azarenko, V.; Park, S.; Tomita-Yamaguchi, M.; Kanno, T.; Brown, K.; Siebenlist, U. The oncoprotein Bcl-3 can facilitate NF-κB-mediated transactivation by removing inhibiting p50 homodimers from select κB sites. EMBO J. 1993, 12, 3893–3901. [Google Scholar] [PubMed]
- Bours, V.; Franzoso, G.; Azarenko, V.; Park, S.; Brown, K.; Siebenlist, U. The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell 1993, 72, 729–739. [Google Scholar] [CrossRef]
- Viatour, P.; Dejardin, E.; Warnier, M.; Lair, F.; Claudio, E.; Bureau, F.; Marine, J.-C.; Merville, M.-P.; Maurer, U.; Green, D.; et al. GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol. Cell 2004, 16, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Paxian, S.; Merkle, H.; Riemann, M.; Wilda, M.; Adler, G.; Hameister, H.; Liptay, S.; Pfeffer, K.; Schmid, R.M. Abnormal organogenesis of Peyer’s patches in mice deficient for NF-κB1, NF-κB2, and Bcl-3. Gastroenterology 2002, 122, 1853–1868. [Google Scholar] [CrossRef] [PubMed]
- Caamaño, J.H.; Perez, P.; Lira, S.A.; Bravo, R. Constitutive Expression of Bcl-3 in Thymocytes Increases the DNA Binding of NF-κB1 (p50) Homodimers in Vivo. Mol. Cell. Biol. 1996, 16, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.J.; Ruan, Q.; Palmer, S.; Hilliard, B.; Chen, Y.H. Negative regulation of Toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science 2007, 317, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Kiely, P.A.; Carmody, R.J. Inhibition of Transcription by B Cell Leukemia 3 (Bcl-3) Protein Requires Interaction with Nuclear Factor κB (NF-κB) p50. J. Biol. Chem. 2014, 289, 7059–7067. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; McEver, R.P. Regulation of the Human P-selectin Promoter by Bcl-3 and Specific Homodimeric Members of the NF-κB/Rel Family. J. Biol. Chem. 1995, 270, 23077–23083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, H.; Claudio, E.; Brown, K.; Siebenlist, U. A Role for the IκB Family Member Bcl-3 in the Control of Central Immunologic Tolerance. Immunity 2007, 27, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Weih, F.; Caamaño, J. Regulation of secondary lymphoid organ development by the nuclear factor-κB signal transduction pathway. Immunol. Rev. 2003, 195, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Dechend, R.; Hirano, F.; Lemann, K.; Heissmeyer, V.; Ansieau, S.; Wulczyn, F.G.; Scheidereit, C.; Leutz, A. The Bcl-3 oncoprotein acts as a bridging factor between NF-κB/Rel and nuclear co-regulators. Oncogene 1999, 18, 3316–3323. [Google Scholar] [CrossRef] [PubMed]
- Corn, R.A.; Hunter, C.; Liou, H.C.; Siebenlist, U.; Boothby, M.R. Opposing Roles for RelB and Bcl-3 in Regulation of T-Box Expressed in T Cells, GATA-3, and Th Effector Differentiation. J. Immunol. 2005, 175, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Bundy, D.L.; McKeithan, T.W. Diverse Effects of BCL3 Phosphorylation on Its Modulation of NF-κB p52 Homodimer Binding to DNA. J. Biol. Chem. 1997, 272, 33132–33139. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R.; Lu, M.; Hai, T.; Lu, Y.; Boldogh, I. NF-κB-inducible BCL-3 Expression is an Autoregulatory Loop Controlling Nuclear p50/NF-κB1 Residence. J. Biol. Chem. 2001, 276, 32080–32093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Harhaj, E.W.; Bell, L.; Sun, S.-C.; Miller, B.A. Bcl-3 Expression and Nuclear Translocation Are Induced by Granulocyte-Macrophage Colony-Stimulating Factor and Erythropoietin in Proliferating Human Erythroid Precursors. Blood 1998, 92, 1225–1234. [Google Scholar] [PubMed]
- Massoumi, R.; Chmielarska, K.; Hennecke, K.; Pfeifer, A.; Fässler, R. Cyld Inhibits Tumor Cell Proliferation by Blocking Bcl-3-Dependent NF-κB Signaling. Cell 2006, 125, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Watanabe, Y.; Miyoshi, H.; Yamamoto, M.; Kaisho, T.; Takeda, K.; Akira, S. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-α production in macrophages. Blood 2003, 102, 4123–4129. [Google Scholar] [CrossRef] [PubMed]
- Wessells, J.; Baer, M.; Young, H.A.; Claudio, E.; Brown, K.; Siebenlist, U.; Johnson, P.F. BCL-3 and NF-κB p50 Attenuate Lipopolysaccharide-induced Inflammatory Responses in Macrophages. J. Biol. Chem. 2004, 279, 49995–50003. [Google Scholar] [CrossRef] [PubMed]
- Riemann, M.; Endres, R.; Liptay, S.; Pfeffer, K.; Schmid, R.M. The IκB Protein Bcl-3 Negatively Regulates Transcription of the IL-10 Gene in Macrophages. J. Immunol. 2005, 175, 3560–3568. [Google Scholar] [CrossRef] [PubMed]
- Pene, F.; Paun, A.; Sønder, S.U.; Rikhi, N.; Wang, H.; Claudio, E.; Siebenlist, U. The IκB Family Member Bcl-3 Coordinates the Pulmonary Defense against Klebsiella. pneumoniae Infection. J. Immunol. 2011, 186, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.; Louahed, J.; Demoulin, J.-B.; Renauld, J.-C. Interleukin-9 Regulates NF-κB Activity through BCL3 Gene Induction. Blood 1999, 93, 4318–4327. [Google Scholar] [PubMed]
- Elliott, S.F.; Coon, C.I.; Hays, E.; Stadheim, T.A.; Vincenti, M.P. Bcl-3 is an interleukin-1-responsive gene in chondrocytes and synovial fibroblasts that activates transcription of the matrix metalloproteinase 1 gene. Arthritis Rheum. 2002, 46, 3230–3239. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Hayakawa, K. B cell development pathways. Annu. Rev. Immunol. 2001, 19, 595–621. [Google Scholar] [CrossRef] [PubMed]
- Hogquist, K.A.; Baldwin, T.A.; Jameson, S.C. Central tolerance: Learning self-control in the thymus. Nat. Rev. Immunol. 2005, 5, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N. T-cell development and the CD4–CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, M.J.; Weisel, F. Germinal center selection and the development of memory B and plasma cells. Immunol. Rev. 2012, 247, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Kyewski, B.; Klein, L. A Central Role for Central Tolerance. Annu. Rev. Immunol. 2006, 24, 571–606. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.M. Anatomical Basis of Tolerance and Immunity to Intestinal Antigens. Nat. Rev. Immunol. 2003, 3, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Ansel, K.M.; Ngo, V.N.; Hyman, P.L.; Luther, S.A.; Förster, R.; Sedgwick, J.D.; Browning, J.L.; Lipp, M.; Cyster, J.G. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 2000, 406, 309–314. [Google Scholar] [PubMed]
- Finke, D.; Acha-Orbea, H.; Mattis, A.; Lipp, M.; Kraehenbuhl, J.P. CD4+CD3− cells induce Peyer’s patch development: Role of α4β1 integrin activation by CXCR5. Immunity 2002, 17, 363–373. [Google Scholar] [CrossRef]
- Franzoso, G.; Carlson, L.; Scharton-Kersten, T.; Shores, E.W.; Epstein, S.; Grinberg, A.; Tran, T.; Shacter, E.; Leonardi, A.; Anver, M.; et al. Critical roles for the Bcl-3 oncoprotein in T cell–mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity 1997, 6, 479–490. [Google Scholar] [CrossRef]
- Poljak, L.; Carlson, L.; Cunningham, K.; Kosco-Vilbois, M.H.; Siebenlist, U. Distinct Activities of p52/NF-κB Required for Proper Secondary Lymphoid Organ Microarchitecture: Functions Enhanced by Bcl-3. J. Immunol. 1999, 163, 6581–6588. [Google Scholar] [PubMed]
- Zhang, X.; Paun, A.; Claudio, E.; Wang, H.; Siebenlist, U. The Tumor Promoter and NF-κB Modulator Bcl-3 Regulates Splenic B Cell Development. J. Immunol. 2013, 191, 5984–5992. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate- like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Cariappa, A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 2009, 9, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.T.; Hackbarth, M.L.; Degenstein, L.C.; Baunoch, D.A.; Anastasi, J.; McKeithan, T.W. Lymphadenopathy, splenomegaly, and altered immunoglobulin production in BCL3 transgenic mice. Oncogene 1998, 16, 2333–2343. [Google Scholar] [CrossRef] [PubMed]
- Hövelmeyer, N.; Wörns, M.A.; Reissig, S.; Adams-Quack, P.; Leclaire, J.; Hahn, M.; Wörtge, S.; Nikolaev, A.; Galle, P.R.; Waisman, A. Overexpression of Bcl-3 inhibits the development of marginal zone B cells. Eur. J. Immunol. 2014, 44, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.M.; Krimpenfort, P.; Berns, A.; Verma, I.M. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997, 11, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G.; Carlson, L.; Poljak, L.; Shores, E.W.; Epstein, S.; Leonardi, A.; Grinberg, A.; Tran, T.; Scharton-Kersten, T.; Anver, M.; et al. Mice Deficient in Nuclear Factor (NF)-κB/p52 Present with Defects in Humoral Responses, Germinal Center Reactions, and Splenic Microarchitecture. J. Exp. Med. 1998, 187, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Caamaño, J.H.; Rizzo, C.A.; Durham, S.K.; Barton, D.S.; Raventós-Suárez, C.; Snapper, C.M.; Bravo, R. Nuclear Factor (NF)-κB2 (p100/p52) Is Required for Normal Splenic Microarchitecture and B Cell–mediated Immune Responses. J. Exp. Med. 1998, 187, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.-Y.; Truitt, M.L.; Ho, I.-C. GATA-3 deficiency abrogates the development and maintenance of T helper type 2 cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1993–1998. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wang, H.; Claudio, E.; Tassi, I.; Ha, H.-L.; Saret, S.; Siebenlist, U. The Oncoprotein and Transcriptional Regulator Bcl-3 Governs Plasticity and Pathogenicity of Autoimmune T Cells. Immunity 2014, 41, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Ruan, Q.; Kameswaran, V.; Zhang, Y.; Zheng, S.; Sun, J.; Wang, J.; DeVirgiliis, J.; Liou, H.-C.; Beg, A.A.; Chen, Y.H. The Th17 immune response is controlled by the Rel–RORγ–RORγT transcriptional axis. J. Exp. Med. 2011, 11, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Meguro, K.; Suzuki, K.; Hosokawa, J.; Sanayama, Y.; Tanaka, S.; Furuta, S.; Ikeda, K.; Takatori, H.; Suto, A.; Sakamoto, A.; et al. Role of Bcl-3 in the Development of Follicular Helper T Cells and in the Pathogenesis of Rheumatoid Arthritis. Arthritis Rheum. 2015, 67, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Follicular Helper CD4 T Cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.C.; Hildeman, D.A.; Kedl, R.M.; Teague, T.K.; Schaefer, B.C.; White, J.; Zhu, Y.; Kappler, J.; Marrack, P. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat. Immunol. 2001, 2, 397–402. [Google Scholar] [PubMed]
- Bauer, A.; Villunger, A.; Labi, V.; Fischer, S.F.; Strasser, A.; Wagner, H.; Schmid, R.M.; Häcker, G. The NF-κB regulator Bcl-3 and the BH3-only proteins Bim and Puma control the death of activated T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10979–10984. [Google Scholar] [CrossRef] [PubMed]
- Rangelova, S.; Kirschnek, S.; Strasser, A.; Häcker, G. FADD and the NF-κB family member Bcl-3 regulate complementary pathways to control T-cell survival and proliferation. Immunology 2008, 125, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Schmidt, C.S.; Mondino, A.; Lins, D.C.; Kedl, R.M.; Jenkins, M.K.; Mescher, M.F. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 1999, 162, 3256–3262. [Google Scholar] [PubMed]
- Valenzuela, J.O.; Hammerbeck, C.D.; Mescher, M.F. Cutting Edge: Bcl-3 Up-Regulation by Signal 3 Cytokine (IL-12) Prolongs Survival of Antigen-Activated CD8 T Cells. J. Immunol. 2005, 174, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Dumoutier, L.; Renauld, J.C.; Zaballos, A.; Ayllón, V.; Martínez-A, C. Bcl-3 expression promotes cell survival following interleukin-4 deprivation and is controlled by AP1 and AP1-like transcription factors. Mol. Cell. Biol. 2000, 20, 3407–3416. [Google Scholar] [CrossRef] [PubMed]
- Grundström, S.; Anderson, P.; Scheipers, P.; Sundstedt, A. Bcl-3 and NFκB p50-p50 Homodimers Act as Transcriptional Repressors in Tolerant CD4+ T Cells. J. Biol. Chem. 2004, 279, 8460–8468. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Fries, H.W.; Scheicher, C.; Keikavoussi, P.; Kolb-Mäurer, A.; Bröcker, E.B.; Serfling, E.; Kämpgen, E. Differential expression of Rel/NF-κB and octamer factors is a hallmark of the generation and maturation of dendritic cells. Blood 2000, 95, 277–285. [Google Scholar] [PubMed]
- Tassi, I.; Claudio, E.; Wang, H.; Tang, W.; Ha, H.-L.; Saret, S.; Ramaswamy, M.; Siegel, R.; Siebenlist, U. The NF-κB Regulator Bcl-3 Governs Dendritic Cell Antigen Presentation Functions in Adaptive Immunity. J. Immunol. 2014, 193, 4303–4311. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Mesman, A.W.; Geijtenbeek, T.B.H. Fucose-specific DC-SIGN signalling directs T helper cell type-2 responses via IKKε- and CYLD-dependent Bcl3 activation. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tassi, I.; Claudio, E.; Wang, H.; Tang, W.; Ha, H.-L.; Saret, S.; Sher, A.; Jankovic, D.; Siebenlist, U. Adaptive immune-mediated host resistance to Toxoplasma gondii is governed by the NF-κB regulator Bcl-3 in dendritic cells. Eur. J. Immunol. 2015, 45, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Fransen, K.; Visschedijk, M.C.; van Sommeren, S.; Fu, J.Y.; Franke, L.; Festen, E.A.M.; Stokkers, P.C.F.; van Bodegraven, A.A.; Crusius, J.B.A.; Hommes, D.W.; et al. Analysis of SNPs with an effect on gene expression identifies UBE2L3 and BCL3 as potential new risk genes for Crohn's disease. Hum. Mol. Gen. 2010, 19, 3482–3488. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, C.; Moloney, G.; Hurley, G.; Melgar, S.; Brint, E.; Nally, K.; Nibbs, R.J.; Shanahan, F.; Carmody, R.J. Bcl-3 deficiency protects against dextran-sodium sulphate-induced colitis in the mouse. Clin. Exp. Immunol. 2013, 173, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Zheng, S.-J.; Palmer, S.; Carmody, R.J.; Chen, Y.H. Roles of Bcl-3 in the Pathogenesis of Murine Type 1 Diabetes. Diabetes 2010, 59, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Lamhamedi-Cherradi, S.-E.; Zheng, S.; Hilliard, B.A.; Xu, L.; Sun, J.; Alsheadat, S.; Liou, H.-C.; Chen, Y.H. Transcriptional Regulation of Type I Diabetes by NF-κB. J. Immunol. 2003, 171, 4886–4892. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.G.; Swan, D.C.; Richardson, S.; Wilson, G.; Hilkens, C.M.U.; Young, D.A.; Isaacs, J.D. A CD4 T cell gene signature for early rheumatoid arthritis implicates interleukin 6-mediated STAT3 signalling, particularly in anti-citrullinated peptide antibody-negative disease. Ann. Rheum. Dis. 2012, 71, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.E.; Pratt, A.G.; Sedhom, M.A.K.; Doran, J.P.; Routledge, C.; Hargreaves, B.; Brown, P.M.; Lê Cao, K.-A.; Isaacs, J.D.; Thomas, R. IL-6-driven STAT signalling in circulating CD4+ lymphocytes is a marker for early anticitrullinated peptide antibody-negative rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhu, C.; Ma, B.; Tian, J.; Baidoo, S.E.; Mao, C.; Wu, W.; Chen, J.; Tong, J.; Yang, M.; et al. Increased Frequency of Circulating Follicular Helper T Cells in Patients with Rheumatoid Arthritis. Clin. Dev. Immunol. 2012, 2012, 827480. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shan, Y.; Jiang, Z.; Feng, J.; Li, C.; Ma, L.; Jiang, Y. High frequencies of activated B cells and follicular helper T cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Exp. Immunol. 2013, 174, 212–220. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrington, F.D.; Nibbs, R.J.B. Regulation of the Adaptive Immune Response by the IκB Family Protein Bcl-3. Cells 2016, 5, 14. https://doi.org/10.3390/cells5020014
Herrington FD, Nibbs RJB. Regulation of the Adaptive Immune Response by the IκB Family Protein Bcl-3. Cells. 2016; 5(2):14. https://doi.org/10.3390/cells5020014
Chicago/Turabian StyleHerrington, Felicity D., and Robert J. B. Nibbs. 2016. "Regulation of the Adaptive Immune Response by the IκB Family Protein Bcl-3" Cells 5, no. 2: 14. https://doi.org/10.3390/cells5020014