
Mitochondrial Impairment May Increase Cellular NAD(P)H: Resazurin Oxidoreductase Activity, Perturbing the NAD(P)H-Based Viability Assays

 ,
, 
Abstract
:1. Introduction
2. Experimental Section
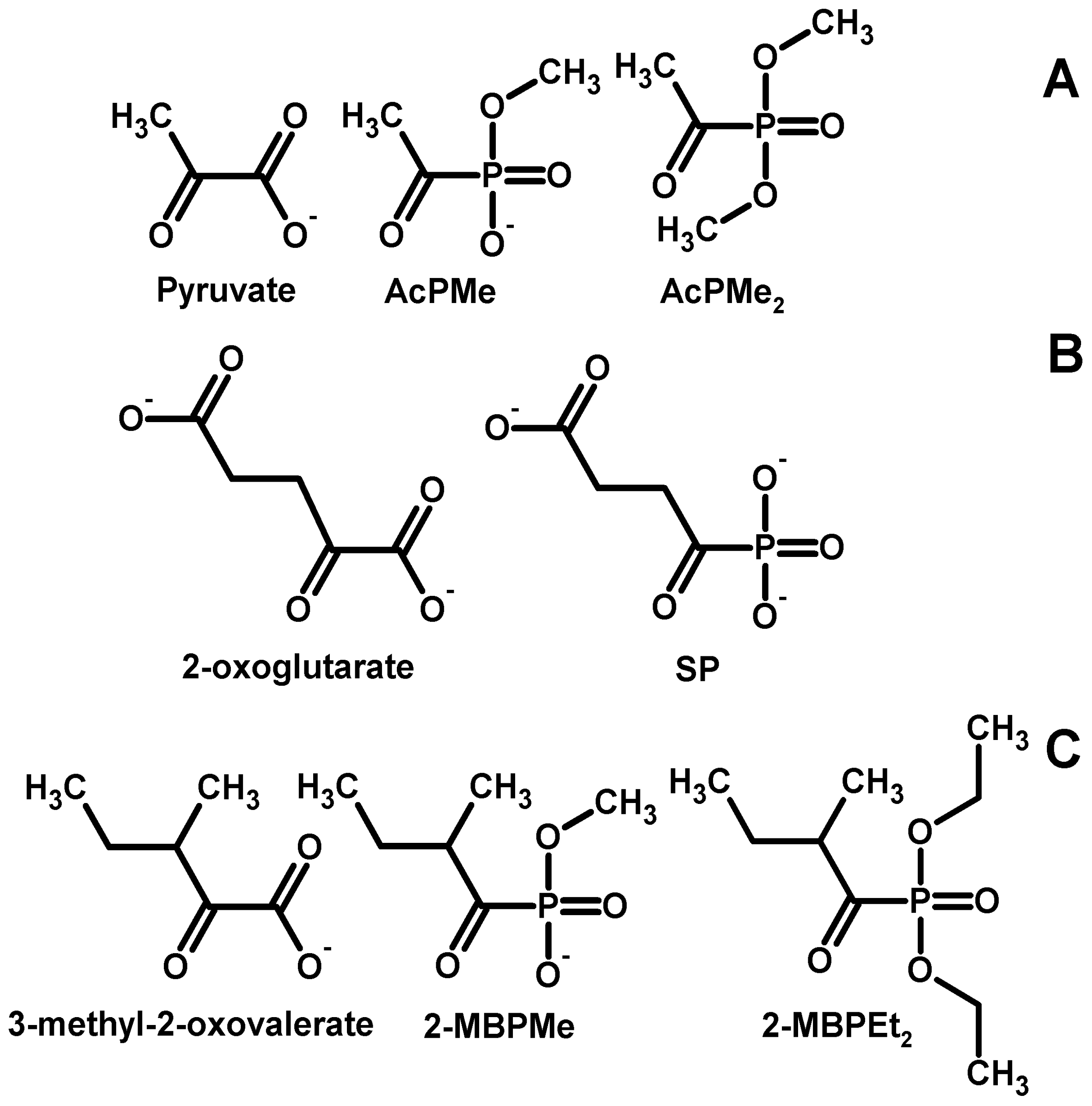
2.1. Synthesis of the Phosphonate Analogs of Pyruvate
2.2. Synthesis of the Phosphonate Analogs of 3-Methyl-2-Oxovalerate (“Ketoisoleucine”)
2.3. Cellular NAD(P)H:Resazurin Oxidoreductase Assay
2.4. Enzyme Assays in Cell Lysates
2.5. Protein Assay in Cell Lysates
2.6. Statistical Analysis
3. Results and Discussion
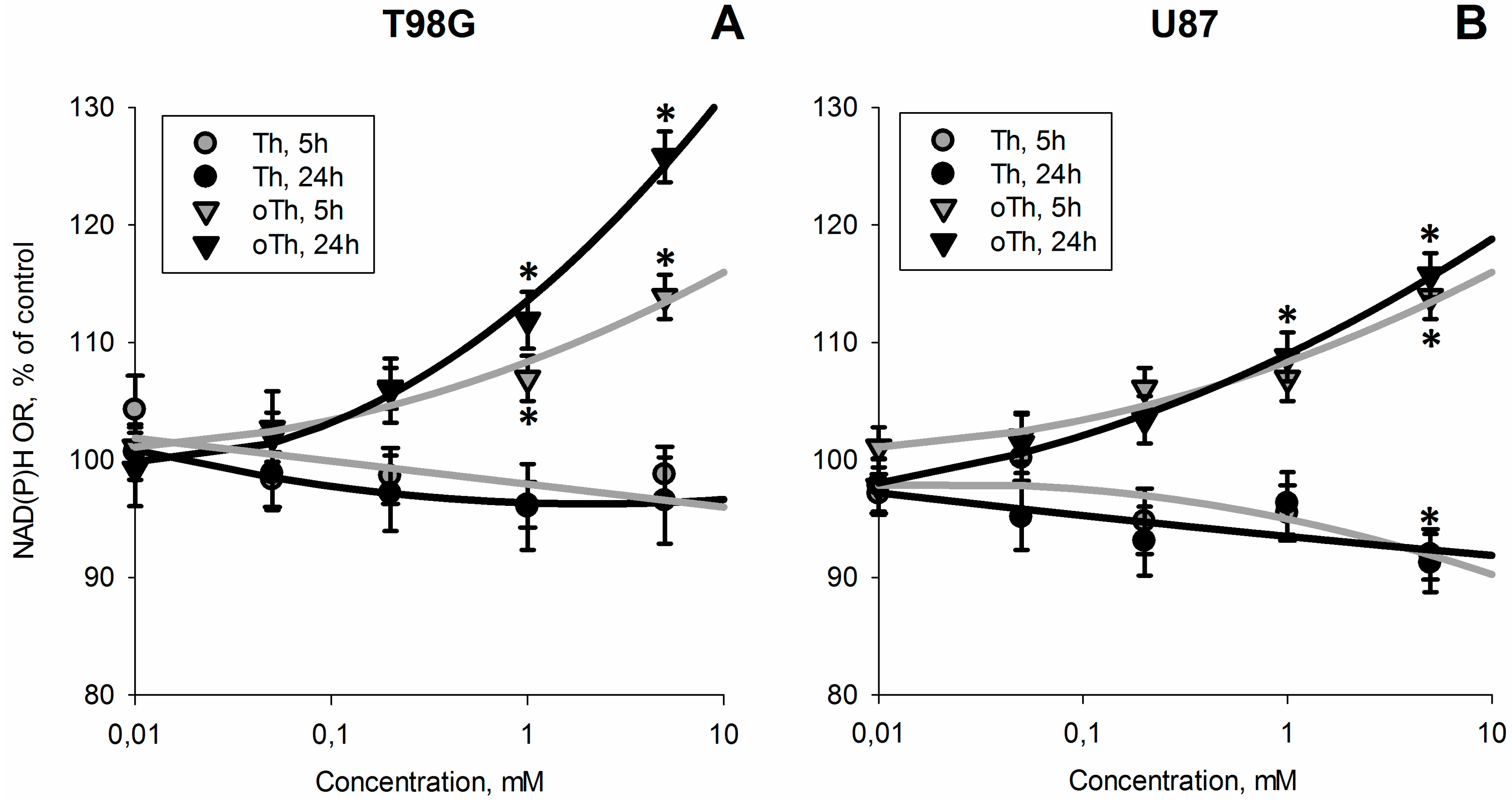
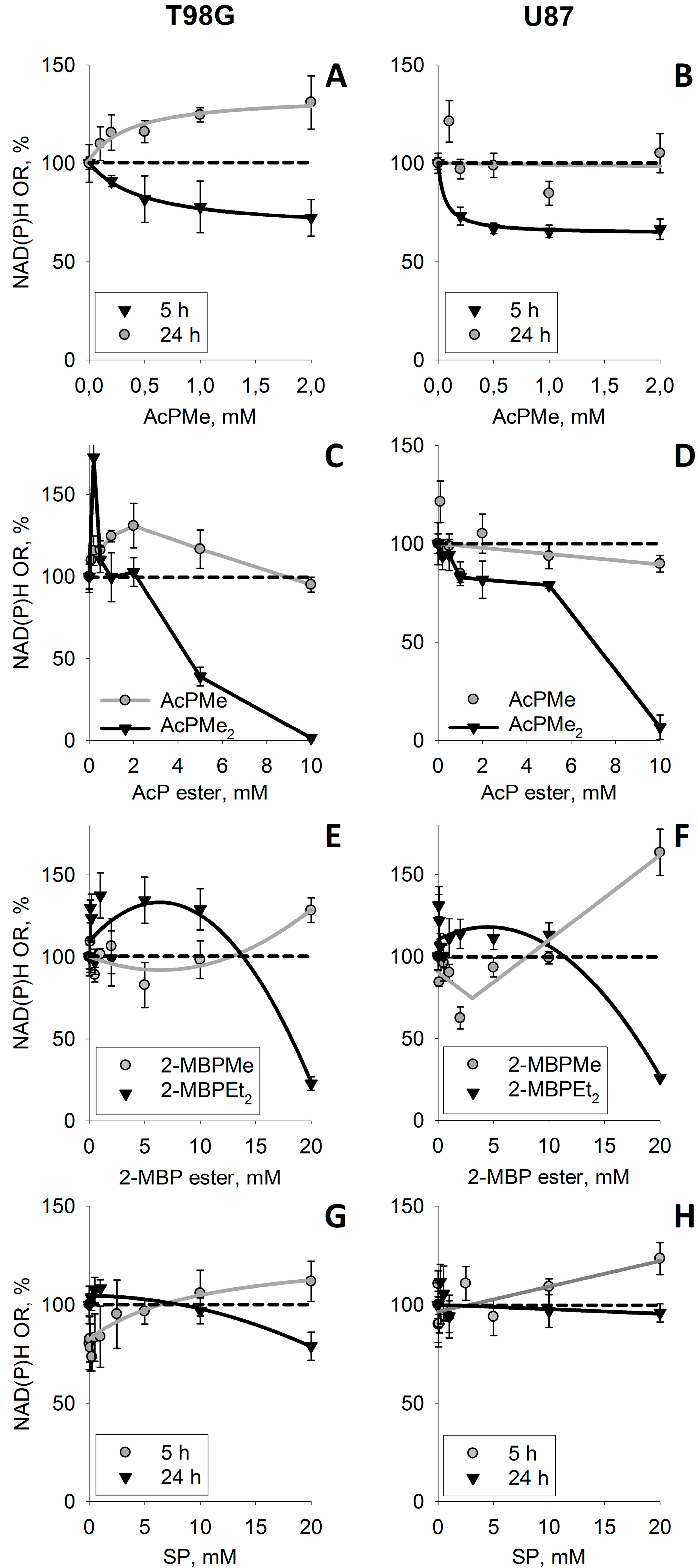
3.1. Influence of Thiamin and Its Antagonist Oxythiamin on Cellular NAD(P)H:Resazurin Oxidoreductase
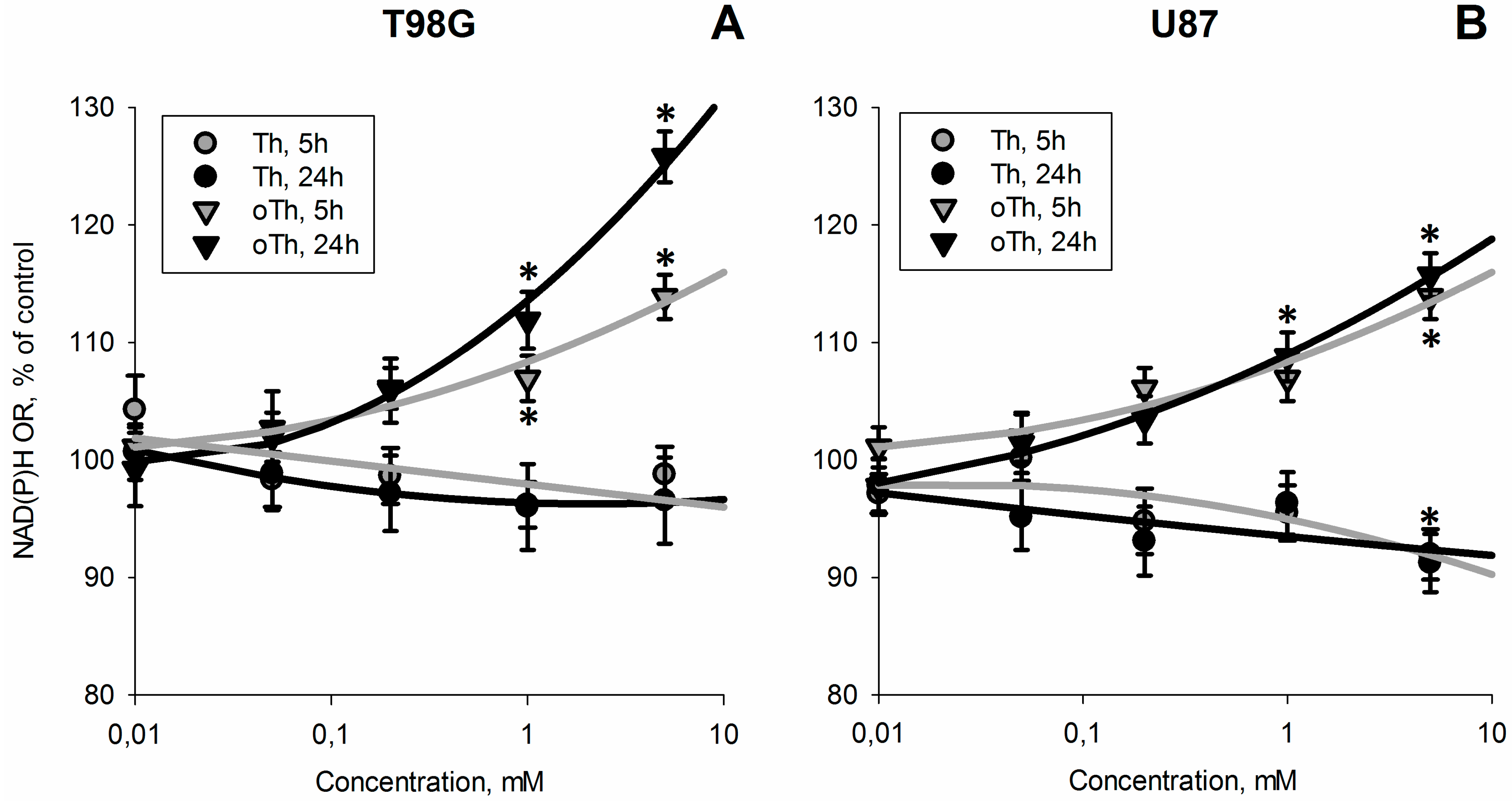
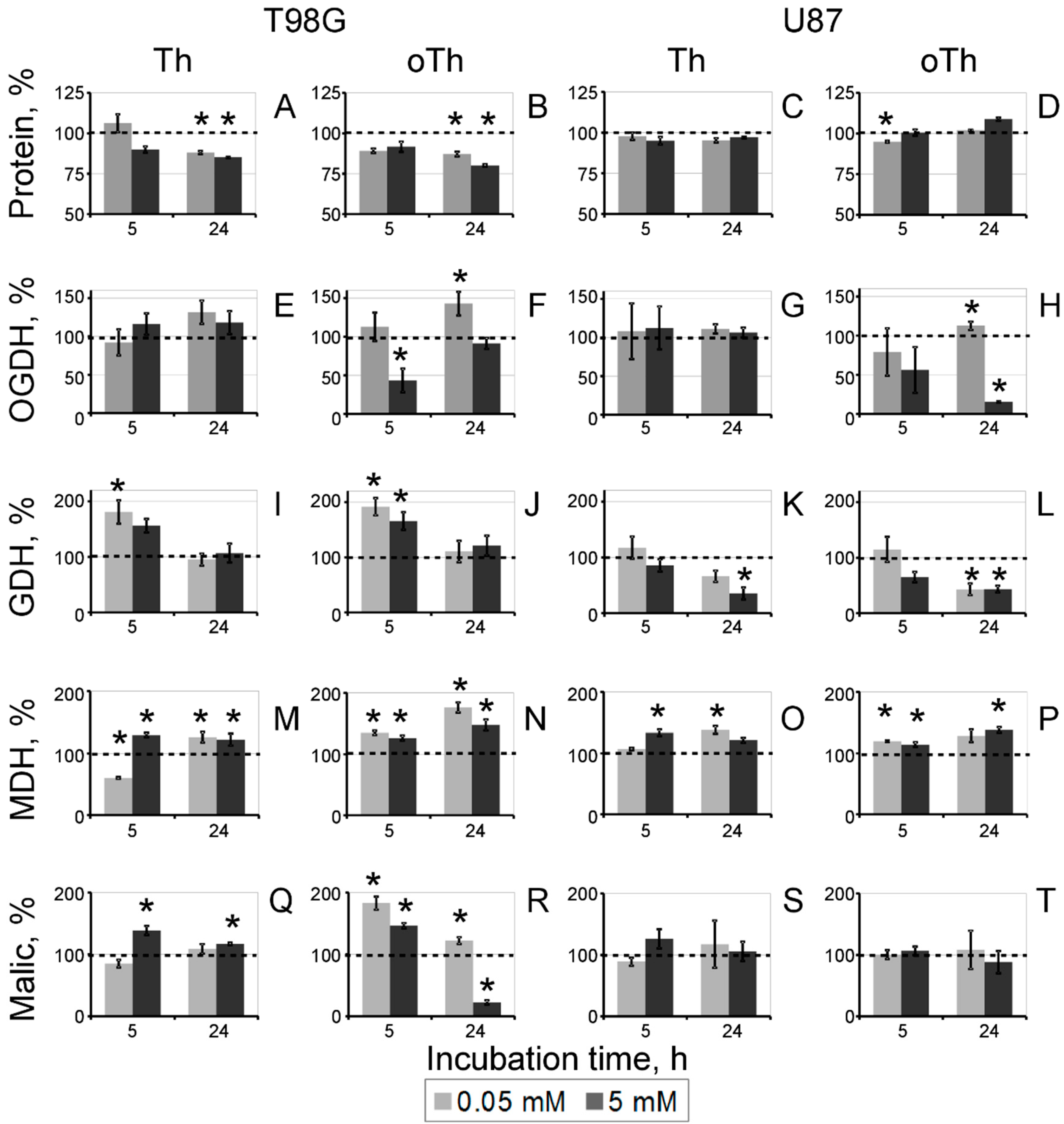
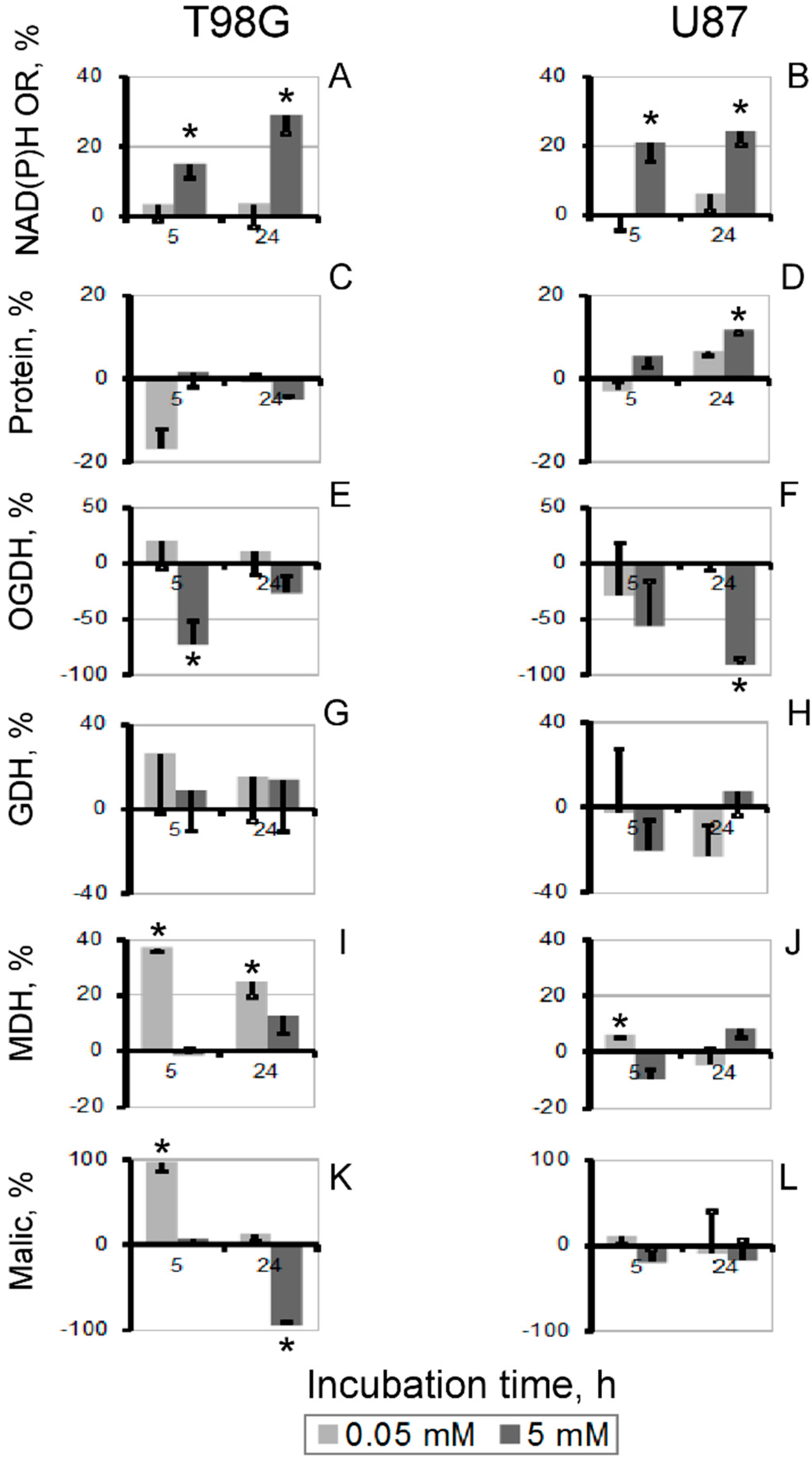
3.2. Biochemical Analysis of the Thiamin and Oxythiamin Effects in Cell Extracts of T98G and U87 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | T98G | U87 | ||
|---|---|---|---|---|
| Time in Medium | 5 h | 24 h | 5 h | 24 h |
| Protein, mg/mL (c,d) | 1.73 ± 0.08 | 1.81 ± 0.00 | 1.33 ± 0.01 | 1.34 ± 0.00 |
| Activity, μmol/(min∙mg) | ||||
| OGDH (b,d) | 0.05 ± 0.01 | 0.03 ± 0.00 | 0.07 ± 0.02 | 0.16 ± 0.01 |
| GDH (b) | 0.016 ± 0.003 | 0.032 ± 0.003 | 0.030 ± 0.002 | 0.052 ± 0.012 |
| MDH (a,b,c,d) | 7.8 ± 0.4 | 13.3 ± 0.3 | 10.6 ± 0.6 | 18.7 ± 0.9 |
| Malic (a,d) | 0.09 ± 0.01 | 0.57 ± 0.01 | 0.06 ± 0.01 | 0.11 ± 0.02 |
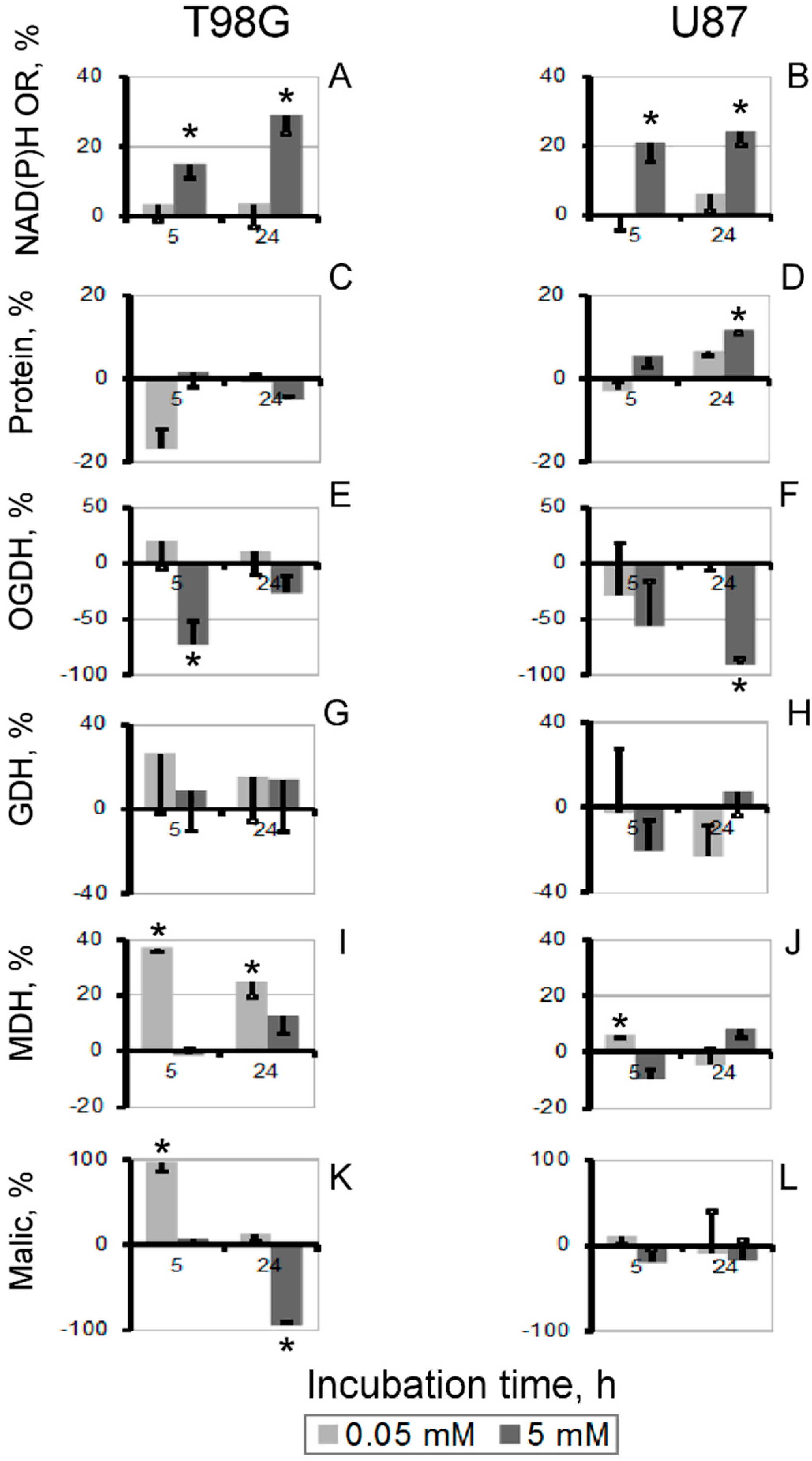
3.3. Changes in Cellular Enzyme Activities, Accompanying the Oxythiamin-Induced Increase in Cellular NAD(P)H:Resazurin Oxidoreductase
3.4. Increase in Cellular NAD(P)H:Resazurin Oxidoreductase by the Site-Directed Inhibitors of Mitochondrial 2-Oxo Acid Dehydrogenases
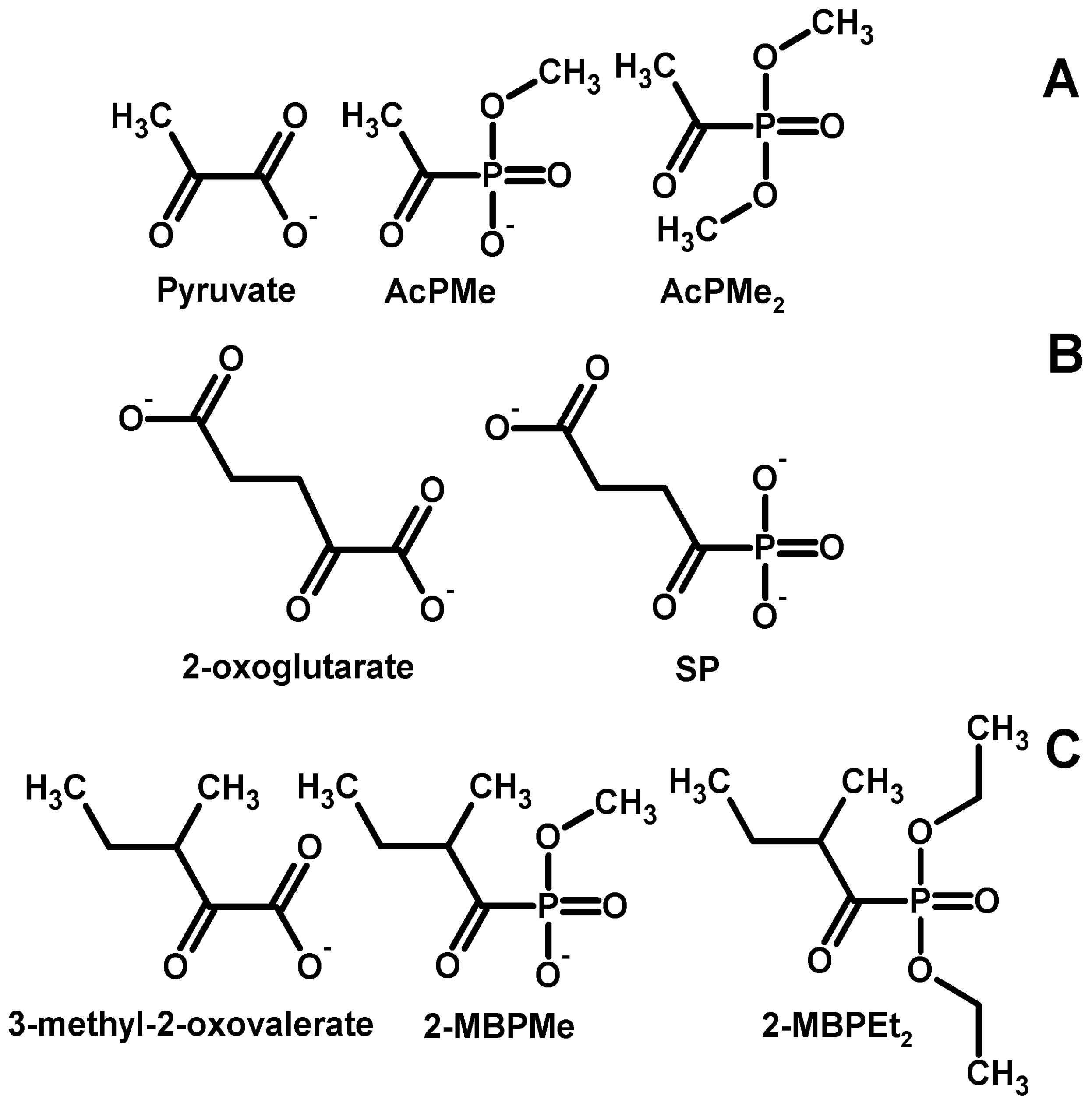
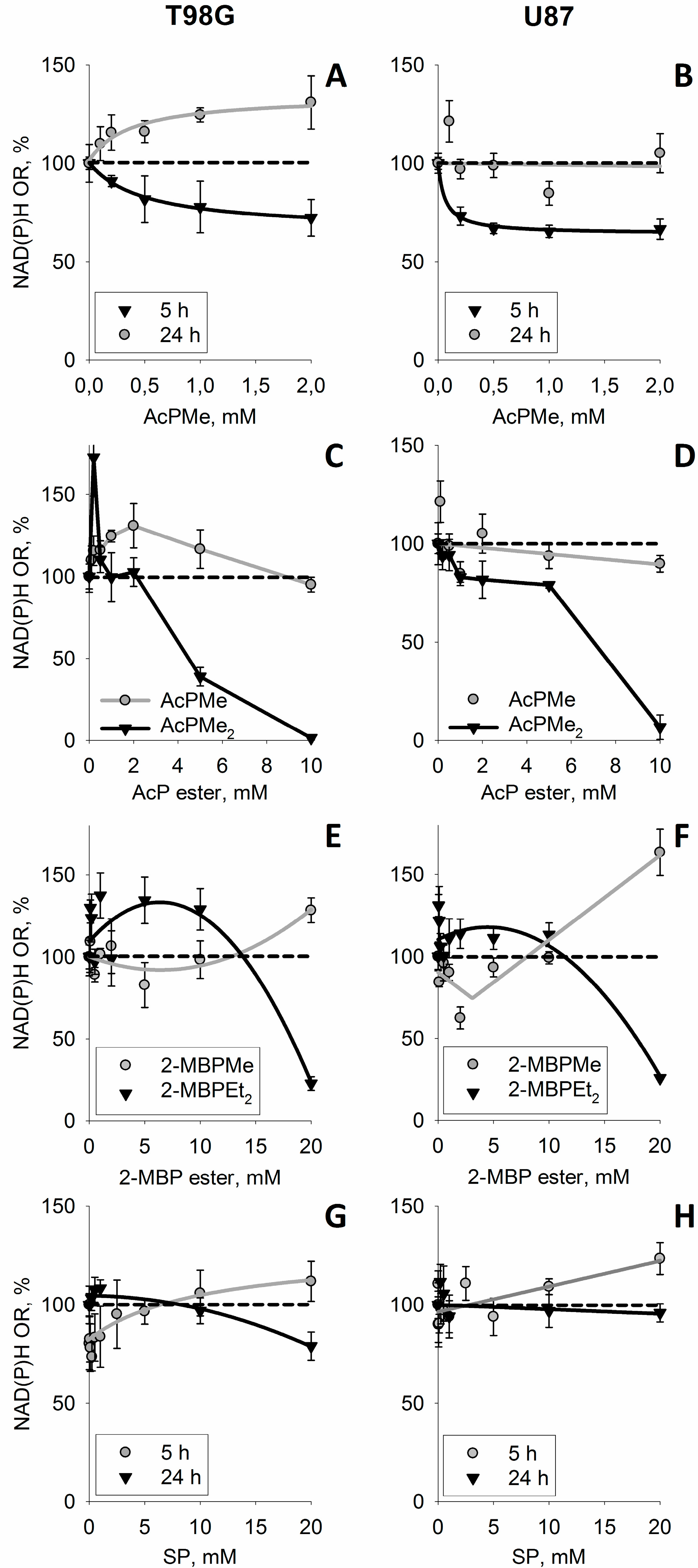
3.5. Discussion
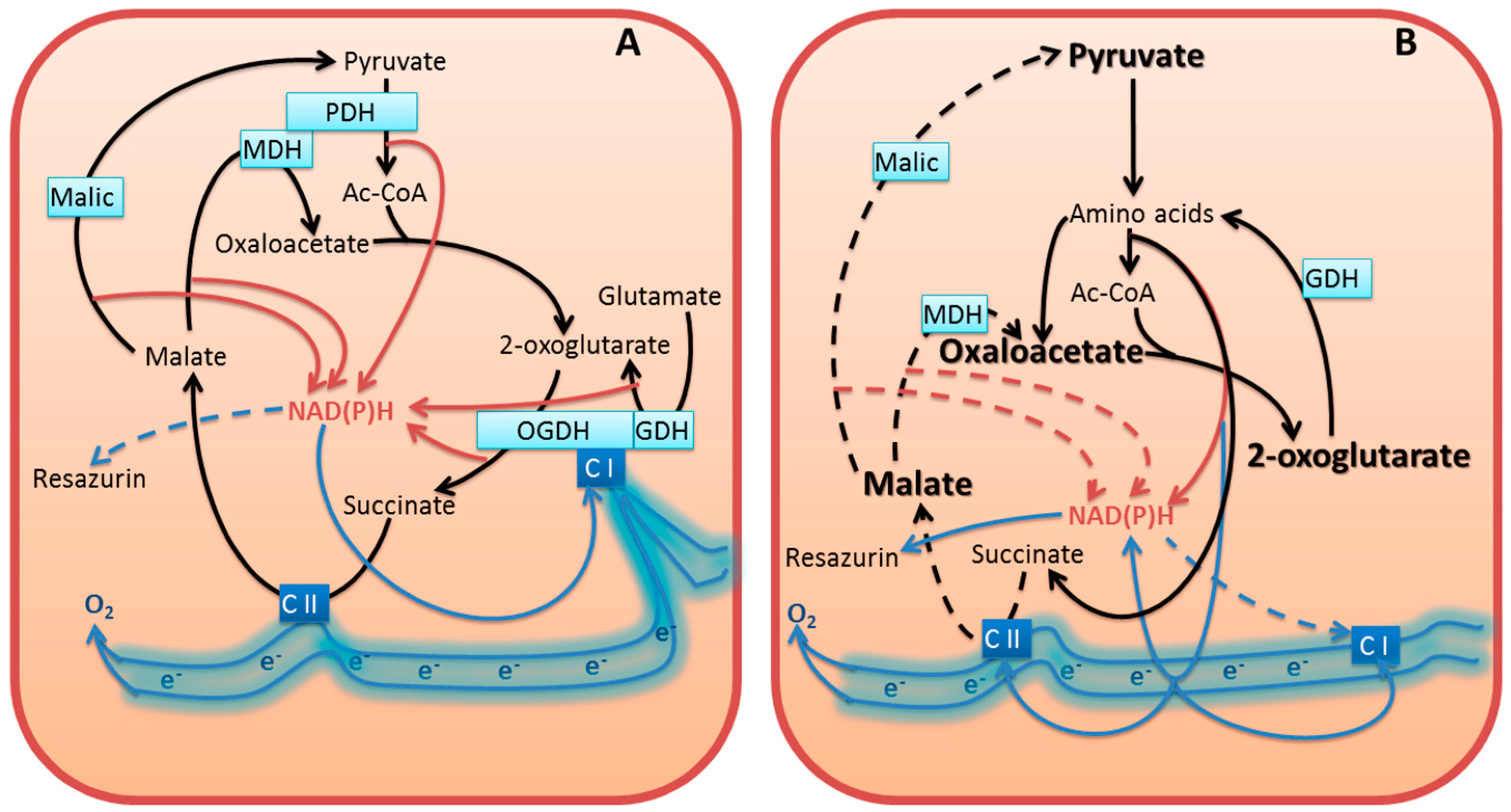
3.5.1. Inhibition of Cellular NAD(P)H Producers May Increase NAD(P)H:Resazurin Oxidoreductase
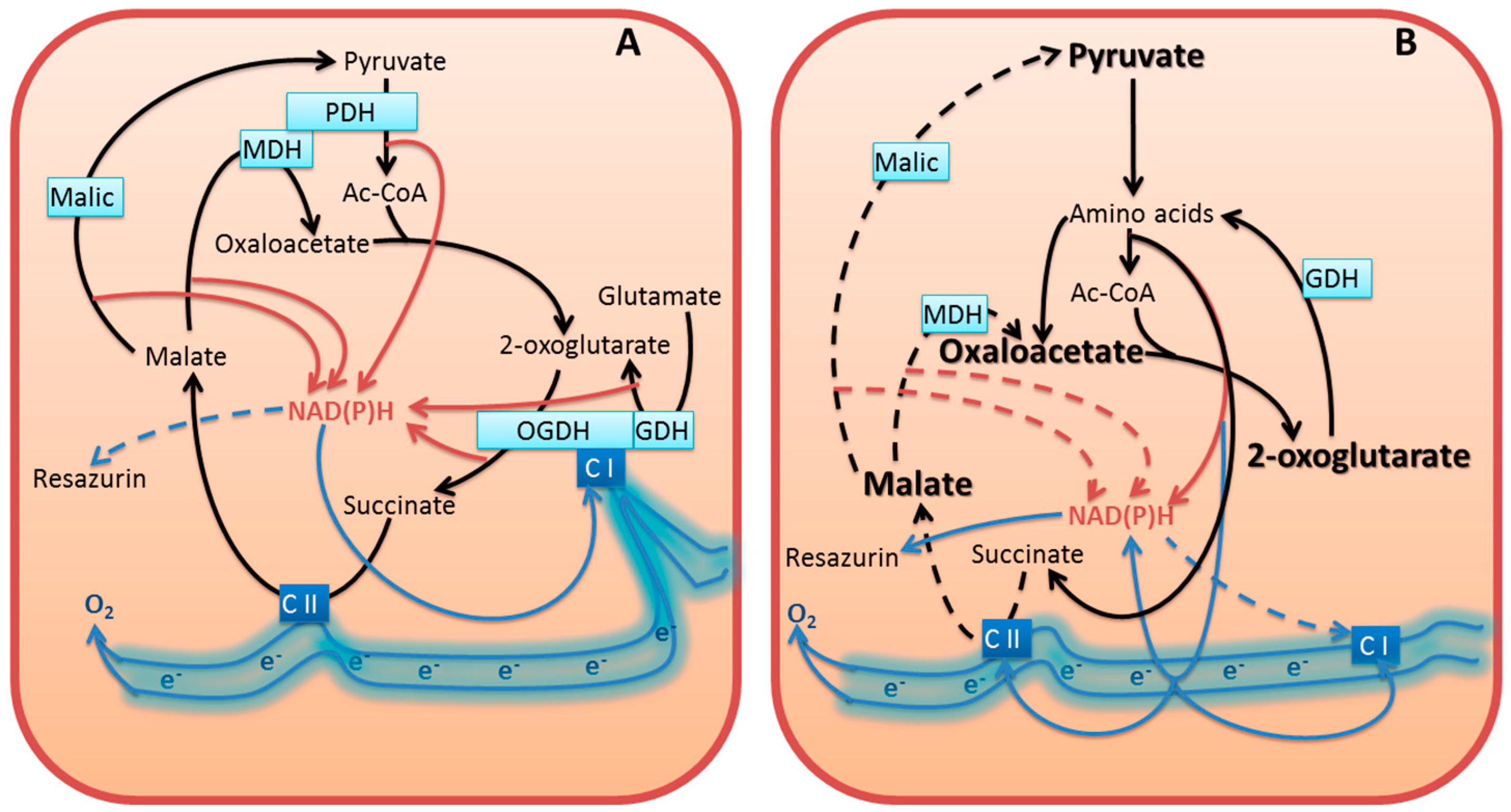
3.5.2. Compensation for Impairment of Metabolic Checkpoints Perturbs the Physiological Flux of Substrates and Electrons
3.5.3. Dysregulated Metabolism Increases Cellular Side Reactions
3.5.4. Similarity between the Action of Thiamin and Oxythiamin on Glioblastoma Cells
3.5.5. Different Resistance of Glioblastoma T98G and U87 Cells to Inhibitors of Central Metabolism is Due to the Cell-Specific Metabolism
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, H.C. Cyclic adp-ribose and nicotinic acid adenine dinucleotide phosphate (naadp) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ros balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Skoge, R.H.; Dolle, C.; Ziegler, M. Regulation of sirt2-dependent alpha-tubulin deacetylation by cellular nad levels. DNA Repair 2014, 23, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Ziegler, M. Pathophysiological relevance of mitochondria in nad(+) metabolism. FEBS Lett. 2001, 492, 4–8. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Sittampalam, G.S., Gal-Edd, N., Arkin, M., Auld, D., Austin, C., Bejcek, B., Glicksman, M., Inglese, J., Lemmon, V., Li, Z., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [PubMed]
- Wu, F.; Minteer, S. Krebs cycle metabolon: Structural evidence of substrate channeling revealed by cross-linking and mass spectrometry. Angew. Chem. 2015, 54, 1851–1854. [Google Scholar] [CrossRef] [PubMed]
- Puchulu-Campanella, E.; Chu, H.; Anstee, D.J.; Galan, J.A.; Tao, W.A.; Low, P.S. Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane. J. Biol. Chem. 2013, 288, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Lyubarev, A.E.; Kurganov, B.I. Supramolecular organization of tricarboxylic acid cycle enzymes. Bio Syst. 1989, 22, 91–102. [Google Scholar] [CrossRef]
- Gazizov, M.B.; Nikolaeva, V.G.; Zakharov, V.M.; Khariullin, R.A.; Moskva, V.V. Reactions of dialkyl hydrogen phosphites with acyl chlorides in presence of alkyl vinyl ethers. Russ. J. Gen. Chem. 1988, 58, 940–941. [Google Scholar]
- Karaman, R.; Goldblum, A.; Breuer, E.; Leader, H.J. Acylphosphonic acids and methyl hydrogen acylphosphonates: Physical and chemical properties and theoretical calculations. J. Chem. Soc. Perkin Trans. 1 1989, 765–774. [Google Scholar] [CrossRef]
- Bunik, V.I.; Tylicki, A.; Lukashev, N.V. Thiamin diphosphate-dependent enzymes: From enzymology to metabolic regulation, drug design and disease models. FEBS J. 2013, 280, 6412–6442. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Di Salvo, M.L.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Trofimova, L.K.; Araujo, W.L.; Strokina, A.A.; Fernie, A.R.; Bettendorff, L.; Bunik, V.I. Consequences of the alpha-ketoglutarate dehydrogenase inhibition for neuronal metabolism and survival: Implications for neurodegenerative diseases. Curr. Med. Chem. 2012, 19, 5895–5906. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.S.; Gibson, G.E.; Cooper, A.J.; Denton, T.T.; Thompson, C.M.; Bunik, V.I.; Alves, P.M.; Sonnewald, U. Inhibitors of the alpha-ketoglutarate dehydrogenase complex alter [1–13c]glucose and [u-13c]glutamate metabolism in cerebellar granule neurons. J. Neurosci. Res. 2006, 83, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Larson, E.M.; Doughman, D.J.; Gregerson, D.S.; Obritsch, W.F. A new, simple, nonradioactive, nontoxic in vitro assay to monitor corneal endothelial cell viability. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1929–1933. [Google Scholar]
- Byth, H.A.; McHunu, B.I.; Dubery, I.A.; Bornman, L. Assessment of a simple, non-toxic alamar blue cell survival assay to monitor tomato cell viability. Phytochem. Anal. PCA 2001, 12, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Schreer, A.; Tinson, C.; Sherry, J.P.; Schirmer, K. Application of alamar blue/5-carboxyfluorescein diacetate acetoxymethyl ester as a noninvasive cell viability assay in primary hepatocytes from rainbow trout. Anal. Biochem. 2005, 344, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Araujo, W.L.; Trofimova, L.; Mkrtchyan, G.; Steinhauser, D.; Krall, L.; Graf, A.; Fernie, A.R.; Bunik, V.I. On the role of the mitochondrial 2-oxoglutarate dehydrogenase complex in amino acid metabolism. Amino Acids 2013, 44, 683–700. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.; Rothschild, D.E.; Quinlan, C.L.; Scott, G.K.; Benz, C.C.; Brand, M.D. Sources of superoxide/h2o2 during mitochondrial proline oxidation. Redox Biol. 2014, 2, 901–909. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. FEBS 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Rampersad, S.N. Multiple applications of alamar blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors 2012, 12, 12347–12360. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.J.; Tarloff, J.B. Evaluation of hepatic subcellular fractions for alamar blue and mtt reductase activity. Toxicol. in Vitro 2001, 15, 257–259. [Google Scholar] [CrossRef]
- The alamarBlue® Assay. Available online: http://tools.thermofisher.com/content/sfs/manuals/PI-DAL1025-1100_TI%20alamarBlue%20Rev%201.1.pdf (accessed on 9 August 2015).
- Shangari, N.; Mehta, R.; O’Brien, P.J. Hepatocyte susceptibility to glyoxal is dependent on cell thiamin content. Chem.-Biol. Interact. 2007, 165, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Raddatz, G.; Strumilo, S.A. Translating enzymology into metabolic regulation: The case of the 2- oxoglutarate dehydrogenase multienzyme complex. Curr. Chem. Biol. 2013, 7, 74–93. [Google Scholar] [CrossRef]
- Bunik, V.I.; Fernie, A.R. Metabolic control exerted by the 2-oxoglutarate dehydrogenase reaction: A cross-kingdom comparison of the crossroad between energy production and nitrogen assimilation. Biochem. J. 2009, 422, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Fahien, L.A.; MacDonald, M.J.; Teller, J.K.; Fibich, B.; Fahien, C.M. Kinetic advantages of hetero-enzyme complexes with glutamate dehydrogenase and the alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 1989, 264, 12303–12312. [Google Scholar] [PubMed]
- Fahien, L.A.; Teller, J.K. Glutamate-malate metabolism in liver mitochondria. A model constructed on the basis of mitochondrial levels of enzymes, specificity, dissociation constants, and stoichiometry of hetero-enzyme complexes. J. Biol. Chem. 1992, 267, 10411–10422. [Google Scholar] [PubMed]
- Sumegi, B.; Srere, P.A. Complex i binds several mitochondrial nad-coupled dehydrogenases. J. Biol. Chem. 1984, 259, 15040–15045. [Google Scholar] [PubMed]
- Porpaczy, Z.; Sumegi, B.; Alkonyi, I. Interaction between nad-dependent isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase complex, and nadh:Ubiquinone oxidoreductase. J. Biol. Chem. 1987, 262, 9509–9514. [Google Scholar] [PubMed]
- Zundorf, G.; Kahlert, S.; Bunik, V.I.; Reiser, G. Alpha-ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience 2009, 158, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chen, H.L.; Xu, H.; Gibson, G.E. Reduction in the e2k subunit of the alpha-ketoglutarate dehydrogenase complex has effects independent of complex activity. J. Biol. Chem. 2005, 280, 10888–10896. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, L.; Yu, C.A. Cross-talk between mitochondrial malate dehydrogenase and the cytochrome bc1 complex. J. Biol. Chem. 2010, 285, 10408–10414. [Google Scholar] [CrossRef] [PubMed]
- Grivennikova, V.G.; Gavrikova, E.V.; Timoshin, A.A.; Vinogradov, A.D. Fumarate reductase activity of bovine heart succinate-ubiquinone reductase. New assay system and overall properties of the reaction. Biochim. Biophys. Acta 1993, 1140, 282–292. [Google Scholar] [CrossRef]
- Araujo, W.L.; Tohge, T.; Osorio, S.; Lohse, M.; Balbo, I.; Krahnert, I.; Sienkiewicz-Porzucek, A.; Usadel, B.; Nunes-Nesi, A.; Fernie, A.R. Antisense inhibition of the 2-oxoglutarate dehydrogenase complex in tomato demonstrates its importance for plant respiration and during leaf senescence and fruit maturation. Plant Cell 2012, 24, 2328–2351. [Google Scholar] [CrossRef] [PubMed]
- Araujo, W.L.; Tohge, T.; Nunes-Nesi, A.; Daloso, D.M.; Nimick, M.; Krahnert, I.; Bunik, V.I.; Moorhead, G.B.; Fernie, A.R. Phosphonate analogs of 2-oxoglutarate perturb metabolism and gene expression in illuminated arabidopsis leaves. Front. Plant Sci. 2012, 3, 114. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, L.H.; Shi, Q.; Gibson, G.E.; Sonnewald, U. Brain [u-13 c]glucose metabolism in mice with decreased alpha-ketoglutarate dehydrogenase complex activity. J. Neurosci. Res. 2011, 89, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Sienkiewicz-Porzucek, A.; Sulpice, R.; Osorio, S.; Krahnert, I.; Leisse, A.; Urbanczyk-Wochniak, E.; Hodges, M.; Fernie, A.R.; Nunes-Nesi, A. Mild reductions in mitochondrial nad-dependent isocitrate dehydrogenase activity result in altered nitrate assimilation and pigmentation but do not impact growth. Mol. Plant 2010, 3, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Sulpice, R.; Sienkiewicz-Porzucek, A.; Osorio, S.; Krahnert, I.; Stitt, M.; Fernie, A.R.; Nunes-Nesi, A. Mild reductions in cytosolic nadp-dependent isocitrate dehydrogenase activity result in lower amino acid contents and pigmentation without impacting growth. Amino Acids 2010, 39, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Fahien, L.A.; Kmiotek, E.H.; MacDonald, M.J.; Fibich, B.; Mandic, M. Regulation of malate dehydrogenase activity by glutamate, citrate, alpha-ketoglutarate, and multienzyme interaction. J. Biol. Chem. 1988, 263, 10687–10697. [Google Scholar] [PubMed]
- Hutson, S.M.; Islam, M.M.; Zaganas, I. Interaction between glutamate dehydrogenase (gdh) and l-leucine catabolic enzymes: Intersecting metabolic pathways. Neurochem. Int. 2011, 59, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Xu, H.; Shi, Q.; Chen, L.H.; Pedrini, S.; Pechman, D.; Baker, H.; Beal, M.F.; Gandy, S.E.; Gibson, G.E. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in alzheimer’s mouse model. Neurobio. Aging 2009, 30, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Rane, A.; Yadava, N.; Andersen, J.K.; Nicholls, D.G.; Polster, B.M. Reactive oxygen species regulation by aif- and complex i-depleted brain mitochondria. Free Radical Biol. Med. 2009, 46, 939–947. [Google Scholar] [CrossRef]
- Gibson, G.E.; Blass, J.P.; Beal, M.F.; Bunik, V. The alpha-ketoglutarate-dehydrogenase complex: A mediator between mitochondria and oxidative stress in neurodegeneration. Mol. Neurobiol. 2005, 31, 43–63. [Google Scholar] [CrossRef]
- Shi, Q.; Gibson, G.E. Up-regulation of the mitochondrial malate dehydrogenase by oxidative stress is mediated by mir-743a. J. Neurochem. 2011, 118, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Lu’o’ng, K.V.; Nguyen, L.T. The role of thiamine in cancer: Possible genetic and cellular signaling mechanisms. Cancer Genom. Proteom. 2013, 10, 169–185. [Google Scholar]
- Richardson, A.D.; Moscow, J.A. Can an enzyme cofactor be a factor in malignant progression? Cancer Biol. Ther. 2010, 10, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; Wojcik, B.E.; Huang, H.J.; Friedmann, T.; de Tribolet, N.; Cavenee, W.K. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994, 54, 649–652. [Google Scholar] [PubMed]
- Yoshida, Y.; Sadata, A.; Zhang, W.; Saito, K.; Shinoura, N.; Hamada, H. Generation of fiber-mutant recombinant adenoviruses for gene therapy of malignant glioma. Hum. Gene Ther. 1998, 9, 2503–2515. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Hatzidaki, E.; Befani, C.D.; Liakos, P.; Papandreou, C.N. Bortezomib overcomes mgmt-related resistance of glioblastoma cell lines to temozolomide in a schedule-dependent manner. Investig. New Drugs 2013, 31, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Massi, P.; Cinquina, V.; Valenti, M.; Bolognini, D.; Gariboldi, M.; Monti, E.; Rubino, T.; Parolaro, D. Cannabidiol, a non-psychoactive cannabinoid compound, inhibits proliferation and invasion in u87-mg and t98g glioma cells through a multitarget effect. PLoS ONE 2013, 8, e76918. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Tan, C.; Brat, D.J.; Post, D.E.; Van Meir, E.G. Genetic and hypoxic regulation of angiogenesis in gliomas. J. Neuro-Oncol. 2004, 70, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Lal, B.; Goodwin, C.R.; Sang, Y.; Foss, C.A.; Cornet, K.; Muzamil, S.; Pomper, M.G.; Kim, J.; Laterra, J. Egfrviii and c-met pathway inhibitors synergize against pten-null/egfrviii+ glioblastoma xenografts. Mol. Cancer Ther. 2009, 8, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.; et al. Molecular determinants of the response of glioblastomas to egfr kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Antonyak, M.A.; Singh, G.; Cerione, R.A. A mechanism for the upregulation of egf receptor levels in glioblastomas. Cell Rep. 2013, 3, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Jena, R.; Burnet, N.G.; Jeynes, J.C.; Merchant, M.J.; Kirkby, K.J.; Kirkby, N.F. In vitro evaluation of combined temozolomide and radiotherapy using x rays and high-linear energy transfer radiation for glioblastoma. Radiat. Res. 2012, 177, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Pyko, I.V.; Nakada, M.; Sabit, H.; Teng, L.; Furuyama, N.; Hayashi, Y.; Kawakami, K.; Minamoto, T.; Fedulau, A.S.; Hamada, J. Glycogen synthase kinase 3beta inhibition sensitizes human glioblastoma cells to temozolomide by affecting o6-methylguanine DNA methyltransferase promoter methylation via c-myc signaling. Carcinogenesis 2013, 34, 2206–2217. [Google Scholar] [CrossRef] [PubMed]
- Graf, A.; Trofimova, L.; Loshinskaja, A.; Mkrtchyan, G.; Strokina, A.; Lovat, M.; Tylicky, A.; Strumilo, S.; Bettendorff, L.; Bunik, V.I. Up-regulation of 2-oxoglutarate dehydrogenase as a stress response. Int. J. Biochem. Cell Biol. 2013, 45, 175–189. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleshin, V.A.; Artiukhov, A.V.; Oppermann, H.; Kazantsev, A.V.; Lukashev, N.V.; Bunik, V.I. Mitochondrial Impairment May Increase Cellular NAD(P)H: Resazurin Oxidoreductase Activity, Perturbing the NAD(P)H-Based Viability Assays. Cells 2015, 4, 427-451. https://doi.org/10.3390/cells4030427
Aleshin VA, Artiukhov AV, Oppermann H, Kazantsev AV, Lukashev NV, Bunik VI. Mitochondrial Impairment May Increase Cellular NAD(P)H: Resazurin Oxidoreductase Activity, Perturbing the NAD(P)H-Based Viability Assays. Cells. 2015; 4(3):427-451. https://doi.org/10.3390/cells4030427
Chicago/Turabian StyleAleshin, Vasily A., Artem V. Artiukhov, Henry Oppermann, Alexey V. Kazantsev, Nikolay V. Lukashev, and Victoria I. Bunik. 2015. "Mitochondrial Impairment May Increase Cellular NAD(P)H: Resazurin Oxidoreductase Activity, Perturbing the NAD(P)H-Based Viability Assays" Cells 4, no. 3: 427-451. https://doi.org/10.3390/cells4030427