
HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities

Abstract
:1. Introduction
2. The Role of Autophagy in Cancer
2.1. Targeted Blockade of Autophagic Flux as a Therapeutic Intervention
2.2. Pitfalls of Using Autophagic Flux Inhibitors as Adjunct Therapy to Anticancer Treatment
3. The Histone Deacetylase Family and Its Role in Cancer
3.1. Class I HDACs and Their Role in Cancer
3.2. Class II HDACs and Their Role in Cancer
3.2.1. Class IIa HDACs
3.2.2. Class IIb HDACs
3.3. Class IV HDAC11 and Its Role in Cancer
3.4. HDAC Inhibitors as Promising Anticancer Agents
4. HDACs and Their Role in Autophagy

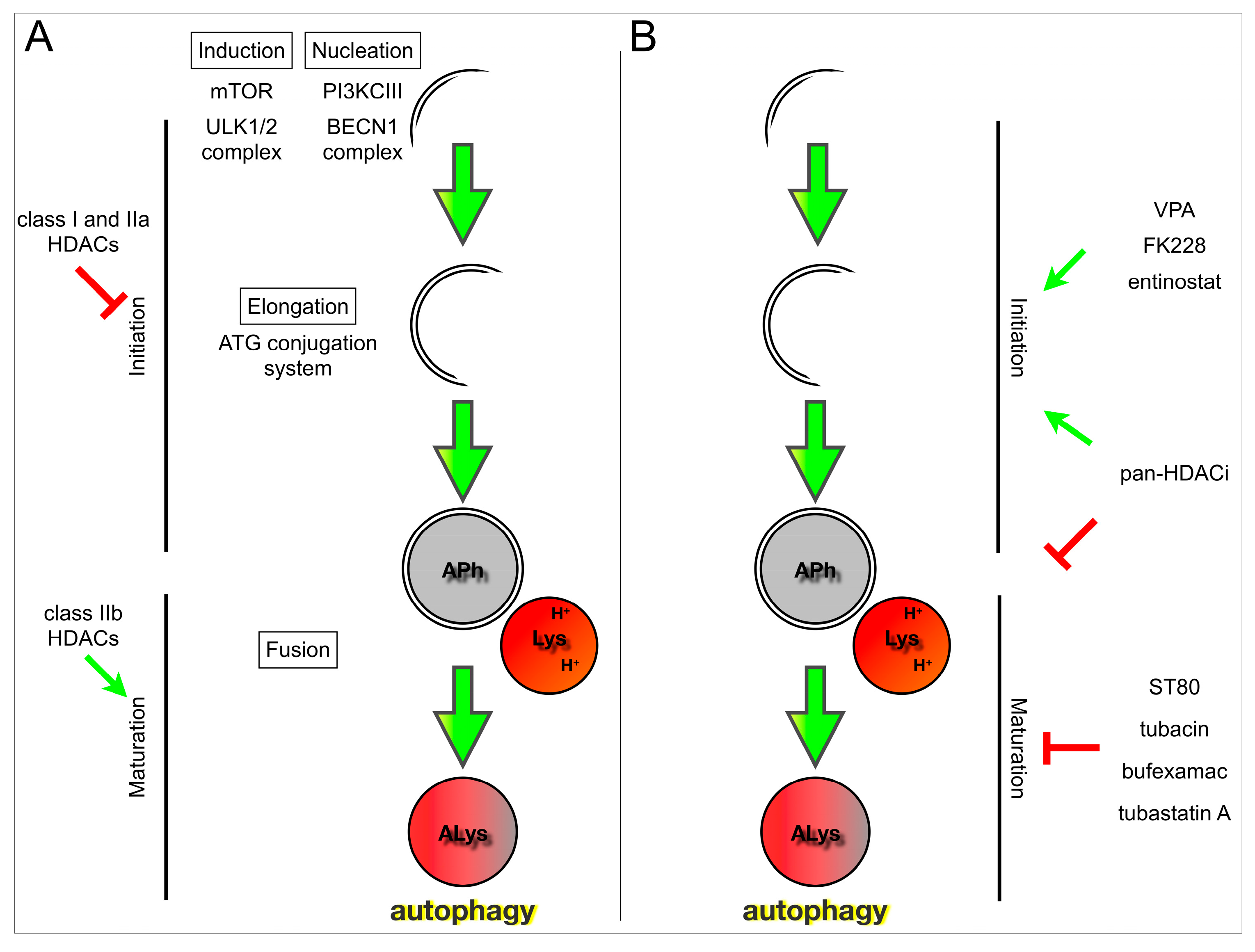{kind=link}
| Methods for the Detection of Autophagy | ||
|---|---|---|
| Method | Description | Technique |
| Morphology | ||
| Autophagosome visualization | ||
| autophagic vesicles ↑ | increase in autophagy-associated structures | EM |
| LC3-GFP ↑ | increase in LC3-containing autophagosomes: characteristic puncta formation | FM |
| LC3-ubiquitin overlapping puncta ↑ | as with LC3 puncta; specific for ubiquitin-tagged proteins targeted for destruction via autophagy | FM |
| Acidic compartment dyes (autolysosomes, lysosomes) | ||
| AO ↑/LTR ↑/MDC puncta ↑ | Acridine Orange/LysoTrackerRed/MonoDansylCadaverine; increase in acidic compartment | FM |
| Components targeted for autophagic degradation | ||
| protein aggregates ↑ | accelerated aggregate formation or impairment of processing | FM |
| mitochondria ↑ | decrease in mitochondrial turnover (e.g., Tom20) | FM/WB |
| Quantification | ||
| Early-stage autophagy | ||
| BECN1 ↑; Vps34 ↑ | accumulation or upregulation of proteins involved in early autophagy (nucleation) | WB/PCR |
| ATG3 ↑; ATG5 ↑; ATG7 ↑ | accumulation or upregulation of protein involved in early autophagy (elongation) | WB/PCR |
| ac-ATG7 ↑ | associated with inhibition of early autophagy | IP-WB |
| Autophagosomes and lysosomes | ||
| autophagosomes (EM) ↑ | enriched number of autophagosomes per square millimeter | EM |
| LC3-II ↑ | increase in LC3-conjugated autophagosomes | WB |
| LC3-GFP ↑; LC3-GFP ↓ | increase in autophagosomes; decrease after fusion with acidic compartment | FACS |
| ac-LC3-II ↑ | associated with decreased autophagy | IP-WB |
| LC3B ↑; GABARAP ↑ | ATG8 homologues; transcriptional upregulation or increase in autophagosomes | WB/PCR |
| RAB7 ↑ | accumulation or upregulation of protein involved in late autophagy (fusion); marker for endosomes | WB/PCR |
| LAMP2 ↑ | lysosome-associated membrane protein 2; increase in lysosomes | WB |
| Acidic compartment dyes (autolysosomes, lysosomes) | ||
| AO ↑; LTR ↑; MDC puncta ↑ | Acridine Orange/LysoTrackerRed/MonoDansylCadaverine; increase in acidic compartment | FACS |
| Cyto-ID ↑ | cationic amphiphilic tracer; increase in autophagic structures | FACS |
| Flux studies | ||
| + early autophagy inhibitor | e.g., 3-methyladenine: early stage autophagy inhibitor; should decrease autophagosomal markers | WB (LC3-II, ATG7)/ FM-quant. (EGFP-LC3, MDC)/ |
| + late autophagy inhibitor | late autophagy inhibitors: NH4Cl, CQ, bafilomycin; should increase autophagosomal markers | WB (LC3-II)/ FACS (EGFP-LC3) |
| + lysosomal protease inhibition | e.g., Pepstatin A/E64d; should increase autophagosomal markers | WB |
| p62/SQSTM1 ↑ | accumulation, marker for inhibition of late stages of autophagy | WB |
| p62 ↓ and + late stage autophagy inhibitor p62 ↑ | increase autophagic flux | WB |
| tandem fluorescent-tagged LC3 | mCherry-EGFP-LC3B or mRFP-GFP-LC3B: Yellow puncta reflect colocalized red and green signals, representing autophagosomes; red puncta represent successful fusion to autolysosomes | FM-quant. |
| Indirect flux measurements | ||
| p-p70S6K ↓ | indicates block of mTOR pathway | WB |
| p-mTOR ↑ | activation of mTOR pathway, leading to an inhibition of autophagy | WB |
| p-AMPK ↑ | activation of AMPK signaling which inhibits mTOR | WB |
| + mitophagy inducer | e.g., Parkin-dependent mitophagy (Tom20 degradation/accumulation) | WB |
| Class | Member | Targeted by | Context | Stress Status | Methods for the Detection of Autophagy | Overall Effect of HDAC KD/KO on Autophagy | Citation | ||
|---|---|---|---|---|---|---|---|---|---|
| Morphology | Quantification | Flux Studies | |||||||
| I | HDAC1 | siRNA | HCC | nutrient rich | autophagic vesicles (EM) ↑ | LC3-II ↑ | −3-MA: LC3-II ↑ +3-MA: LC3-II ↓ | Induced. 72 h post-transfection | [126] |
| HDAC1 | shRNA | MM | nutrient rich +p73 | n.d. | BECN1 ↑ ATG3 ↑ LC3-II ↑ | n.d. | Induced. 48 h post-transfection | [127] | |
| Hdac1 and Hdac2 | double knockout | mouse (skeletal muscle) | nutrient rich | LC3 puncta (FM) ↔ | ATG5 ↓ ATG7 ↓ LC3-II ↑ | p-AMPK ↑ p62 ↑ | Inhibition of initiation | [128] | |
| starvation | LC3 puncta (FM) ↓ | LC3-I and LC3-II ↑ | p62 ↑↑ | Inhibition of initiation | |||||
| IIa | HDAC4 | siRNA | GC | nutrient rich | LC3 puncta (FM) ↑ | BECN1 ↑ ATG7 ↑ LC3-II ↑ | −3-MA: ATG7 ↑ +3-MA: ATG7 ↓ −3-MA: LC3-II ↑ +3-MA: LC3-II ↓ | Induction. 48–72 h post-transfection | [82] |
| HDACs 4 and 5 | miRNA9 * | WM | nutrient rich | n.d. | RAB7 ↑ LC3B ↑ | n.d. | Induced. 24 h post-transfection | [129] | |
| HDAC5 | siRNA | mixed | nutrient rich | autophagic vesicles (EM) ↑ LC3 puncta (FM) ↑ | LC3-II ↑ | +NH4Cl: LC3-II ↑↑ | Induced. 24–72 h post-transfection | [130] | |
| IIb | HDAC6 | siRNA | HeLa cells | starvation | LC3 puncta (FM) ↑ | LC3-II ↑ | p62 ↑ ac-LC3-II ↑ | Blocked. 72 h post-transfection | [131] |
| Hdac6 | knockout | MEF | nutrient rich | autophagic vesicles (EM) ↑ | LC3-II ↑ | p62 ↑ mCherry-GFP-LC3 (FM): yellow ↑ | Blocked—QC autophagy only | [132] | |
| starvation | n.d. | LC3-II ↑ | mCherry-GFP-LC3 (FM): yellow ↔ | Blocked—QC autophagy only | |||||
| nutrient rich + proteasome inhibition | LC3 puncta (FM) ↑ LC3-ubiquitin overlapping puncta (FM) ↑ | +MG132: LC3-II ↑↑ | n.d. | Blocked—QC autophagy only | |||||
| HDAC6 | siRNA | breast cancer | nutrient rich | LC3 puncta (FM) ↑ | LC3B ↔ | n.d. | Blocked. | [134] | |
| nutrient rich + proteasome inhibition | +BORT: LC3 puncta (FM) ↓ | +BORT: LC3B ↓ | n.d. | Blocked. | |||||
| Hdac6 | knockout | mouse | nutrient rich + smoke | protein aggregates (FM) ↑ | autophagosomes (EM) ↑ a | +lysosomal protease inhibition: LC3B-II ↔ | Blocked. | [135] | |
| Hdac6 | knockout; siRNA | MEF | nutrient rich + mitochond. dysfunction | mitochondria (FM) ↑ | n.d. | mitochondrial marker (Tom20;WB) ↑ | Blocked. Impaired mitophagy | [136] | |
| HDAC10 | siRNA | NB | nutrient rich | autophagic vesicles (EM) ↑ LC3 puncta (FM) ↑ | LC3-II ↑ AO ↑ LAMP2 ↑ | +CQ: LC3-II ↔ p62↑ EGFP-LC3 (FACS) ↓ mCherry-EGFP-LC3 (FM): yellow ↑ | Blocked. 72–144 h post-transfection | [11] | |
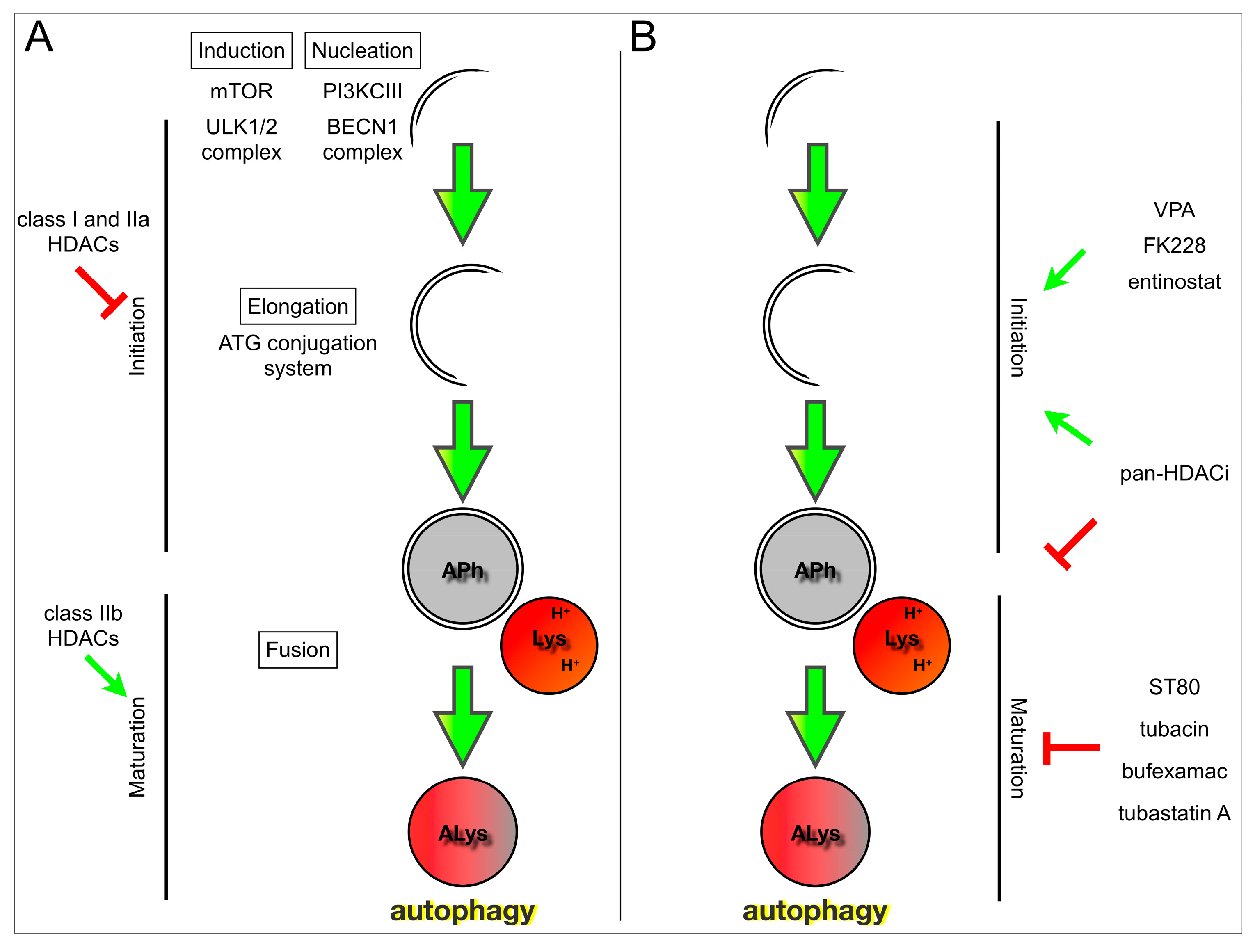
5. Targeting Autophagy with HDAC Inhibition in Cancer
| Inhibitor | Target(s) | Conc. (µM) | Context | Stress Status | Methods for the Detection of Autophagy | Overall Effect on Autophagy | Citation | ||
|---|---|---|---|---|---|---|---|---|---|
| Morphology | Quantification | Flux Studies | |||||||
| vorinostat | pan | 5–20 | MEF | nutrient rich | LC3 puncta (FM) ↑ | LC3-II ↑ LC3 mRNA ↑ | mTOR blocked: p-p70S6K ↓ p62 ↓ +BAF: LC3-II ↑↑ | Induced. 8–48 h treatment | [141] |
| vorinostat | pan | 1–2 | ovarian cancer | nutrient rich + chemo | vacuoles (EM) ↑ LC3 puncta (FM) ↑ | AO ↑ | n.d. | Induced. 24–120 h treatment | [137] |
| vorinostat | pan | 2–50 | chondro-sarcoma | nutrient rich | vacuoles (EM) ↑ | LC3-II ↑ | +3-MA: LC3-II ↓ | Induced. 24–48 h treatment | [138] |
| vorinostat | pan | 8 | cervical cancer | nutrient rich | LTR (FM) ↑ LC3 puncta (FM) ↑ | LC3-II ↑ | n.d. | Induced. 24 h treatment | [139] |
| vorinostat | pan | 1 | DS-AMKL | nutrient rich | n.d. | ROS ↑ Cyto-ID (FACS) ↓ | +CQ: LC3-GFP (FACS) ↓ | Blocked. | [57] |
| TSA | pan | 0.04–1 | colon cancer | nutrient rich | n.d. | LC3-II ↑ ATG5 ↑ AO ↑ | n.d. | Induced. 24 h treatment | [42] |
| nutrient rich + radiation | n.d. | LC3-II ↑ AO ↑↑ | n.d. | Induced. 24 h treatment | |||||
| TSA | pan | 0.4 | DS-AMKL | nutrient rich | n.d. | ROS ↑ LC3-GFP (FACS) ↑ | n.d. | Blocked. | [57] |
| panobinostat | pan | 0.02–0.05 | triple negative breast cancer | nutrient rich | LC3 puncta (FM) ↑ | BECN1 ↑ LC3-II ↑ | p62 ↓ +CQ: p62 ↑ | Induced. 16 h treatment | [143] |
| panobinostat | pan | 0.1–0.4 | DS-AMKL | nutrient rich | n.d. | Cyto-ID (FACS) ↓ | +CQ: LC3-GFP (FACS) ↓ | Blocked. | [57] |
| panobinostat | pan | 0.1 | breast cancer | nutrient rich | LC3 puncta (FM) ↑ | BECN1 ↑ Vps34 ↑ LC3-II ↑ | p62 ↓ m-RFP-GFP-LC3 (FM): red ↑ | Induced. 24–48 h treatment | [142] |
| panobinostat | pan | 0.05 | colon cancer | nutrient rich | LC3 puncta (FM) ↑ AO (FM) ↑ | LC3-II ↑ | p62 ↓ +BAF: p62 ↑ | Induced. 24–48 h treatment | [144] |
| nutrient rich + DAPK | LC3 puncta (FM) ↑↑ AO (FM) ↑↑ | LC3-II ↑↑ | p62 ↓ +BAF: p62 ↑ | Induced. 24–48 h treatment | |||||
| PCI-24781 | pan | 0.5 | MPNST | nutrient rich | vacuoles (EM) ↑ AO (FM) ↑ LC3 puncta (FM) ↑ | AO ↑ LC3-II ↑ | +BAF: LC3-II ↑↑ +CQ: LC3-II ↑↑ | Induced. 24 h treatment | [140] |
| VPA | Class I | 2000 | DS-AMKL | nutrient rich | n.d. | ROS ↑ ac-ATG7 ↑ | 12–17 h: LC3-GFP (FACS) ↓ 17–24 h: LC3-GFP (FACS) ↑ | Induced early (12–17 h) Blocked later (17–48 h) | [57] |
| starvation | n.d. | ROS ↔ | LC3-GFP (FACS) ↔ | No effect. 24 h treatment | |||||
| VPA | Class I | 1000 | glioma | nutrient rich | vacuoles (EM) ↑ LC3 puncta (FM) ↑ MDC puncta (FM)↑ | LC3-II ↑ MDC ↑ | +3-MA: LC3-GFP (FM) ↓ +3-MA: LC3-II ↓ +3-MA: MDC (FM) ↓ | Induced. 48–96 h treatment | [145] |
| FK228 | 1, 2 | 0.148 | cervical cancer | nutrient rich | vacuoles (EM) ↑ LC3 puncta (FM) ↑ MDC (FM) ↑ LTR (FM) ↑ | LC3-II ↑ | n.d. | Induced. 24 h treatment | [139] |
| FK228 | 1, 2 | 0.0025 | MRT | nutrient rich | vacuoles (EM) ↑ | LC3-II ↑ | n.d. | Induced. 24–48 h treatment | [146] |
| Entinostat | 1, 2, 3 | 3–5 | colon cancer | nutrient rich | LC3 puncta (FM) ↑ vacuoles (EM) ↑ | LC3-II ↑ ATG7 ↑ p-ERK ↑ | n.d. | Induced. 2–24 h treatment | [147] |
| MGCD0103 | 1, 2, 3, 11 | 0.5 and 3 | CLL | nutrient rich | n.d. | mRNA: ATG7 ↓ GABARAP ↓ WB: BECN1 ↓ ATG5 ↓ Cyto-ID (FACS) ↓ | p-mTOR, early ↑ later ↓ Time-dependent: LC3-II ↓ p62 ↓ +CQ: LC3-II ↓ +CQ: p62 ↓ | Inhibition of initiation. 2–48 h treatment | [148] |
| bufexamac | Class IIb | 30 | NB | nutrient rich | n.d. | AO ↑ | mCherry-EGFP-LC3 (FM): yellow ↑ | Blocked. 24 h treatment | [11] |
| bufexamac | Class IIb | 30 | MB | nutrient rich | n.d. | n.d. | p62 ↑ after 6 h mCherry-EGFP-LC3 (FM): yellow ↑ | Blocked. 24 h treatment | Oehme, unpublished data |
| ST80 | 6 | 50 | RMS | nutrient rich + proteasome inhibition | n.d. | +/− BORT: LTR (FACS) ↔ | p62 ↑ +BORT: p62 ↑↑ | Blocked—PQC. No change in flux. 48 h treatment | [149] |
| tubacin | 6 | 2 | cervical cancer | nutrient rich | LC3 puncta (FM) ↑ | LC3-II ↑ ac-LC3-II ↑ | p62 ↑ +CQ: LC3-II ↔ | Blocked. 2–24 h treatment | [131] |
| starvation | LC3 puncta (FM) ↑↑ | ac-LC3-II ↑ (partial) | p62 ↑ | ||||||
6. Conclusion
Acknowledgments
Author Contribution
Conflicts of Interest
References
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Fulda, S. Receptor proteins in selective autophagy. Int. J. Cell Biol. 2012, 2012, 673290. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Linke, J.P.; Bock, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 2004, 112, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, B.; Cai, D.; Zou, X. Therapeutic potential of histone deacetylase inhibitors in pancreatic cancer. Cancer Lett. 2014, 347, 183–190. [Google Scholar] [CrossRef] [PubMed]
- United States Food and Drug Administration. Zolinza (R) (Vorinostat) Product Label. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021991s002lbl.pdf (accessed on 30 September 2014).
- United States Food and Drug Administration. Istodax (R) (Romidepsin) Product Label. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022393s013lbl.pdf (accessed on 2 March 2015).
- Therapeutic Goods Administration, A. Search: “Vorinostat” and “Romidepsin”. Available online: https://www.tga.gov.au/orphan-drugs#summary-v (accessed on 30 September 2014).
- ClinicalTrialsRegister.eu Search: “Vorinostat”. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=vorinostat (accessed on 30 September 2014).
- AustralianClinicalTrials.gov.au Search: “Vorinostat”. Available online: https://www.australianclinicaltrials.gov.au/trials?populate=vorinostat (accessed on 30 September 2014).
- ClinicalTrials.gov Search: “Vorinostat” or “HDAC Inhibitor”. Available online: http://www.clinicaltrials.gov/ct2/results?term=vorinostat&Search=Search (accessed on 20 February 2014).
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.; Levine, B.; Debnath, J. Autophagy and cancer metabolism. Methods Enzymol. 2014, 542, 25–57. [Google Scholar] [PubMed]
- Hou, W.; Zhang, Q.; Yan, Z.; Chen, R.; Zeh Iii, H.J.; Kang, R.; Lotze, M.T.; Tang, D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013, 4, e966. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Cescon, M.; Bonaldo, P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014, 10, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, T. Autophagy: A regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 2004, 313, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N.; et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Belaid, A.; Cerezo, M.; Chargui, A.; Corcelle-Termeau, E.; Pedeutour, F.; Giuliano, S.; Ilie, M.; Rubera, I.; Tauc, M.; Barale, S.; et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013, 73, 4311–4322. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.B.; Hui, B.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Gu, C.Y.; Yang, H.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin. Cancer Res. 2011, 17, 6229–6238. [Google Scholar] [CrossRef] [PubMed]
- Hundeshagen, P.; Hamacher-Brady, A.; Eils, R.; Brady, N.R. Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol. 2011, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Bock, B.C.; Tagscherer, K.E.; Fassl, A.; Kramer, A.; Oehme, I.; Zentgraf, H.W.; Keith, M.; Roth, W. The PEA-15 protein regulates autophagy via activation of JNK. J. Biol. Chem. 2010, 285, 21644–21654. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gu, C.; Zhong, D.; Shi, L.; Kong, Y.; Zhou, Z.; Liu, S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014, 355, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Mo, N.; Lu, Y.K.; Xie, W.M.; Liu, Y.; Zhou, W.X.; Wang, H.X.; Nong, L.; Jia, Y.X.; Tan, A.H.; Chen, Y.; et al. Inhibition of autophagy enhances the radiosensitivity of nasopharyngeal carcinoma by reducing Rad51 expression. Oncol. Rep. 2014, 32, 1905–1912. [Google Scholar] [PubMed]
- He, G.; Wang, Y.; Pang, X.; Zhang, B. Inhibition of autophagy induced by TSA sensitizes colon cancer cell to radiation. Tumour Biol. 2014, 35, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Poklepovic, A.; Gewirtz, D.A. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 2014, 10, 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- Puustinen, P.; Rytter, A.; Mortensen, M.; Kohonen, P.; Moreira, J.M.; Jaattela, M. CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J. Cell Biol. 2014, 204, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, V.O.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Zhivotovsky, B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy 2012, 8, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhang, X.; Sun, H.; Zhang, J.; Yan, M.; Zhang, H. Autophagy inhibition promotes 5-fluorouraci-induced apoptosis by stimulating ROS formation in human non-small cell lung cancer A549 cells. PLoS One 2013, 8, e56679. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; el Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qi, H.; Taylor, R.; Xu, W.; Liu, L.F.; Jin, S. The role of autophagy in mitochondria maintenance: Characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy 2007, 3, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Metivier, D.; Galluzzi, L.; Perfettini, J.L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Aguilera, M.O.; Colombo, M.I. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy 2012, 8, 1741–1756. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Tang, B.L. Sirtuins’ modulation of autophagy. J. Cell. Physiol. 2013, 228, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J.; et al. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Udeshi, N.D.; Wolfson, N.A.; Pitcairn, C.A.; Sullivan, E.D.; Jaffe, J.D.; Svinkina, T.; Natoli, T.; Lu, X.; Paulk, J.; et al. An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol. 2014, 9, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009, 23, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Santoro, F.; Botrugno, O.A.; dal Zuffo, R.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Barozzi, I.; Senese, S.; Fornasari, L.; Moretti, S.; et al. A dual role for Hdac1: Oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.H.; Wang, C.Y.; Zhang, W.L.; Zhang, J.T.; Yuan, C.H.; Zhao, P.W.; Lin, Y.Y.; Hong, S.; Li, C.Y.; Wang, L.; et al. Histone deacetylase HDAC4 promotes gastric cancer SGC-7901 cells progression via p21 repression. PLoS One 2014, 9, e98894. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P.; et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- De Zoeten, E.F.; Wang, L.; Butler, K.; Beier, U.H.; Akimova, T.; Sai, H.; Bradner, J.E.; Mazitschek, R.; Kozikowski, A.P.; Matthias, P.; et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol. Cell. Biol. 2011, 31, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W., Jr.; Castle, V.P.; Kwok, R.P. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 2011, 13, 726–734. [Google Scholar] [PubMed]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, A.R.; Yao, T.P. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem. 2002, 277, 3350–3356. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.Y.; Lee, C.H.; Komarov, A.; Han, C.C.; Evans, R.M. Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J. Biol. Chem. 2002, 277, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.D.; Cai, R.; Bhatia, U.; Asselbergs, F.A.; Song, C.; Terry, R.; Trogani, N.; Widmer, R.; Atadja, P.; Cohen, D.; et al. Isolation and characterization of a novel class II histone deacetylase, HDAC10. J. Biol. Chem. 2002, 277, 6656–6666. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.J.; Liu, J.; Bertos, N.R.; Yang, X.J. Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 2002, 30, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Kotian, S.; Liyanarachchi, S.; Zelent, A.; Parvin, J.D. Histone deacetylases 9 and 10 are required for homologous recombination. J. Biol. Chem. 2011, 286, 7722–7726. [Google Scholar] [CrossRef] [PubMed]
- Lai, I.L.; Lin, T.P.; Yao, Y.L.; Lin, C.Y.; Hsieh, M.J.; Yang, W.M. Histone deacetylase 10 relieves repression on the melanogenic program by maintaining the deacetylation status of repressors. J. Biol. Chem. 2010, 285, 7187–7196. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jeong, E.G.; Choi, M.C.; Kim, S.H.; Park, J.H.; Song, S.H.; Park, J.; Bang, Y.J.; Kim, T.Y. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol. Cells 2010, 30, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Park, B.L.; Kim, Y.J.; Cheong, H.S.; Lee, S.O.; Han, C.S.; Yoon, J.H.; Park, J.H.; Chang, H.S.; Park, C.S.; Lee, H.S.; et al. HDAC10 promoter polymorphism associated with development of HCC among chronic HBV patients. Biochem. Biophys. Res. Commun. 2007, 363, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, M.; Crompot, E.; Meuleman, N.; Mineur, P.; Bron, D.; Lagneaux, L.; Stamatopoulos, B. HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics 2012, 7, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhu, S.; Wu, C.; Kang, J. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J. Biol. Chem. 2013, 288, 28021–28033. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Voelter-Mahlknecht, S.; Ho, A.D.; Mahlknecht, U. Chromosomal organization and localization of the novel class IV human histone deacetylase 11 gene. Int. J. Mol. Med. 2005, 16, 589–598. [Google Scholar] [PubMed]
- Liu, H.; Hu, Q.; Kaufman, A.; D’Ercole, A.J.; Ye, P. Developmental expression of histone deacetylase 11 in the murine brain. J. Neurosci. Res. 2008, 86, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Q.; D’Ercole, A.J.; Ye, P. Histone deacetylase 11 regulates oligodendrocyte-specific gene expression and cell development in OL-1 oligodendroglia cells. Glia 2009, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bagui, T.K.; Sharma, S.S.; Ma, L.; Pledger, W.J. Proliferative status regulates HDAC11 mRNA abundance in nontransformed fibroblasts. Cell Cycle 2013, 12, 3433–3441. [Google Scholar] [CrossRef] [PubMed]
- Deubzer, H.E.; Schier, M.C.; Oehme, I.; Lodrini, M.; Haendler, B.; Sommer, A.; Witt, O. HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 2013, 132, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.; Akerstrom, G.; Westin, G. Mutational analyses of WNT7A and HDAC11 as candidate tumour suppressor genes in sporadic malignant pancreatic endocrine tumours. Clin. Endocrinol. 2007, 66, 110–114. [Google Scholar]
- Sahakian, E.; Powers, J.J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Friend, C.; Scher, W.; Holland, J.G.; Sato, T. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: Stimulation of erythroid differentiation by dimethyl sulfoxide. Proc. Natl. Acad. Sci. USA 1971, 68, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Marks, B.D.; Fakhoury, S.A.; Frazee, W.J.; Eliason, H.C.; Riddle, S.M. A substrate-independent TR-FRET histone deacetylase inhibitor assay. J. Biomol. Screen 2011, 16, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Wagner, F.F.; Kaya, T.; Gale, J.P.; Aidoud, N.; Davoine, E.L.; Lazzaro, F.; Weiwer, M.; Zhang, Y.L.; Holson, E.B.; et al. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013, 56, 4816–4820. [Google Scholar] [CrossRef] [PubMed]
- Krennhrubec, K.; Marshall, B.L.; Hedglin, M.; Verdin, E.; Ulrich, S.M. Design and evaluation of “Linkerless” hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Saunders, L.; Schlimme, S.; Valkov, V.; Wagner, J.M.; Kratz, F.; Sippl, W.; Verdin, E.; Jung, M. Pyridylalanine-containing hydroxamic acids as selective HDAC6 inhibitors. ChemMedChem 2009, 4, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Fullgrabe, J.; Heldring, N.; Hermanson, O.; Joseph, B. Cracking the survival code: Autophagy-related histone modifications. Autophagy 2014, 10, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.J.; Noh, J.H.; Kim, J.K.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Lee, J.Y.; Park, H.; et al. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS ONE 2012, 7, e34265. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.; Alla, V.; Meier, C.; Nettelbeck, D.M.; Herchenroder, O.; Putzer, B.M. Eradication of metastatic melanoma through cooperative expression of RNA-based HDAC1 inhibitor and p73 by oncolytic adenovirus. Oncotarget 2014, 5, 5893–5907. [Google Scholar] [PubMed]
- Moresi, V.; Carrer, M.; Grueter, C.E.; Rifki, O.F.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Jia, X.; Azab, A.K.; Maiso, P.; Ngo, H.T.; Azab, F.; Runnels, J.; Quang, P.; Ghobrial, I.M.; et al. microRNA-dependent modulation of histone acetylation in Waldenstrom macroglobulinemia. Blood 2010, 116, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.; Castronovo, V.; Matheus, N.; Polese, C.; Peulen, O.; Gonzalez, A.; Boxus, M.; Verdin, E.; Thiry, M.; Dequiedt, F.; et al. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differ. 2012, 19, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.P.; Zhou, D.; Ouyang, D.Y.; Xu, L.H.; Wang, Y.; Wang, L.X.; Pan, H.; He, X.H. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 2013, 441, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.C.; Tan, B.C.; Chen, W.H.; Lin, Y.H.; Huang, J.Y.; Chang, H.Y.; Sun, H.Y.; Hsu, P.H.; Liou, G.G.; Shen, J.; et al. Reversible acetylation regulates salt-inducible kinase (SIK2) and its function in autophagy. J. Biol. Chem. 2013, 288, 6227–6237. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Liao, W.S.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C., Jr.; et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tanaka, K.; Sakimura, R.; Okada, T.; Nakamura, T.; Li, Y.; Takasaki, M.; Nakabeppu, Y.; Iwamoto, Y. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 2008, 28, 1585–1591. [Google Scholar] [PubMed]
- Oh, M.; Choi, I.K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Torres, K.; Liu, J.; Hernandez, B.; Young, E.; Belousov, R.; Bolshakov, S.; Lazar, A.J.; Slopis, J.M.; McCutcheon, I.E.; et al. Autophagic survival in resistance to histone deacetylase inhibitors: Novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011, 71, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fiskus, W.; Yong, B.; Atadja, P.; Takahashi, Y.; Pandita, T.K.; Wang, H.G.; Bhalla, K.N. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 6841–6846. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Balusu, R.; Fiskus, W.; Mudunuru, U.; Venkannagari, S.; Chauhan, L.; Smith, J.E.; Hembruff, S.L.; Ha, K.; Atadja, P.; et al. Combination of pan-histone deacetylase inhibitor and autophagy inhibitor exerts superior efficacy against triple-negative human breast cancer cells. Mol. Cancer Ther. 2012, 11, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Gandesiri, M.; Chakilam, S.; Ivanovska, J.; Benderska, N.; Ocker, M.; di Fazio, P.; Feoktistova, M.; Gali-Muhtasib, H.; Rave-Frank, M.; Prante, O.; et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2012, 17, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Shao, C.J.; Chen, F.R.; Ng, H.K.; Chen, Z.P. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol. 2010, 12, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Adachi, S.; Matsubara, H.; Imai, T.; Yui, Y.; Mizushima, Y.; Hiraumi, Y.; Watanabe, K.; Kamitsuji, Y.; Toyokuni, S.Y.; et al. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer 2009, 124, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Gong, K.; Chen, C.; Wang, H.; Li, W. P38 MAP kinase functions as a switch in MS-275-induced reactive oxygen species-dependent autophagy and apoptosis in human colon cancer cells. Free Radic. Biol. Med. 2012, 53, 532–543. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.; Roland, O.; Cherrier-de Wilde, S.; Plawny, L.; van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Rapino, F.; Jung, M.; Fulda, S. BAG3 induction is required to mitigate proteotoxicity via selective autophagy following inhibition of constitutive protein degradation pathways. Oncogene 2014, 33, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Kong, Y.; Tan, W.; May, H.; Battiprolu, P.K.; Pedrozo, Z.; Wang, Z.V.; Morales, C.; Luo, X.; Cho, G.; et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 2014, 129, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Mahendran, A.; Yao, Y.; Ngo, L.; Venta-Perez, G.; Choy, M.L.; Kim, N.; Ham, W.S.; Breslow, R.; Marks, P.A.; et al. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. USA 2013, 110, 15704–15709. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koeneke, E.; Witt, O.; Oehme, I. HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities. Cells 2015, 4, 135-168. https://doi.org/10.3390/cells4020135
Koeneke E, Witt O, Oehme I. HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities. Cells. 2015; 4(2):135-168. https://doi.org/10.3390/cells4020135
Chicago/Turabian StyleKoeneke, Emily, Olaf Witt, and Ina Oehme. 2015. "HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities" Cells 4, no. 2: 135-168. https://doi.org/10.3390/cells4020135