Physiological Function and Characterization of TRPCs in Neurons

Abstract
:1. Introduction
2. TRPC Channels Properties and Their Mode of Activation
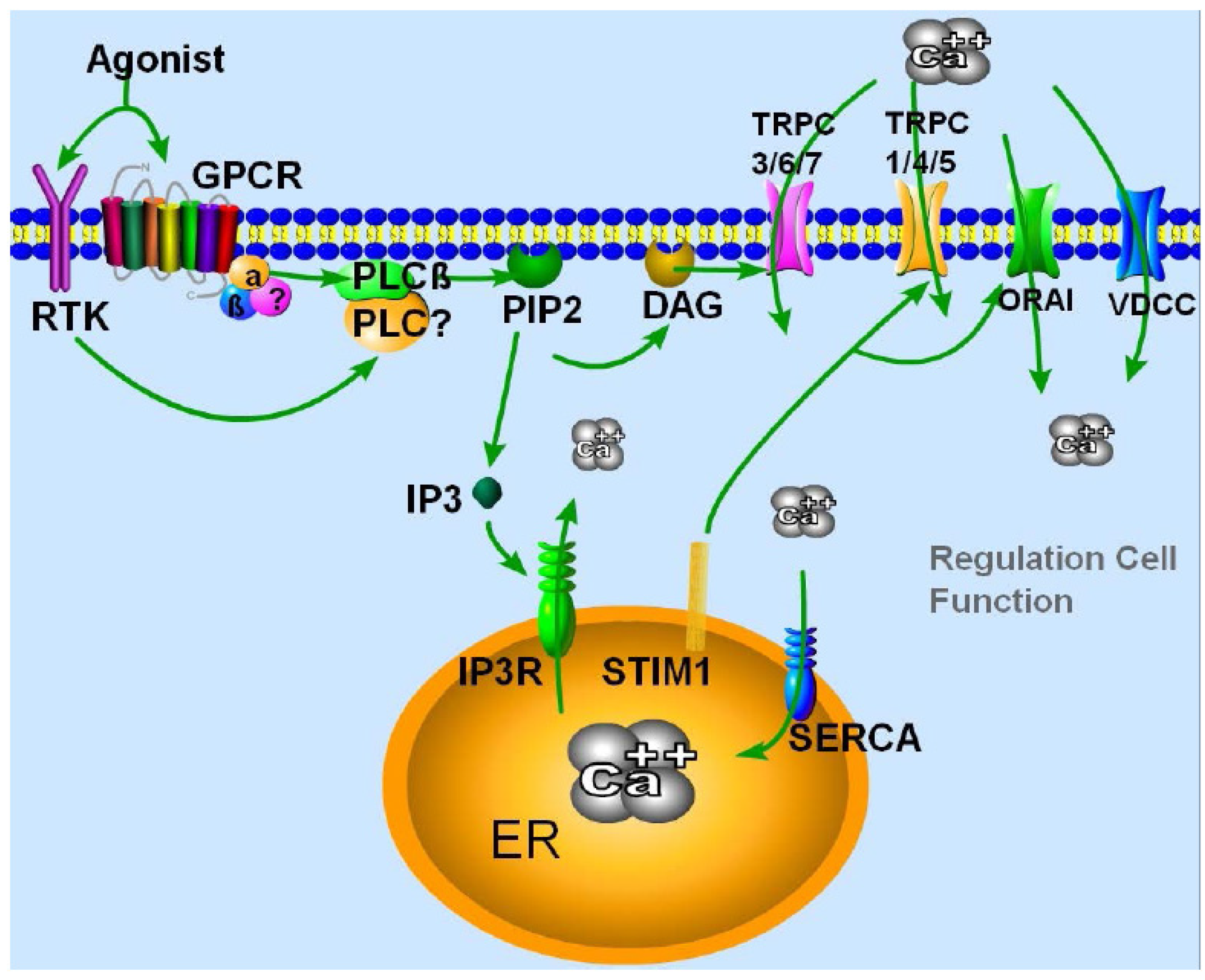

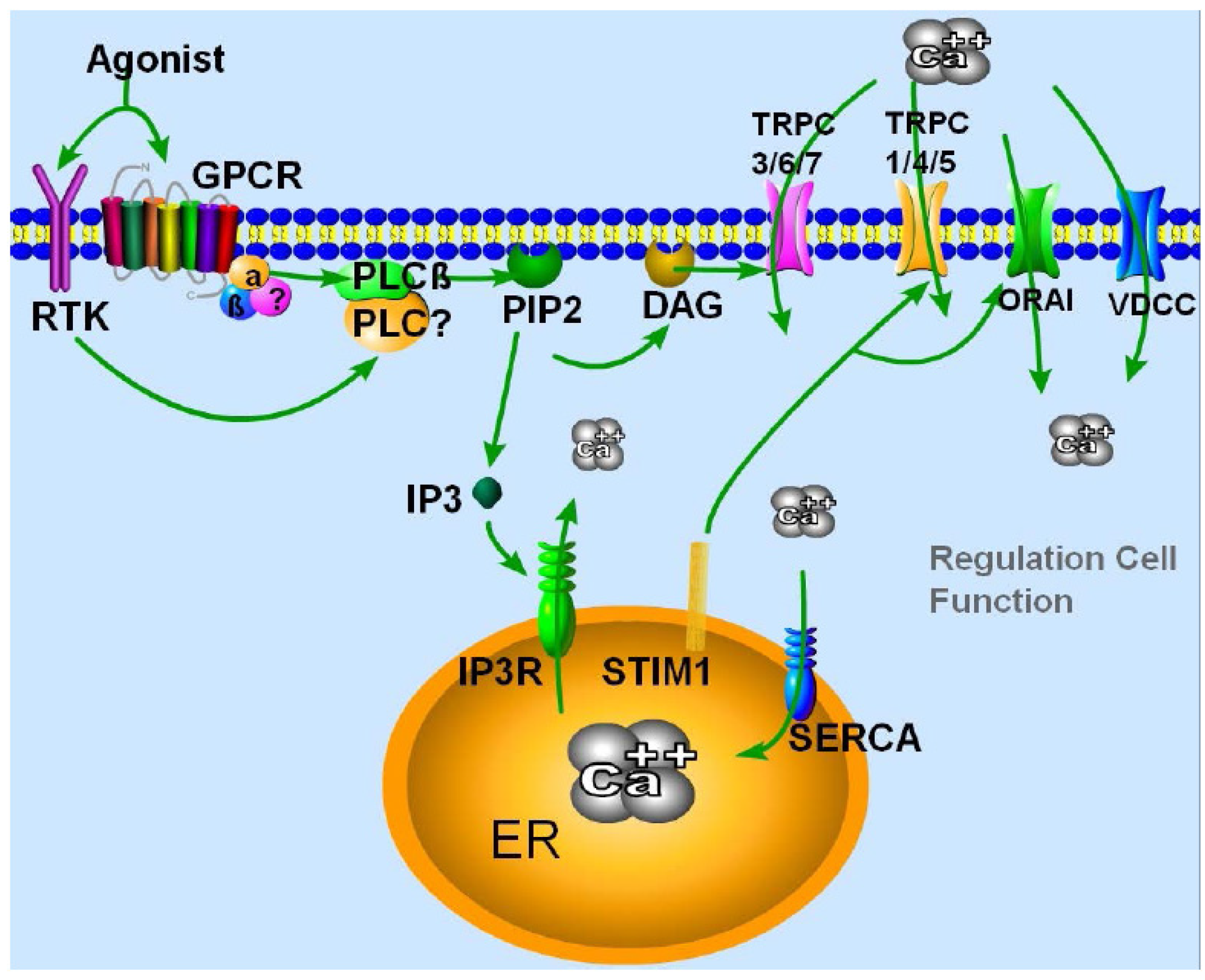{kind=link}
| Subfamily | Cellular expression in Neurons | Ion channel properties | References |
|---|---|---|---|
| TRPC1 | Brain, retina, peripheral axons and the mechanosensory terminals | Non selective, 16 pS current conductance, non-rectifying or mildly inward rectifying with a reverse potential of about +10 mV | [27,28,29,30] |
| TRPC2 | Dendritic tips of the vomeronasal sensory neurons and spermatozoa (mouse) | Partially selective with a Pca/PNa ratio of 2.7, 42 pS current conductance, non-rectifying | [31,32] |
| TRPC3 | Central nervous system (CNS) | Non selective, 60–66 pS current conductance, slightly dual (inward and outward) rectifying with a reverse potential of +5 mV | [33,34,35,36] |
| TRPC4 | CNS, retina | Non selective, 30–42 pS current conductance, dual (inward and outward) rectifying with a reverse potential of about +10 mV | [37,38,39,40] |
| TRPC5 | Brain, especially in fetal brain | Partially selective with a Pca/PNa ratio of 9, 47–66 pS current conductance, dual (inward and outward) rectifying as a homomer, outwardly rectifying when expressed with TRPC1 or TRPC4 | [30,38,40,41,42,43] |
| TRPC6 | Brain, retina | Partially selective with a Pca/PNa ratio of 5, 28–37 pS current conductance, dual (inward and outward) rectifying or inward rectifying | [33,35,44,45,46] |
| TRPC7 | CNS (human); weak in CNS (mouse) | Partially selective with a Pca/PNa ratio of 5.9, 25–50 pS current conductance, slightly outward rectifying | [35,44,47,48] |
3. Physiological Function of TRPCs in Neuronal Cells
3.1. TRPC1
3.2. TRPC2
3.3. TRPC3
3.4. TRPC4
3.5. TRPC5
3.6. TRPC6
3.7. TRPC7
4. TRPC Channels in Neurodegenerative Diseases
5. Conclusions
Abbreviations
| TRPC | transient receptor potential canonical cation channels |
| STIM | stromal interaction molecule |
| PKC | protein kinase C |
| ATP | adenosine triphosphate |
| PLC | phospholipase C |
| IP3R | inositol trisphosphate receptor |
| SERCA | sarco/endoplasmic reticulum Ca2+-ATPase |
| ER | endoplasmic reticulum |
| PM | plasma membrane |
| DAG | diacylglycerol |
| ORAI | calcium release-activated calcium channel protein |
| SOCE | store-operated calcium entry |
| GPCR | G-protein coupled receptor |
| BDNF | brain-derived neurotrophic factor |
| CNS | central nervous system |
| PD | Parkinson’s disease |
| AD | Alzheimer’s disease |
| HD | Huntington’s disease |
| RTKs | Receptor Tyrosine Kinases |
| Pca/PNa | Possible ratio. |
Acknowledgments
Conflicts of Interest
References
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Singh, B.B. TRPC channels and their implication in neurological diseases. CNS Neurol. Disord. Drug Targets 2010, 9, 94–104. [Google Scholar] [CrossRef]
- Yamamoto, S.; Wajima, T.; Hara, Y.; Nishida, M.; Mori, Y. Transient receptor potential channels in Alzheimer’s disease. Biochim. Biophys. Acta 2007, 1772, 958–967. [Google Scholar] [CrossRef]
- Reboreda, A.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. TRP channels and neural persistent activity. Adv. Exp. Med. Biol. 2011, 704, 595–613. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Putney, J.W. The physiological function of store-operated calcium entry. Neurochem. Res. 2011, 36, 1157–1165. [Google Scholar] [CrossRef]
- Smyth, J.T.; Hwang, S.Y.; Tomita, T.; DeHaven, W.I.; Mercer, J.C.; Putney, J.W. Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 2010, 14, 2337–2349. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, K.T.; Bandyopadhyay, B.C.; Pani, B.; Dietrich, A.; Paria, B.C.; Swaim, W.D.; Beech, D.; Yildrim, E.; Singh, B.B.; et al. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(–/–) mice. Proc. Natl. Acad. Sci. USA 2007, 104, 17542–17547. [Google Scholar] [CrossRef]
- Liao, Y.; Plummer, N.W.; George, M.D.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. USA 2009, 106, 3202–3206. [Google Scholar]
- Pan, Z.; Yang, H.; Reinach, P.S. Transient receptor potential (TRP) gene superfamily encoding cation channels. Hum. Genomics 2011, 5, 108–116. [Google Scholar] [CrossRef]
- Putney, J.W. Physiological mechanisms of TRPC activation. Pflugers Archiv 2005, 451, 29–34. [Google Scholar] [CrossRef]
- Hao, B.; Lu, Y.; Wang, Q.; Guo, W.; Cheung, K.H.; Yue, J. Role of STIM1 in survival and neural differentiation of mouse embryonic stem cells independent of Orai1-mediated Ca entry. Stem Cell Res. 2013, 12, 452–466. [Google Scholar]
- Pani, B.; Ong, H.L.; Liu, X.; Rauser, K.; Ambudkar, I.S.; Singh, B.B. Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE). J. Biol. Chem. 2008, 283, 17333–17340. [Google Scholar]
- Pani, B.; Bollimuntha, S.; Singh, B.B. The TR (i)P to Ca(2)(+) signaling just got STIMy: An update on STIM1 activated TRPC channels. Front. Biosci. 2012, 17, 805–823. [Google Scholar] [CrossRef]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef]
- Liu, X.; Ong, H.L.; Pani, B.; Johnson, K.; Swaim, W.B.; Singh, B.; Ambudkar, I. Effect of cell swelling on ER/PM junctional interactions and channel assembly involved in SOCE. Cell Calcium 2010, 47, 491–499. [Google Scholar] [CrossRef]
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116. [Google Scholar] [CrossRef]
- Minke, B.; Cook, B. TRP channel proteins and signal transduction. Physiol. Rev. 2002, 82, 429–472. [Google Scholar]
- Kiselyov, K.; van Rossum, D.B.; Patterson, R.L. TRPC channels in pheromone sensing. Vitam. Horm. 2010, 83, 197–213. [Google Scholar] [CrossRef]
- Bandyopadhyay, B.C.; Swaim, W.D.; Liu, X.; Redman, R.S.; Patterson, R.L.; Ambudkar, I.S. Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells. Role in apical Ca2+ influx. J. Biol. Chem. 2005, 280, 12908–12916. [Google Scholar]
- Liu, X.; Bandyopadhyay, B.C.; Singh, B.B.; Groschner, K.; Ambudkar, I.S. Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J. Biol. Chem. 2005, 280, 21600–21606. [Google Scholar]
- Andrade-Talavera, Y.; Duque-Feria, P.; Sihra, T.S.; Rodriguez-Moreno, A. Pre-synaptic kainate receptor-mediated facilitation of glutamate release involves PKA and Ca(2+) -calmodulin at thalamocortical synapses. J. Neurochem. 2013, 126, 565–578. [Google Scholar] [CrossRef]
- Wong, A.C.; Birnbaumer, L.; Housley, G.D. Canonical transient receptor potential channel subtype 3-mediated hair cell Ca(2+) entry regulates sound transduction and auditory neurotransmission. Eur. J. Neurosci. 2013, 37, 1478–1486. [Google Scholar] [CrossRef]
- Kerstein, P.C.; Jacques-Fricke, B.T.; Rengifo, J.; Mogen, B.J.; Williams, J.C.; Gottlieb, P.A.; Sachs, F.; Gomez, T.M. Mechanosensitive TRPC1 channels promote calpain proteolysis of talin to regulate spinal axon outgrowth. J. Neurosci. 2013, 33, 273–285. [Google Scholar] [CrossRef]
- Lohmann, C.; Bonhoeffer, T. A role for local calcium signaling in rapid synaptic partner selection by dendritic filopodia. Neuron 2008, 59, 253–260. [Google Scholar] [CrossRef]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef]
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656. [Google Scholar] [CrossRef]
- Liu, X.; Wang, W.; Singh, B.B.; Lockwich, T.; Jadlowiec, J.; O’Connell, B.; Wellner, R.; Zhu, M.X.; Ambudkar, I.S. Trp1, a candidate protein for the store-operated Ca(2+) influx mechanism in salivary gland cells. J. Biol. Chem. 2000, 275, 3403–3411. [Google Scholar] [CrossRef]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001, 29, 645–655. [Google Scholar] [CrossRef]
- Lucas, P.; Ukhanov, K.; Leinders-Zufall, T.; Zufall, F. A diacylglycerol-gated cation channel in vomeronasal neuron dendrites is impaired in TRPC2 mutant mice: Mechanism of pheromone transduction. Neuron 2003, 40, 551–561. [Google Scholar] [CrossRef]
- Liman, E.R.; Corey, D.P.; Dulac, C. TRP2: A candidate transduction channel for mammalian pheromone sensory signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 5791–5796. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Clapham, D.E.; Julius, D.; Montell, C.; Schultz, G. International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol. Rev. 2005, 57, 427–450. [Google Scholar] [CrossRef]
- Lemonnier, L.; Trebak, M.; Putney, J.W., Jr. Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium 2008, 43, 506–514. [Google Scholar] [CrossRef]
- Li, Y.; Jia, Y.C.; Cui, K.; Li, N.; Zheng, Z.Y.; Wang, Y.Z.; Yuan, X.B. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature 2005, 434, 894–898. [Google Scholar] [CrossRef]
- Philipp, S.; Cavalie, A.; Freichel, M.; Wissenbach, U.; Zimmer, S.; Trost, C.; Marquart, A.; Murakami, M.; Flockerzi, V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J. 1996, 15, 6166–6171. [Google Scholar]
- Schaefer, M.; Plant, T.D.; Obukhov, A.G.; Hofmann, T.; Gudermann, T.; Schultz, G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J. Biol. Chem. 2000, 275, 17517–17526. [Google Scholar]
- Warnat, J.; Philipp, S.; Zimmer, S.; Flockerzi, V.; Cavalie, A. Phenotype of a recombinant store-operated channel: Highly selective permeation of Ca2+. J. Physiol. 1999, 518, 631–638. [Google Scholar] [CrossRef]
- Philipp, S.; Hambrecht, J.; Braslavski, L.; Schroth, G.; Freichel, M.; Murakami, M.; Cavalie, A.; Flockerzi, V. A novel capacitative calcium entry channel expressed in excitable cells. EMBO J. 1998, 17, 4274–4282. [Google Scholar] [CrossRef]
- Okada, T.; Shimizu, S.; Wakamori, M.; Maeda, A.; Kurosaki, T.; Takada, N.; Imoto, K.; Mori, Y. Molecular cloning and functional characterization of a novel receptor-activated TRP Ca2+ channel from mouse brain. J. Biol. Chem. 1998, 273, 10279–10287. [Google Scholar]
- Yamada, H.; Wakamori, M.; Hara, Y.; Takahashi, Y.; Konishi, K.; Imoto, K.; Mori, Y. Spontaneous single-channel activity of neuronal TRP5 channel recombinantly expressed in HEK293 cells. Neurosci. Lett. 2000, 285, 111–114. [Google Scholar]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar]
- Shi, J.; Mori, E.; Mori, Y.; Mori, M.; Li, J.; Ito, Y.; Inoue, R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J. Physiol. 2004, 561, 415–432. [Google Scholar] [CrossRef]
- Huang, W.C.; Young, J.S.; Glitsch, M.D. Changes in TRPC channel expression during postnatal development of cerebellar neurons. Cell Calcium 2007, 42, 1–10. [Google Scholar] [CrossRef]
- Riccio, A.; Medhurst, A.D.; Mattei, C.; Kelsell, R.E.; Calver, A.R.; Randall, A.D.; Benham, C.D.; Pangalos, M.N. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res. Mol. Brain Res. 2002, 109, 95–104. [Google Scholar] [CrossRef]
- Okada, T.; Inoue, R.; Yamazaki, K.; Maeda, A.; Kurosaki, T.; Yamakuni, T.; Tanaka, I.; Shimizu, S.; Ikenaka, K.; Imoto, K.; et al. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca(2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J. Biol. Chem. 1999, 274, 27359–27370. [Google Scholar] [CrossRef]
- Riccio, A.; Mattei, C.; Kelsell, R.E.; Medhurst, A.D.; Calver, A.R.; Randall, A.D.; Davis, J.B.; Benham, C.D.; Pangalos, M.N. Cloning and functional expression of human short TRP7, a candidate protein for store-operated Ca2+ influx. J. Biol. Chem. 2002, 277, 12302–12309. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Hiesinger, P.R. The synaptic maintenance problem: Membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol. Neurodegener 2013, 8. [Google Scholar] [CrossRef]
- Bollimuntha, S.; Selvaraj, S.; Singh, B.B. Emerging roles of canonical TRP channels in neuronal function. Adv. Exp. Med. Biol. 2011, 704, 573–593. [Google Scholar] [CrossRef]
- Wu, X.; Zagranichnaya, T.K.; Gurda, G.T.; Eves, E.M.; Villereal, M.L. A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J. Biol. Chem. 2004, 279, 43392–43402. [Google Scholar]
- Boudes, M.; Uvin, P.; Pinto, S.; Freichel, M.; Birnbaumer, L.; Voets, T.; de Ridder, D.; Vennekens, R. Crucial role of TRPC1 and TRPC4 in cystitis-induced neuronal sprouting and bladder overactivity. PLoS One 2013, 8, e69550. [Google Scholar] [CrossRef]
- Alessandri-Haber, N.; Dina, O.A.; Chen, X.; Levine, J.D. TRPC1 and TRPC6 channels cooperate with TRPV4 to mediate mechanical hyperalgesia and nociceptor sensitization. J. Neurosci. 2009, 29, 6217–6228. [Google Scholar] [CrossRef]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Wu, H.; Rhee, S.W.; Howell, M.D.; Gottschall, P.E.; Freichel, M.; Flockerzi, V.; Birnbaumer, L.; et al. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol. 2013, 83, 429–438. [Google Scholar] [CrossRef]
- Ariano, P.; Dalmazzo, S.; Owsianik, G.; Nilius, B.; Lovisolo, D. TRPC channels are involved in calcium-dependent migration and proliferation in immortalized GnRH neurons. Cell Calcium 2011, 49, 387–394. [Google Scholar] [CrossRef]
- Fiorio Pla, A.; Maric, D.; Brazer, S.-C.; Giacobini, P.; Liu, X.; Chang, Y.H.; Ambudkar, I.S.; Barker, J.L. Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2+ entry and embryonic rat neural stem cell proliferation. J. Neurosci. 2005, 25, 2687–2701. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, Y.S.; Yuan, J.P.; Petralia, R.S.; Worley, P.F.; Linden, D.J. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 2003, 426, 285–291. [Google Scholar] [CrossRef]
- Liman, E.R. Regulation by voltage and adenine nucleotides of a Ca2+-activated cation channel from hamster vomeronasal sensory neurons. J. Physiol. 2003, 548, 777–787. [Google Scholar] [CrossRef]
- Zhang, J.; Webb, D.M. Evolutionary deterioration of the vomeronasal pheromone transduction pathway in catarrhine primates. Proc. Natl. Acad. Sci. USA 2003, 100, 8337–8341. [Google Scholar] [CrossRef]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009, 23, 297–328. [Google Scholar] [CrossRef]
- Yildirim, E.; Birnbaumer, L. TRPC2: Molecular biology and functional importance. Handb. Exp. Pharmacol. 2007, 179, 53–75. [Google Scholar] [CrossRef]
- Leypold, B.G.; Yu, C.R.; Leinders-Zufall, T.; Kim, M.M.; Zufall, F.; Axel, R. Altered sexual and social behaviors in trp2 mutant mice. Proc. Natl. Acad. Sci. USA 2002, 99, 6376–6381. [Google Scholar]
- Stowers, L.; Holy, T.E.; Meister, M.; Dulac, C.; Koentges, G. Loss of sex discrimination and male-male aggression in mice deficient for TRP2. Science 2002, 295, 1493–1500. [Google Scholar] [CrossRef]
- Kimchi, T.; Xu, J.; Dulac, C. A functional circuit underlying male sexual behaviour in the female mouse brain. Nature 2007, 448, 1009–1014. [Google Scholar] [CrossRef]
- Wu, M.V.; Manoli, D.S.; Fraser, E.J.; Coats, J.K.; Tollkuhn, J.; Honda, S.-I.; Harada, N.; Shah, N.M. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell 2009, 139, 61–72. [Google Scholar] [CrossRef]
- Jungnickel, M.K.; Marrero, H.; Birnbaumer, L.; Lémos, J.R.; Florman, H.M. Trp2 regulates entry of Ca2+ into mouse sperm triggered by egg ZP3. Nat. Cell Biol. 2001, 3, 499–502. [Google Scholar] [CrossRef]
- Sutton, K.A.; Jungnickel, M.K.; Wang, Y.; Cullen, K.; Lambert, S.; Florman, H.M. Enkurin is a novel calmodulin and TRPC channel binding protein in sperm. Dev. Biol. 2004, 274, 426–435. [Google Scholar] [CrossRef]
- Li, H.S.; Xu, X.Z.; Montell, C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron 1999, 24, 261–273. [Google Scholar] [CrossRef]
- Amaral, M.D.; Pozzo-Miller, L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J. Neurosci. 2007, 27, 5179–5189. [Google Scholar] [CrossRef]
- Amaral, M.D.; Pozzo-Miller, L. BDNF induces calcium elevations associated with ibdnf, a nonselective cationic current mediated by TRPC channels. J. Neurophysiol. 2007, 98, 2476–2482. [Google Scholar] [CrossRef]
- Freichel, M.; Vennekens, R.; Olausson, J.; Stolz, S.; Philipp, S.E.; Weissgerber, P.; Flockerzi, V. Functional role of TRPC proteins in native systems: Implications from knockout and knock-down studies. J. Physiol. 2005, 567, 59–66. [Google Scholar] [CrossRef]
- Congar, P.; Leinekugel, X.; Ben-Ari, Y.; Crépel, V. A long-lasting calcium-activated nonselective cationic current is generated by synaptic stimulation or exogenous activation of group I metabotropic glutamate receptors in CA1 pyramidal neurons. J. Neurosci. 1997, 17, 5366–5379. [Google Scholar]
- Linden, R. The survival of developing neurons: A review of afferent control. Neuroscience 1994, 58, 671–682. [Google Scholar] [CrossRef]
- Ruat, M.; Traiffort, E. Roles of the calcium sensing receptor in the central nervous system. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 429–442. [Google Scholar] [CrossRef]
- Harteneck, C.; Gollasch, M. Pharmacological modulation of diacylglycerol-sensitive TRPC3/6/7 channels. Curr. Pharm. Biotechnol. 2011, 12, 35–41. [Google Scholar] [CrossRef]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef]
- Su, B.; Ji, Y.-S.; Sun, X.-L.; Liu, X.-H.; Chen, Z.-Y. Brain-derived neurotrophic factor (BDNF)-induced mitochondrial motility arrest and presynaptic docking contribute to BDNF-enhanced synaptic transmission. J. Biol. Chem. 2014, 289, 1213–1226. [Google Scholar] [CrossRef]
- Rodríguez-Santiago, M.; Mendoza-Torres, M.; Jiménez-Bremont, J.F.; López-Revilla, R. Knockout of the trcp3 gene causes a recessive neuromotor disease in mice. Biochem. Biophys. Res. Commun. 2007, 360, 874–879. [Google Scholar] [CrossRef]
- Singh, B.B.; Lockwich, T.P.; Bandyopadhyay, B.C.; Liu, X.; Bollimuntha, S.; Brazer, S.-C.; Combs, C.; Das, S.; Leenders, A.G.M.; Sheng, Z.-H.; et al. VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol. Cell 2004, 15, 635–646. [Google Scholar] [CrossRef]
- Bandyopadhyay, B.C.; Ong, H.L.; Lockwich, T.P.; Liu, X.; Paria, B.C.; Singh, B.B.; Ambudkar, I.S. TRPC3 controls agonist-stimulated intracellular Ca2+ release by mediating the interaction between inositol 1,4,5-trisphosphate receptor and RACK1. J. Biol. Chem. 2008, 283, 32821–32830. [Google Scholar]
- Streifel, K.M.; Miller, J.; Mouneimne, R.; Tjalkens, R.B. Manganese inhibits ATP-induced calcium entry through the transient receptor potential channel TRPC3 in astrocytes. Neurotoxicology 2013, 34, 160–166. [Google Scholar] [CrossRef]
- Obukhov, A.G.; Nowycky, M.C. TRPC4 can be activated by G-protein-coupled receptors and provides sufficient Ca(2+) to trigger exocytosis in neuroendocrine cells. J. Biol. Chem. 2002, 277, 16172–16178. [Google Scholar] [CrossRef]
- Lee, K.P.; Jun, J.Y.; Chang, I.-Y.; Suh, S.-H.; So, I.; Kim, K.W. TRPC4 is an essential component of the nonselective cation channel activated by muscarinic stimulation in mouse visceral smooth muscle cells. Mol. Cells 2005, 20, 435–441. [Google Scholar]
- Zhang, S.; Remillard, C.V.; Fantozzi, I.; Yuan, J.X.J. ATP-induced mitogenesis is mediated by cyclic AMP response element-binding protein-enhanced TRPC4 expression and activity in human pulmonary artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1192–C1201. [Google Scholar] [CrossRef]
- Gao, Y.-Q.; Gao, H.; Zhou, Z.-Y.; Lu, S.-D.; Sun, F.-Y. Expression of transient receptor potential channel 4 in striatum and hippocampus of rats is increased after focal cerebral ischemia. Sheng Li Xue Bao 2004, 56, 153–157. [Google Scholar]
- Greka, A.; Navarro, B.; Oancea, E.; Duggan, A.; Clapham, D.E. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat. Neurosci. 2003, 6, 837–845. [Google Scholar] [CrossRef]
- Hui, H.; McHugh, D.; Hannan, M.; Zeng, F.; Xu, S.-Z.; Khan, S.-U.-H.; Levenson, R.; Beech, D.J.; Weiss, J.L. Calcium-sensing mechanism in TRPC5 channels contributing to retardation of neurite outgrowth. J. Physiol. 2006, 572, 165–172. [Google Scholar]
- Davare, M.A.; Fortin, D.A.; Saneyoshi, T.; Nygaard, S.; Kaech, S.; Banker, G.; Soderling, T.R.; Wayman, G.A. Transient receptor potential canonical 5 channels activate Ca2+/calmodulin kinase Igamma to promote axon formation in hippocampal neurons. J.Neurosci. 2009, 29, 9794–9808. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.-H.; Obukhov, A.G.; Nowycky, M.C.; Ledeen, R.W. Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with alpha5beta1 integrin initiates neurite outgrowth. J. Neurosci. 2007, 27, 7447–7458. [Google Scholar] [CrossRef]
- Riccio, A.; Li, Y.; Moon, J.; Kim, K.-S.; Smith, K.S.; Rudolph, U.; Gapon, S.; Yao, G.L.; Tsvetkov, E.; Rodig, S.J.; et al. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 2009, 137, 761–772. [Google Scholar] [CrossRef]
- Yan, H.-D.; Villalobos, C.; Andrade, R. TRPC channels mediate a muscarinic receptor-induced afterdepolarization in cerebral cortex. J. Neurosci. 2009, 29, 10038–10046. [Google Scholar] [CrossRef]
- Calupca, M.A.; Locknar, S.A.; Parsons, R.L. TRPC6 immunoreactivity is colocalized with neuronal nitric oxide synthase in extrinsic fibers innervating guinea pig intrinsic cardiac ganglia. J. Comp. Neurol. 2002, 450, 283–291. [Google Scholar] [CrossRef]
- Warren, E.J.; Allen, C.N.; Brown, R.L.; Robinson, D.W. The light-activated signaling pathway in SCN-projecting rat retinal ganglion cells. Eur. J. Neurosci. 2006, 23, 2477–2487. [Google Scholar] [CrossRef]
- Elsaesser, R.; Montani, G.; Tirindelli, R.; Paysan, J. Phosphatidyl-inositide signalling proteins in a novel class of sensory cells in the mammalian olfactory epithelium. Eur. J. Neurosci. 2005, 21, 2692–2700. [Google Scholar] [CrossRef]
- Chung, Y.H.; Sun Ahn, H.; Kim, D.; Hoon Shin, D.; Su Kim, S.; Yong Kim, K.; Bok Lee, W.; Ik Cha, C. Immunohistochemical study on the distribution of TRPC channels in the rat hippocampus. Brain Res. 2006, 1085, 132–137. [Google Scholar] [CrossRef]
- Giampà, C.; DeMarch, Z.; Patassini, S.; Bernardi, G.; Fusco, F.R. Immunohistochemical localization of TRPC6 in the rat substantia nigra. Neurosci. Lett. 2007, 424, 170–174. [Google Scholar] [CrossRef]
- Tai, Y.; Feng, S.; Ge, R.; Du, W.; Zhang, X.; He, Z.; Wang, Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J. Cell. Sci. 2008, 121, 2301–2307. [Google Scholar] [CrossRef]
- Zhou, J.; Du, W.; Zhou, K.; Tai, Y.; Yao, H.; Jia, Y.; Ding, Y.; Wang, Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat. Neurosci. 2008, 11, 741–743. [Google Scholar] [CrossRef]
- Min, M.-Y.; Shih, P.-Y.; Wu, Y.-W.; Lu, H.-W.; Lee, M.-L.; Yang, H.-W. Neurokinin 1 receptor activates transient receptor potential-like currents in noradrenergic A7 neurons in rats. Mol. Cell. Neurosci. 2009, 42, 56–65. [Google Scholar] [CrossRef]
- Babich, L.G.; Ku, C.-Y.; Young, H.W.J.; Huang, H.; Blackburn, M.R.; Sanborn, B.M. Expression of capacitative calcium TrpC proteins in rat myometrium during pregnancy. Biol. Reprod. 2004, 70, 919–924. [Google Scholar]
- Elg, S.; Marmigere, F.; Mattsson, J.P.; Ernfors, P. Cellular subtype distribution and developmental regulation of TRPC channel members in the mouse dorsal root ganglion. J. Comp. Neurol. 2007, 503, 35–46. [Google Scholar]
- Satoh, S.; Tanaka, H.; Ueda, Y.; Oyama, J.-I.; Sugano, M.; Sumimoto, H.; Mori, Y.; Makino, N. Transient receptor potential (TRP) protein 7 acts as a G protein-activated Ca2+ channel mediating angiotensin II-induced myocardial apoptosis. Mol. Cell. Biochem. 2007, 294, 205–215. [Google Scholar] [CrossRef]
- Föller, M.; Kasinathan, R.S.; Duranton, C.; Wieder, T.; Huber, S.M.; Lang, F. PGE2-induced apoptotic cell death in K562 human leukaemia cells. Cell. Physiol. Biochem. 2006, 17, 201–210. [Google Scholar] [CrossRef]
- Xue, T.; Do, M.T.; Riccio, A.; Jiang, Z.; Hsieh, J.; Wang, H.C.; Merbs, S.L.; Welsbie, D.S.; Yoshioka, T.; Weissgerber, P.; et al. Melanopsin signalling in mammalian iris and retina. Nature 2011, 479, 67–73. [Google Scholar]
- Putney, J.W., Jr. Capacitative calcium entry in the nervous system. Cell Calcium 2003, 34, 339–344. [Google Scholar] [CrossRef]
- Chen, B.T.; Rice, M.E. Novel Ca2+ dependence and time course of somatodendritic dopamine release: Substantia nigra vs. striatum. J. Neurosci. 2001, 21, 7841–7847. [Google Scholar]
- Patel, J.C.; Witkovsky, P.; Avshalumov, M.V.; Rice, M.E. Mobilization of calcium from intracellular stores facilitates somatodendritic dopamine release. J. Neurosci. 2009, 29, 6568–6579. [Google Scholar] [CrossRef]
- Small, D.H. Dysregulation of calcium homeostasis in Alzheimer’s disease. Neurochem. Res. 2009, 34, 1824–1829. [Google Scholar] [CrossRef]
- Yu, J.T.; Chang, R.C.; Tan, L. Calcium dysregulation in Alzheimer’s disease: From mechanisms to therapeutic opportunities. Progr. Neurobiol. 2009, 89, 240–255. [Google Scholar] [CrossRef]
- Giacomello, M.; Oliveros, J.C.; Naranjo, J.R.; Carafoli, E. Neuronal Ca(2+) dyshomeostasis in Huntington disease. Prion 2013, 7, 76–84. [Google Scholar] [CrossRef]
- Suzuki, M.; Nagai, Y.; Wada, K.; Koike, T. Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochem. Biophys. Res. Commun. 2012, 429, 18–23. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef]
- Henke, N.; Albrecht, P.; Pfeiffer, A.; Toutzaris, D.; Zanger, K.; Methner, A. Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J. Biol. Chem. 2012, 287, 42042–42052. [Google Scholar] [CrossRef]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar]
- Timmons, J.A.; Rao, J.N.; Turner, D.J.; Zou, T.; Liu, L.; Xiao, L.; Wang, P.Y.; Wang, J.Y. Induced expression of STIM1 sensitizes intestinal epithelial cells to apoptosis by modulating store-operated Ca2+ influx. J. Gastrointest. Surg. 2012, 16, 1397–1405. [Google Scholar] [CrossRef]
- Song, M.Y.; Makino, A.; Yuan, J.X. Role of reactive oxygen species and redox in regulating the function of transient receptor potential channels. Antioxidants Redox Signal. 2011, 15, 1549–1565. [Google Scholar] [CrossRef]
- Miller, B.A.; Zhang, W. TRP channels as mediators of oxidative stress. Adv. Exp. Med. Biol. 2011, 704, 531–544. [Google Scholar] [CrossRef]
- Chen, T.; Yang, Y.F.; Luo, P.; Liu, W.; Dai, S.H.; Zheng, X.R.; Fei, Z.; Jiang, X.F. Homer1 knockdown protects dopamine neurons through regulating calcium homeostasis in an in vitro model of Parkinson’s disease. Cell. Signalling 2013, 25, 2863–2870. [Google Scholar] [CrossRef]
- Przedborski, S.; Tieu, K.; Perier, C.; Vila, M. MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J. Bioenerget. Biomembr. 2004, 36, 375–379. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef]
- Zeng, X.; Pan, Z.G.; Shao, Y.; Wu, X.N.; Liu, S.X.; Li, N.L.; Wang, W.M. SKF-96365 attenuates toxin-induced neuronal injury through opposite regulatory effects on Homer1a and Homer1b/c in cultured rat mesencephalic cells. Neurosci. Lett. 2013, 543, 183–188. [Google Scholar] [CrossRef]
- Bollimuntha, S.; Singh, B.B.; Shavali, S.; Sharma, S.K.; Ebadi, M. TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J. Biol. Chem. 2005, 280, 2132–2140. [Google Scholar]
- Selvaraj, S.; Watt, J.A.; Singh, B.B. TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+). Cell Calcium 2009, 46, 209–218. [Google Scholar] [CrossRef]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Wu, J.; Shih, H.P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef]
- Briones, N.; Dinu, V. Data mining of high density genomic variant data for prediction of Alzheimer’s disease risk. BMC Med. Genet. 2012, 13, 7. [Google Scholar] [CrossRef]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; de Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Laferla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Lessard, C.B.; Lussier, M.P.; Cayouette, S.; Bourque, G.; Boulay, G. The overexpression of presenilin2 and Alzheimer’s-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell. Signalling. 2005, 17, 437–445. [Google Scholar] [CrossRef]
- Giacomello, M.; Barbiero, L.; Zatti, G.; Squitti, R.; Binetti, G.; Pozzan, T.; Fasolato, C.; Ghidoni, R.; Pizzo, P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol. Dis. 2005, 18, 638–648. [Google Scholar] [CrossRef]
- Cheung, K.H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef]
- Bandara, S.; Malmersjo, S.; Meyer, T. Regulators of calcium homeostasis identified by inference of kinetic model parameters from live single cells perturbed by siRNA. Sci. Signal. 2013, 6, ra56. [Google Scholar]
- Stranahan, A.M.; Mattson, M.P. Recruiting adaptive cellular stress responses for successful brain ageing. Nat. Rev. Neurosci. 2012, 13, 209–216. [Google Scholar]
- McGurk, J.S.; Shim, S.; Kim, J.Y.; Wen, Z.; Song, H.; Ming, G.L. Postsynaptic TRPC1 function contributes to BDNF-induced synaptic potentiation at the developing neuromuscular junction. J. Neurosci. 2011, 31, 14754–14762. [Google Scholar] [CrossRef]
- Wang, H.J.; Cao, J.P.; Yu, J.K.; Zhang, L.C.; Jiang, Z.J.; Gao, D.S. Calbindin-D28K expression induced by glial cell line-derived neurotrophic factor in substantia nigra neurons dependent on PI3K/Akt/NF-kappaB signaling pathway. Eur. J. Pharmacol. 2008, 595, 7–12. [Google Scholar] [CrossRef]
- Mattson, M.P. Parkinson’s disease: Don’t mess with calcium. J. Clin. Investig. 2012, 122, 1195–1198. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.J.; Feske, S.; White, C.L., 3rd; Bezprozvanny, I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef]
- Bojarski, L.; Pomorski, P.; Szybinska, A.; Drab, M.; Skibinska-Kijek, A.; Gruszczynska-Biegala, J.; Kuznicki, J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1793, 1050–1057. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, Y.; Sukumaran, P.; Bandyopadhyay, B.C.; Singh, B.B. Physiological Function and Characterization of TRPCs in Neurons. Cells 2014, 3, 455-475. https://doi.org/10.3390/cells3020455
Sun Y, Sukumaran P, Bandyopadhyay BC, Singh BB. Physiological Function and Characterization of TRPCs in Neurons. Cells. 2014; 3(2):455-475. https://doi.org/10.3390/cells3020455
Chicago/Turabian StyleSun, Yuyang, Pramod Sukumaran, Bidhan C. Bandyopadhyay, and Brij B. Singh. 2014. "Physiological Function and Characterization of TRPCs in Neurons" Cells 3, no. 2: 455-475. https://doi.org/10.3390/cells3020455
APA StyleSun, Y., Sukumaran, P., Bandyopadhyay, B. C., & Singh, B. B. (2014). Physiological Function and Characterization of TRPCs in Neurons. Cells, 3(2), 455-475. https://doi.org/10.3390/cells3020455