When Good Turns Bad: Regulation of Invasion and Metastasis by ErbB2 Receptor Tyrosine Kinase


Abstract
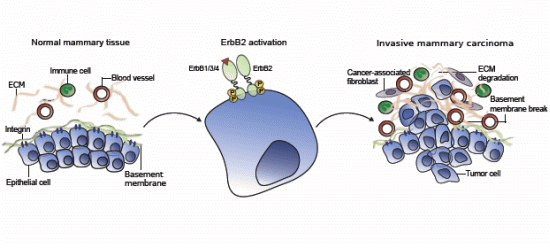
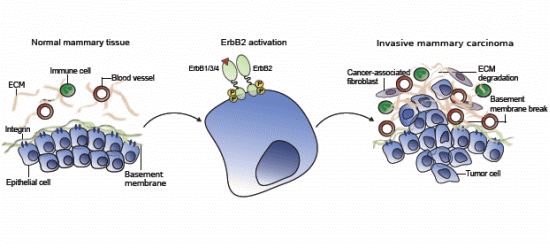
:
1. ErbB2 Function and Basis of the ErbB2-Induced Malignancy
1.1. ErbB2 as a Potent Oncogene, Biomarker and a Therapeutic Target
1.2. ErbB Family Connections
1.3. Two Major Functional Forms of ErbB2: p185 and p95
1.4. ErbB2 Downstream Signaling and its Physiological and Cellular Responses
2. Invasive Signaling of ErbB2
2.1. ErbB2 is not Enough
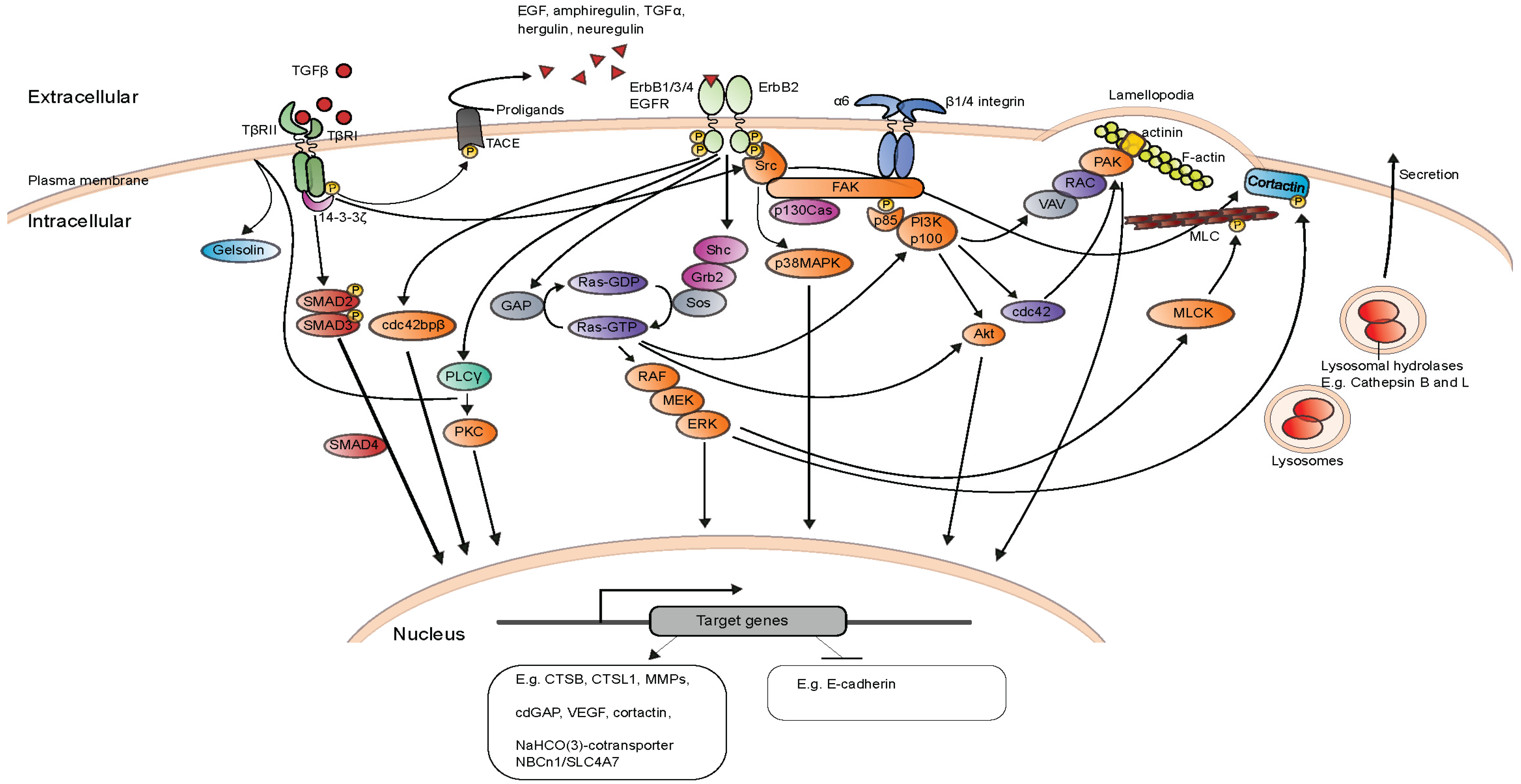
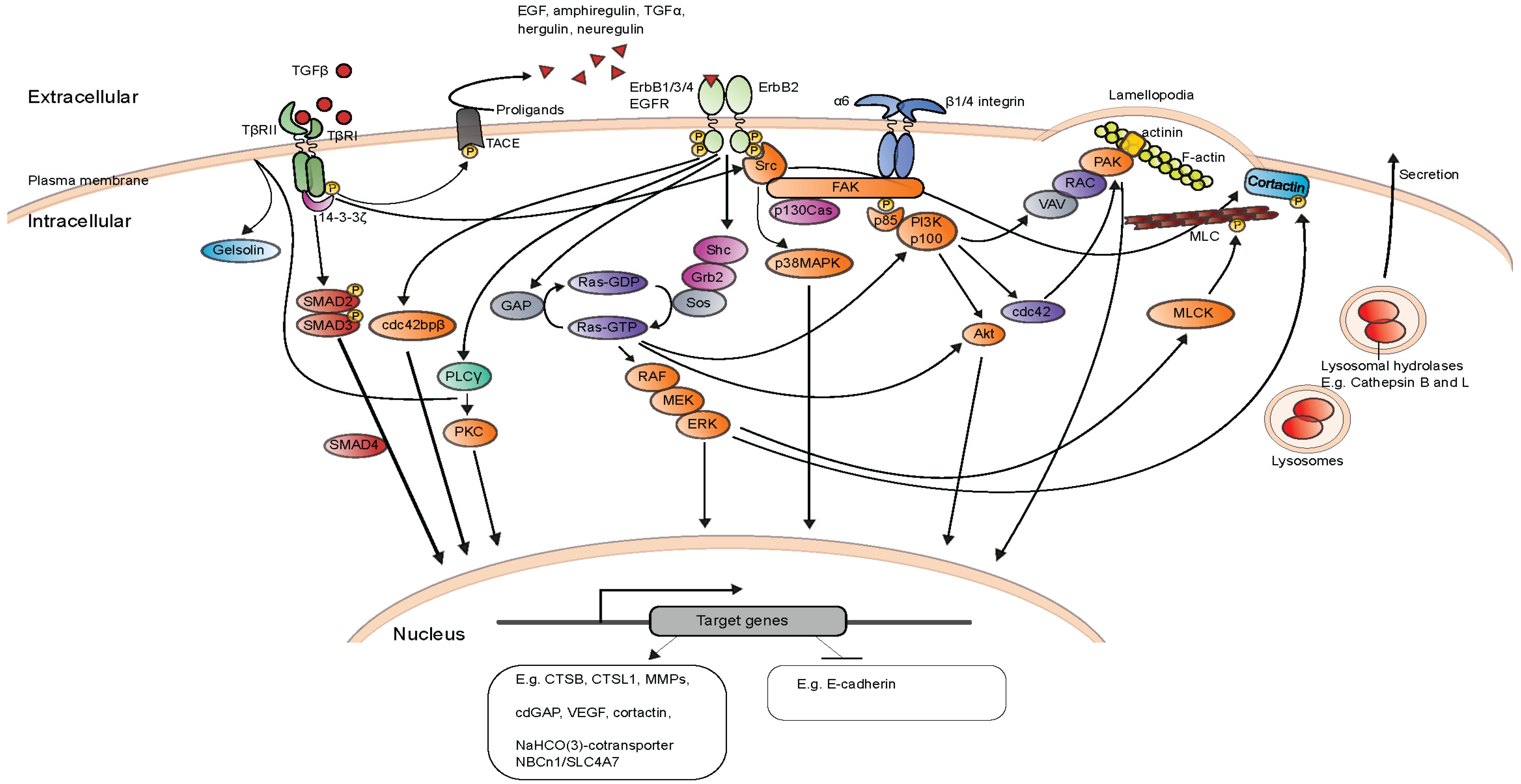
{kind=link}
{kind=link}
2.2. Transforming Growth Factor β and ErbB2-Induced Invasion and Metastasis
2.3. More Players for the Game
2.4. Alternative Approaches to Identify Invasion Mechanisms
2.5. Invasion and Metastasis Inducing Targets of ErbB2 Downstream Signaling
2.6. Other Important Mediators and Targets of ErbB2-Induced Invasion
2.7. ErbB2 Overexpressing Mice and ErbB2-Induced Metastasis
2.8. ErbB2, Cancer Initiating Cells and EMT in Breast Cancer Invasion
3. Conclusions
Acknowledgements
Author Contributions
Conflicts of interest
References
- Berchuck, A.; Kamel, A.; Whitaker, R.; Kerns, B.; Olt, G.; Kinney, R.; Soper, J.T.; Dodge, R.; Clarke-Pearson, D.L.; Marks, P.; et al. Overexpression of HER-2/neu is associated with poor survival in advanced epithelial ovarian cancer. Cancer Res. 1990, 50, 4087–4091. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Kern, J.A.; Schwartz, D.A.; Nordberg, J.E.; Weiner, D.B.; Greene, M.I.; Torney, L.; Robinson, R.A. p185neu expression in human lung adenocarcinomas predicts shortened survival. Cancer Res. 1990, 50, 5184–5187. [Google Scholar]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar]
- Zeillinger, R.; Kury, F.; Czerwenka, K.; Kubista, E.; Sliutz, G.; Knogler, W.; Huber, J.; Zielinski, C.; Reiner, G.; Jakesz, R.; et al. HER-2 amplification, steroid receptors and epidermal growth factor receptor in primary breast cancer. Oncogene 1989, 4, 109–114. [Google Scholar]
- Fukushige, S.; Matsubara, K.; Yoshida, M.; Sasaki, M.; Suzuki, T.; Semba, K.; Toyoshima, K.; Yamamoto, T. Localization of a novel v-erbB-related gene, c-erbB-2, on human chromosome 17 and its amplification in a gastric cancer cell line. Mol. Cell. Biol. 1986, 6, 955–958. [Google Scholar]
- Yokota, J.; Yamamoto, T.; Toyoshima, K.; Terada, M.; Sugimura, T.; Battifora, H.; Cline, M.J. Amplification of c-erbB-2 oncogene in human adenocarcinomas in vivo. Lancet 1986, 1, 765–767. [Google Scholar]
- Zhau, H.E.; Zhang, X.; von Eschenbach, A.C.; Scorsone, K.; Babaian, R.J.; Ro, J.Y.; Hung, M.C. Amplification and expression of the c-erb B-2/neu proto-oncogene in human bladder cancer. Mol. Carcinogen 1990, 3, 254–257. [Google Scholar] [CrossRef]
- McCann, A.; Dervan, P.A.; Johnston, P.A.; Gullick, W.J.; Carney, D.N. c-erbB-2 oncoprotein expression in primary human tumors. Cancer 1990, 65, 88–92. [Google Scholar] [CrossRef]
- Brunner, K.; Fischer, C.A.; Driemel, O.; Hartmann, A.; Brockhoff, G.; Schwarz, S. EGFR (HER) family protein expression and cytogenetics in 219 squamous cell carcinomas of the upper respiratory tract: ERBB2 overexpression independent prediction of poor prognosis. Anal. Quant. Cytol. Histol. 2010, 32, 78–89. [Google Scholar]
- Ross, J.S.; Fletcher, J.A. The HER-2/neu oncogene in breast cancer: Prognostic factor, predictive factor, and target for therapy. Stem Cells 1998, 16, 413–428. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufi, S.; Moreno, J.M.; Corominas-Faja, B.; Urruticoechea, A.; Martin, A.G.; Lopez-Bonet, E.; Menendez, J.A. Basal/HER2 breast carcinomas: Integrating molecular taxonomy with cancer stem cell dynamics to predict primary resistance to trastuzumab (Herceptin). Cell Cycle 2013, 12, 225–245. [Google Scholar]
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef]
- Nielsen, D.L.; Kumler, I.; Palshof, J.A.; Andersson, M. Efficacy of HER2-targeted therapy in metastatic breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Breast 2013, 22, 1–12. [Google Scholar]
- Moasser, M.M. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene 2007, 26, 6577–6592. [Google Scholar] [CrossRef]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Nat. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef]
- Gullo, G.; Zuradelli, M.; Sclafani, F.; Santoro, A.; Crown, J. Durable complete response following chemotherapy and trastuzumab for metastatic HER2-positive breast cancer. Ann. Oncol. 2012, 23, 2204–2205. [Google Scholar] [CrossRef]
- Kumler, I.; Tuxen, M.K.; Nielsen, D.L. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treatment Rev. 2014, 40, 259–270. [Google Scholar] [CrossRef]
- Blumenthal, G.M.; Scher, N.S.; Cortazar, P.; Chattopadhyay, S.; Tang, S.; Song, P.; Liu, Q.; Ringgold, K.; Pilaro, A.M.; Tilley, A.; et al. First FDA approval of dual anti-her2 regimen: Pertuzumab in combination with trastuzumab and docetaxel for her2-positive metastatic breast cancer. Clin. Cancer Res. 2013, 19, 4911–4916. [Google Scholar] [CrossRef]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Molecular cell 2003, 11, 495–505. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Nat. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef]
- Hsieh, M.Y.; Yang, S.; Raymond-Stinz, M.A.; Steinberg, S.; Vlachos, D.G.; Shu, W.; Wilson, B.; Edwards, J.S. Stochastic simulations of ErbB homo and heterodimerisation: potential impacts of receptor conformational state and spatial segregation. IET Syst. Biol. 2008, 2, 256–272. [Google Scholar] [CrossRef]
- Lonardo, F.; di Marco, E.; King, C.R.; Pierce, J.H.; Segatto, O.; Aaronson, S.A.; di Fiore, P.P. The normal erbB-2 product is an atypical receptor-like tyrosine kinase with constitutive activity in the absence of ligand. New Biol. 1990, 2, 992–1003. [Google Scholar]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287. [Google Scholar]
- Jones, R.B.; Gordus, A.; Krall, J.A.; MacBeath, G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 2006, 439, 168–174. [Google Scholar] [CrossRef]
- Baulida, J.; Kraus, M.H.; Alimandi, M.; di Fiore, P.P.; Carpenter, G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 1996, 271, 5251–5257. [Google Scholar] [CrossRef]
- Worthylake, R.; Opresko, L.K.; Wiley, H.S. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J. Biol. Chem. 1999, 274, 8865–8874. [Google Scholar] [CrossRef]
- Lenferink, A.E.; Pinkas-Kramarski, R.; van de Poll, M.L.; van Vugt, M.J.; Klapper, L.N.; Tzahar, E.; Waterman, H.; Sela, M.; van Zoelen, E.J.; Yarden, Y. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 1998, 17, 3385–3397. [Google Scholar] [CrossRef]
- Dankort, D.; Jeyabalan, N.; Jones, N.; Dumont, D.J.; Muller, W.J. Multiple ErbB-2/Neu phosphorylation sites mediate transformation through distinct effector proteins. J. Biol. Chem. 2001, 276, 38921–38928. [Google Scholar] [CrossRef]
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Alroy, I.; Yarden, Y. The ErbB signaling network in embryogenesis and oncogenesis: Signal diversification through combinatorial ligand-receptor interactions. FEBS Lett. 1997, 410, 83–86. [Google Scholar] [CrossRef]
- Christianson, T.A.; Doherty, J.K.; Lin, Y.J.; Ramsey, E.E.; Holmes, R.; Keenan, E.J.; Clinton, G.M. NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res. 1998, 58, 5123–5129. [Google Scholar]
- Molina, M.A.; Saez, R.; Ramsey, E.E.; Garcia-Barchino, M.J.; Rojo, F.; Evans, A.J.; Albanell, J.; Keenan, E.J.; Lluch, A.; Garcia-Conde, J.; et al. NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin. Cancer Res. 2002, 8, 347–353. [Google Scholar]
- Xia, W.; Liu, Z.; Zong, R.; Liu, L.; Zhao, S.; Bacus, S.S.; Mao, Y.; He, J.; Wulfkuhle, J.D.; Petricoin, E.F., 3rd; et al. Truncated ErbB2 expressed in tumor cell nuclei contributes to acquired therapeutic resistance to ErbB2 kinase inhibitors. Mol. Cancer Ther. 2011, 10, 1367–1374. [Google Scholar] [CrossRef]
- Saez, R.; Molina, M.A.; Ramsey, E.E.; Rojo, F.; Keenan, E.J.; Albanell, J.; Lluch, A.; Garcia-Conde, J.; Baselga, J.; Clinton, G.M. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin. Cancer Res. 2006, 12, 424–431. [Google Scholar] [CrossRef]
- Sperinde, J.; Jin, X.; Banerjee, J.; Penuel, E.; Saha, A.; Diedrich, G.; Huang, W.; Leitzel, K.; Weidler, J.; Ali, S.M.; et al. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin. Cancer Res. 2010, 16, 4226–4235. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Clinton, G.M. A soluble protein related to the HER-2 proto-oncogene product is released from human breast carcinoma cells. Oncogene 1991, 6, 639–643. [Google Scholar]
- Pupa, S.M.; Menard, S.; Morelli, D.; Pozzi, B.; de Palo, G.; Colnaghi, M.I. The extracellular domain of the c-erbB-2 oncoprotein is released from tumor cells by proteolytic cleavage. Oncogene 1993, 8, 2917–2923. [Google Scholar]
- Yuan, C.X.; Lasut, A.L.; Wynn, R.; Neff, N.T.; Hollis, G.F.; Ramaker, M.L.; Rupar, M.J.; Liu, P.; Meade, R. Purification of Her-2 extracellular domain and identification of its cleavage site. Protein Expres. Purif. 2003, 29, 217–222. [Google Scholar] [CrossRef]
- Zabrecky, J.R.; Lam, T.; McKenzie, S.J.; Carney, W. The extracellular domain of p185/neu is released from the surface of human breast carcinoma cells, SK-BR-3. J. Biol. Chem. 1991, 266, 1716–1720. [Google Scholar]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. [Google Scholar]
- Kwong, K.Y.; Hung, M.C. A novel splice variant of HER2 with increased transformation activity. Mol. Carcinogen 1998, 23, 62–68. [Google Scholar] [CrossRef]
- Anido, J.; Scaltriti, M.; Bech Serra, J.J.; Santiago Josefat, B.; Todo, F.R.; Baselga, J.; Arribas, J. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006, 25, 3234–3244. [Google Scholar] [CrossRef]
- Scott, G.K.; Robles, R.; Park, J.W.; Montgomery, P.A.; Daniel, J.; Holmes, W.E.; Lee, J.; Keller, G.A.; Li, W.L.; Fendly, B.M.; et al. A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Mol. Cell. Biol. 1993, 13, 2247–2257. [Google Scholar]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and breast cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef]
- Pedersen, K.; Angelini, P.D.; Laos, S.; Bach-Faig, A.; Cunningham, M.P.; Ferrer-Ramon, C.; Luque-Garcia, A.; Garcia-Castillo, J.; Parra-Palau, J.L.; Scaltriti, M.; et al. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol. Cell. Biol. 2009, 29, 3319–3331. [Google Scholar] [CrossRef]
- Xia, W.; Liu, L.H.; Ho, P.; Spector, N.L. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene 2004, 23, 646–653. [Google Scholar] [CrossRef]
- Segatto, O.; King, C.R.; Pierce, J.H.; di Fiore, P.P.; Aaronson, S.A. Different structural alterations upregulate in vitro tyrosine kinase activity and transforming potency of the erbB-2 gene. Mol. Cell. Biol. 1988, 8, 5570–5574. [Google Scholar]
- Egeblad, M.; Mortensen, O.H.; Jaattela, M. Truncated ErbB2 receptor enhances ErbB1 signaling and induces reversible, ERK-independent loss of epithelial morphology. Int. J. Cancer 2001, 94, 185–191. [Google Scholar] [CrossRef]
- Rafn, B.; Nielsen, C.F.; Andersen, S.H.; Szyniarowski, P.; Corcelle-Termeau, E.; Valo, E.; Fehrenbacher, N.; Olsen, C.J.; Daugaard, M.; Egebjerg, C.; et al. ErbB2-driven breast cancer cell invasion depends on a complex signaling network activating myeloid zinc finger-1-dependent cathepsin B expression. Mol. Cell 2012, 45, 764–776. [Google Scholar] [CrossRef]
- Garcia-Castillo, J.; Pedersen, K.; Angelini, P.D.; Bech-Serra, J.J.; Colome, N.; Cunningham, M.P.; Parra-Palau, J.L.; Canals, F.; Baselga, J.; Arribas, J. HER2 carboxyl-terminal fragments regulate cell migration and cortactin phosphorylation. J. Biol. Chem. 2009, 284, 25302–25313. [Google Scholar] [CrossRef]
- Lauritzen, G.; Stock, C.M.; Lemaire, J.; Lund, S.F.; Jensen, M.F.; Damsgaard, B.; Petersen, K.S.; Wiwel, M.; Ronnov-Jessen, L.; Schwab, A.; et al. The Na+/H+ exchanger NHE1, but not the Na+, HCO3(-) cotransporter NBCn1, regulates motility of MCF7 breast cancer cells expressing constitutively active ErbB2. Cancer Lett. 2012, 317, 172–183. [Google Scholar] [CrossRef]
- Alajati, A.; Sausgruber, N.; Aceto, N.; Duss, S.; Sarret, S.; Voshol, H.; Bonenfant, D.; Bentires-Alj, M. Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res. 2013, 73, 5320–5327. [Google Scholar] [CrossRef]
- Angelini, P.D.; Fluck, M.F.Z.; Pedersen, K.; Parra-Palau, J.L.; Guiu, M.; Morales, C.B.; Vicario, R.; Luque-Garcia, A.; Navalpotro, N.P.; Giralt, J.; et al. Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res. 2013, 73, 450–458. [Google Scholar] [CrossRef]
- Broad-Novartis Cancer Cell Line Encyclopedia (CCLE). Available online: http://www.Broadinstitute.org/ccle/home (accessed on 1 November 2013).
- Zagozdzon, R.; Gallagher, W.M.; Crown, J. Truncated HER2: implications for HER2-targeted therapeutics. Drug discovery today 2011, 16, 810–816. [Google Scholar] [CrossRef]
- Scaltriti, M.; Chandarlapaty, S.; Prudkin, L.; Aura, C.; Jimenez, J.; Angelini, P.D.; Sanchez, G.; Guzman, M.; Parra, J.L.; Ellis, C.; et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin. Cancer Res. 2010, 16, 2688–2695. [Google Scholar] [CrossRef]
- Lee, K.F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar] [CrossRef]
- Garratt, A.N.; Ozcelik, C.; Birchmeier, C. ErbB2 pathways in heart and neural diseases. Trend. Cardiovasc. Med. 2003, 13, 80–86. [Google Scholar] [CrossRef]
- Park, S.K.; Miller, R.; Krane, I.; Vartanian, T. The erbB2 gene is required for the development of terminally differentiated spinal cord oligodendrocytes. J. Cell Biol. 2001, 154, 1245–1258. [Google Scholar] [CrossRef]
- Schmid, R.S.; McGrath, B.; Berechid, B.E.; Boyles, B.; Marchionni, M.; Sestan, N.; Anton, E.S. Neuregulin 1-erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex. Proc. Nat. Acad. Sci. USA. 2003, 100, 4251–4256. [Google Scholar]
- Andrechek, E.R.; Hardy, W.R.; Girgis-Gabardo, A.A.; Perry, R.L.; Butler, R.; Graham, F.L.; Kahn, R.C.; Rudnicki, M.A.; Muller, W.J. ErbB2 is required for muscle spindle and myoblast cell survival. Mol. Cell. Biol. 2002, 22, 4714–4722. [Google Scholar] [CrossRef]
- Jackson-Fisher, A.J.; Bellinger, G.; Ramabhadran, R.; Morris, J.K.; Lee, K.F.; Stern, D.F. ErbB2 is required for ductal morphogenesis of the mammary gland. Proc. Nat. Acad. Sci. USA. 2004, 101, 17138–17143. [Google Scholar]
- Morris, J.K.; Lin, W.; Hauser, C.; Marchuk, Y.; Getman, D.; Lee, K.F. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 1999, 23, 273–283. [Google Scholar] [CrossRef]
- Negro, A.; Brar, B.K.; Lee, K.F. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog. Hormone Res. 2004, 59, 1–12. [Google Scholar] [CrossRef]
- Crone, S.A.; Zhao, Y.Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar]
- Grazette, L.P.; Boecker, W.; Matsui, T.; Semigran, M.; Force, T.L.; Hajjar, R.J.; Rosenzweig, A. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: Implications for herceptin-induced cardiomyopathy. J. Amer. Coll. Cardiol. 2004, 44, 2231–2238. [Google Scholar] [CrossRef]
- Liu, F.F.; Stone, J.R.; Schuldt, A.J.; Okoshi, K.; Okoshi, M.P.; Nakayama, M.; Ho, K.K.; Manning, W.J.; Marchionni, M.A.; Lorell, B.H.; et al. Heterozygous knockout of neuregulin-1 gene in mice exacerbates doxorubicin-induced heart failure. Am. J. Physiol-Heart. Circ. Phy. 2005, 289, H660–H666. [Google Scholar] [CrossRef]
- Cote, G.M.; Sawyer, D.B.; Chabner, B.A. ERBB2 inhibition and heart failure. N. Engl. J. Med. 2012, 367, 2150–2153. [Google Scholar] [CrossRef]
- Roca-Alonso, L.; Pellegrino, L.; Castellano, L.; Stebbing, J. Breast cancer treatment and adverse cardiac events: What are the molecular mechanisms? Cardiology 2012, 122, 253–259. [Google Scholar] [CrossRef]
- Sendur, M.A.; Aksoy, S.; Altundag, K. Cardiotoxicity of novel HER2-targeted therapies. Curr. Med. Res. Opin. 2013, 29, 1015–1024. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell. Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell. Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Lee, R.J.; Albanese, C.; Fu, M.; D’Amico, M.; Lin, B.; Watanabe, G.; Haines, G.K., 3rd; Siegel, P.M.; Hung, M.C.; Yarden, Y.; et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol. Cell. Biol. 2000, 20, 672–683. [Google Scholar] [CrossRef]
- Neve, R.M.; Sutterluty, H.; Pullen, N.; Lane, H.A.; Daly, J.M.; Krek, W.; Hynes, N.E. Effects of oncogenic ErbB2 on G1 cell cycle regulators in breast tumour cells. Oncogene 2000, 19, 1647–1656. [Google Scholar] [CrossRef]
- Neve, R.M.; Holbro, T.; Hynes, N.E. Distinct roles for phosphoinositide 3-kinase, mitogen-activated protein kinase and p38 MAPK in mediating cell cycle progression of breast cancer cells. Oncogene 2002, 21, 4567–4576. [Google Scholar] [CrossRef]
- Lenferink, A.E.; Busse, D.; Flanagan, W.M.; Yakes, F.M.; Arteaga, C.L. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 2001, 61, 6583–6591. [Google Scholar]
- Zhang, W.; Tan, W.; Wu, X.; Poustovoitov, M.; Strasner, A.; Li, W.; Borcherding, N.; Ghassemian, M.; Karin, M. A NIK-IKKalpha module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 2013, 23, 647–659. [Google Scholar]
- Pianetti, S.; Arsura, M.; Romieu-Mourez, R.; Coffey, R.J.; Sonenshein, G.E. Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene 2001, 20, 1287–1299. [Google Scholar] [CrossRef]
- Feldner, J.C.; Brandt, B.H. Cancer cell motility--on the road from c-erbB-2 receptor steered signaling to actin reorganization. Exp. Cell Res. 2002, 272, 93–108. [Google Scholar] [CrossRef]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; de Lanerolle, P.; Cheresh, D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef]
- Bryce, N.S.; Clark, E.S.; Leysath, J.L.; Currie, J.D.; Webb, D.J.; Weaver, A.M. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr. Biol. 2005, 15, 1276–1285. [Google Scholar] [CrossRef]
- Hill, A.; McFarlane, S.; Mulligan, K.; Gillespie, H.; Draffin, J.E.; Trimble, A.; Ouhtit, A.; Johnston, P.G.; Harkin, D.P.; McCormick, D.; et al. Cortactin underpins CD44-promoted invasion and adhesion of breast cancer cells to bone marrow endothelial cells. Oncogene 2006, 25, 6079–6091. [Google Scholar] [CrossRef]
- Rothschild, B.L.; Shim, A.H.; Ammer, A.G.; Kelley, L.C.; Irby, K.B.; Head, J.A.; Chen, L.; Varella-Garcia, M.; Sacks, P.G.; Frederick, B.; et al. Cortactin overexpression regulates actin-related protein 2/3 complex activity, motility, and invasion in carcinomas with chromosome 11q13 amplification. Cancer Res. 2006, 66, 8017–8025. [Google Scholar] [CrossRef]
- Lodato, R.F.; Maguire, H.C., Jr.; Greene, M.I.; Weiner, D.B.; LiVolsi, V.A. Immunohistochemical evaluation of c-erbB-2 oncogene expression in ductal carcinoma in situ and atypical ductal hyperplasia of the breast. Mod. Pathol. 1990, 3, 449–454. [Google Scholar]
- Van de Vijver, M.J.; Peterse, J.L.; Mooi, W.J.; Wisman, P.; Lomans, J.; Dalesio, O.; Nusse, R. Neu-protein overexpression in breast cancer. Association with comedo-type ductal carcinoma in situ and limited prognostic value in stage II breast cancer. N. Engl. J. Med. 1988, 319, 1239–1245. [Google Scholar] [CrossRef]
- Seton-Rogers, S.E.; Lu, Y.; Hines, L.M.; Koundinya, M.; LaBaer, J.; Muthuswamy, S.K.; Brugge, J.S. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Nat. Acad. Sci. USA 2004, 101, 1257–1262. [Google Scholar]
- Pradeep, C.R.; Zeisel, A.; Kostler, W.J.; Lauriola, M.; Jacob-Hirsch, J.; Haibe-Kains, B.; Amariglio, N.; Ben-Chetrit, N.; Emde, A.; Solomonov, I.; et al. Modeling invasive breast cancer: Growth factors propel progression of HER2-positive premalignant lesions. Oncogene 2012, 31, 3569–3583. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Schade, B.; Cardiff, R.D.; Muller, W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 2007, 7, 389–397. [Google Scholar] [CrossRef]
- Chan, R.; Muller, W.J.; Siegel, P.M. Oncogenic activating mutations in the neu/erbB-2 oncogene are involved in the induction of mammary tumors. Ann. N. Y. Acad. Sci. 1999, 889, 45–51. [Google Scholar] [CrossRef]
- Siegel, P.M.; Dankort, D.L.; Hardy, W.R.; Muller, W.J. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol. Cell. Biol. 1994, 14, 7068–7077. [Google Scholar]
- Siegel, P.M.; Muller, W.J. Mutations affecting conserved cysteine residues within the extracellular domain of Neu promote receptor dimerization and activation. Proc. Nat. Acad. Sci. USA 1996, 93, 8878–8883. [Google Scholar] [CrossRef]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef]
- Zito, C.I.; Riches, D.; Kolmakova, J.; Simons, J.; Egholm, M.; Stern, D.F. Direct resequencing of the complete ERBB2 coding sequence reveals an absence of activating mutations in ERBB2 amplified breast cancer. Genes Chromosomes Canc. 2008, 47, 633–638. [Google Scholar] [CrossRef]
- Ueda, Y.; Wang, S.; Dumont, N.; Yi, J.Y.; Koh, Y.; Arteaga, C.L. Overexpression of HER2 (erbB2) in human breast epithelial cells unmasks transforming growth factor beta-induced cell motility. J. Biol. Chem. 2004, 279, 24505–24513. [Google Scholar] [CrossRef]
- Wang, S.E.; Xiang, B.; Guix, M.; Olivares, M.G.; Parker, J.; Chung, C.H.; Pandiella, A.; Arteaga, C.L. Transforming growth factor beta engages TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Mol. Cell. Biol. 2008, 28, 5605–5620. [Google Scholar] [CrossRef]
- Wang, S.E.; Xiang, B.; Zent, R.; Quaranta, V.; Pozzi, A.; Arteaga, C.L. Transforming growth factor beta induces clustering of HER2 and integrins by activating Src-focal adhesion kinase and receptor association to the cytoskeleton. Cancer Res. 2009, 69, 475–482. [Google Scholar] [CrossRef]
- Wang, S.E.; Shin, I.; Wu, F.Y.; Friedman, D.B.; Arteaga, C.L. HER2/Neu (ErbB2) signaling to Rac1-Pak1 is temporally and spatially modulated by transforming growth factor beta. Cancer Res. 2006, 66, 9591–9600. [Google Scholar] [CrossRef]
- Lu, J.; Guo, H.; Treekitkarnmongkol, W.; Li, P.; Zhang, J.; Shi, B.; Ling, C.; Zhou, X.; Chen, T.; Chiao, P.J.; et al. 14–3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer cell 2009, 16, 195–207. [Google Scholar]
- Giunciuglio, D.; Culty, M.; Fassina, G.; Masiello, L.; Melchiori, A.; Paglialunga, G.; Arand, G.; Ciardiello, F.; Basolo, F.; Thompson, E.W.; et al. Invasive phenotype of MCF10A cells overexpressing c-Ha-ras and c-erbB-2 oncogenes. Int. J. Cancer 1995, 63, 815–822. [Google Scholar] [CrossRef]
- Connolly, J.; Rose, D. Expression of the invasive phenotype by MCF-7 human breast cancer cells transfected to overexpress protein kinase C-alpha or the erbB2 proto-oncogene. Int. J. Oncol. 1997, 10, 71–76. [Google Scholar]
- Lauritzen, G.; Jensen, M.B.; Boedtkjer, E.; Dybboe, R.; Aalkjaer, C.; Nylandsted, J.; Pedersen, S.F. NBCn1 and NHE1 expression and activity in DeltaNErbB2 receptor-expressing MCF-7 breast cancer cells: contributions to pHi regulation and chemotherapy resistance. Exp. Cell Res. 2010, 316, 2538–2553. [Google Scholar] [CrossRef]
- Gorbatenko, A.; Olesen, C.W.; Morup, N.; Thiel, G.; Kallunki, T.; Valen, E.; Pedersen, S.F. ErbB2 upregulates the Na+,HCO3--cotransporter NBCn1/SLC4A7 in human breast cancer cells via Akt, ERK, Src, and Kruppel-like factor 4. FASEB J. 2014, 28, 350–363. [Google Scholar] [CrossRef]
- Zu, X.; Zhang, Q.; Cao, R.; Liu, J.; Zhong, J.; Wen, G.; Cao, D. Transforming growth factor-beta signaling in tumor initiation, progression and therapy in breast cancer: An update. Cell Tissue Res. 2012, 347, 73–84. [Google Scholar] [CrossRef]
- Imamura, T.; Hikita, A.; Inoue, Y. The roles of TGF-beta signaling in carcinogenesis and breast cancer metastasis. Breast Cancer 2012, 19, 118–124. [Google Scholar] [CrossRef]
- Taylor, M.A.; Lee, Y.H.; Schiemann, W.P. Role of TGF-beta and the tumor microenvironment during mammary tumorigenesis. Gene Expression 2011, 15, 117–132. [Google Scholar] [CrossRef]
- Muraoka, R.S.; Koh, Y.; Roebuck, L.R.; Sanders, M.E.; Brantley-Sieders, D.; Gorska, A.E.; Moses, H.L.; Arteaga, C.L. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol. Cell. Biol. 2003, 23, 8691–8703. [Google Scholar] [CrossRef]
- Siegel, P.M.; Shu, W.; Cardiff, R.D.; Muller, W.J.; Massague, J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Nat. Acad. Sci. USA 2003, 100, 8430–8435. [Google Scholar] [CrossRef]
- Muraoka-Cook, R.S.; Shin, I.; Yi, J.Y.; Easterly, E.; Barcellos-Hoff, M.H.; Yingling, J.M.; Zent, R.; Arteaga, C.L. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene 2006, 25, 3408–3423. [Google Scholar] [CrossRef]
- Eswaran, J.; Soundararajan, M.; Kumar, R.; Knapp, S. UnPAKing the class differences among p21-activated kinases. Trends Biochem. Sci. 2008, 33, 394–403. [Google Scholar] [CrossRef]
- Whale, A.; Hashim, F.N.; Fram, S.; Jones, G.E.; Wells, C.M. Signalling to cancer cell invasion through PAK family kinases. Front. Biosci. 2011, 16, 849–864. [Google Scholar] [CrossRef]
- Arias-Romero, L.E.; Villamar-Cruz, O.; Pacheco, A.; Kosoff, R.; Huang, M.; Muthuswamy, S.K.; Chernoff, J. A Rac-Pak signaling pathway is essential for ErbB2-mediated transformation of human breast epithelial cancer cells. Oncogene 2010, 29, 5839–5849. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, N.; Cui, X.; Zheng, X.; Deng, L.; Price, S.; Karantza, V.; Minden, A. The protein kinase Pak4 disrupts mammary acinar architecture and promotes mammary tumorigenesis. Oncogene 2010, 29, 5883–5894. [Google Scholar] [CrossRef]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef]
- Mason, S.D.; Joyce, J.A. Proteolytic networks in cancer. Trends Cell Biol. 2011, 21, 228–237. [Google Scholar] [CrossRef]
- Jedeszko, C.; Sloane, B.F. Cysteine cathepsins in human cancer. Biol. Chem. 2004, 385, 1017–1027. [Google Scholar]
- Neal, C.L.; Yao, J.; Yang, W.; Zhou, X.; Nguyen, N.T.; Lu, J.; Danes, C.G.; Guo, H.; Lan, K.H.; Ensor, J.; et al. 14–3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009, 69, 3425–3432. [Google Scholar] [CrossRef]
- Nylandsted, J.; Becker, A.C.; Bunkenborg, J.; Andersen, J.S.; Dengjel, J.; Jaattela, M. ErbB2-associated changes in the lysosomal proteome. Proteomics 2011, 11, 2830–2838. [Google Scholar] [CrossRef]
- Tang, Y.; Mackey, J.; Lai, R.; Ghosh, S.; Santos, C.; Graham, K.; Damaraju, S.; Pasdar, M.; Li, L. Quantitative proteomic analysis of HER2 normal and overexpressing MCF-7 breast cancer cells revealed proteomic changes accompanied with HER2 gene amplification. J. Proteomics 2013, 91C, 200–209. [Google Scholar]
- Mackay, A.; Jones, C.; Dexter, T.; Silva, R.L.; Bulmer, K.; Jones, A.; Simpson, P.; Harris, R.A.; Jat, P.S.; Neville, A.M.; et al. cDNA microarray analysis of genes associated with ERBB2 (HER2/neu) overexpression in human mammary luminal epithelial cells. Oncogene 2003, 22, 2680–2688. [Google Scholar] [CrossRef]
- Brandt, R.; Eisenbrandt, R.; Leenders, F.; Zschiesche, W.; Binas, B.; Juergensen, C.; Theuring, F. Mammary gland specific hEGF receptor transgene expression induces neoplasia and inhibits differentiation. Oncogene 2000, 19, 2129–2137. [Google Scholar] [CrossRef]
- Zhan, L.; Xiang, B.; Muthuswamy, S.K. Controlled activation of ErbB1/ErbB2 heterodimers promote invasion of three-dimensional organized epithelia in an ErbB1-dependent manner: implications for progression of ErbB2-overexpressing tumors. Cancer Res. 2006, 66, 5201–5208. [Google Scholar] [CrossRef]
- Chang, J.; Nicolau, M.M.; Cox, T.R.; Wetterskog, D.; Martens, J.W.; Barker, H.E.; Erler, J.T. LOXL2 induces aberrant acinar morphogenesis via ErbB2 signaling. Breast Cancer Res. 2013, 15, R67. [Google Scholar] [CrossRef]
- Watabe, T.; Yoshida, K.; Shindoh, M.; Kaya, M.; Fujikawa, K.; Sato, H.; Seiki, M.; Ishii, S.; Fujinaga, K. The Ets-1 and Ets-2 transcription factors activate the promoters for invasion-associated urokinase and collagenase genes in response to epidermal growth factor. Int. J. Cancer. 1998, 77, 128–137. [Google Scholar] [CrossRef]
- Kim, S.; Han, J.; Shin, I.; Kil, W.H.; Lee, J.E.; Nam, S.J. A functional comparison between the HER2(high)/HER3 and the HER2(low)/HER3 dimers on heregulin-beta1-induced MMP-1 and MMP-9 expression in breast cancer cells. Exp. Mol. Med. 2012, 44, 473–482. [Google Scholar] [CrossRef]
- Park, Y.H.; Jung, H.H.; Ahn, J.S.; Im, Y.H. Ets-1 upregulates HER2-induced MMP-1 expression in breast cancer cells. Biochem. Biophys. Res. Commun. 2008, 377, 389–394. [Google Scholar] [CrossRef]
- Brisson, L.; Reshkin, S.J.; Gore, J.; Roger, S. pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur. J. Cell. Biol. 2012, 91, 847–860. [Google Scholar] [CrossRef]
- Marcotte, R.; Muller, W.J. Signal transduction in transgenic mouse models of human breast cancer--implications for human breast cancer. J. Mammary Gland Biol. Neoplasi. 2008, 13, 323–335. [Google Scholar] [CrossRef]
- Oshima, R.G.; Lesperance, J.; Munoz, V.; Hebbard, L.; Ranscht, B.; Sharan, N.; Muller, W.J.; Hauser, C.A.; Cardiff, R.D. Angiogenic acceleration of Neu induced mammary tumor progression and metastasis. Cancer Res. 2004, 64, 169–179. [Google Scholar] [CrossRef]
- Barbieri, I.; Pensa, S.; Pannellini, T.; Quaglino, E.; Maritano, D.; Demaria, M.; Voster, A.; Turkson, J.; Cavallo, F.; Watson, C.J.; et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010, 70, 2558–2567. [Google Scholar] [CrossRef]
- Wakefield, A.; Soukupova, J.; Montagne, A.; Ranger, J.; French, R.; Muller, W.J.; Clarkson, R.W. Bcl3 selectively promotes metastasis of ERBB2-driven mammary tumors. Cancer Res. 2013, 73, 745–755. [Google Scholar] [CrossRef]
- Hutchinson, J.N.; Jin, J.; Cardiff, R.D.; Woodgett, J.R.; Muller, W.J. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004, 64, 3171–3178. [Google Scholar] [CrossRef]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer cell 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Schade, B.; Rao, T.; Dourdin, N.; Lesurf, R.; Hallett, M.; Cardiff, R.D.; Muller, W.J. PTEN deficiency in a luminal ErbB-2 mouse model results in dramatic acceleration of mammary tumorigenesis and metastasis. J. Biol. Chem. 2009, 284, 19018–19026. [Google Scholar] [CrossRef]
- Ling, C.; Su, V.M.; Zuo, D.; Muller, W.J. Loss of the 14–3-3sigma tumor suppressor is a critical event in ErbB2-mediated tumor progression. Cancer Discov. 2012, 2, 68–81. [Google Scholar] [CrossRef]
- Ramirez, N.E.; Zhang, Z.; Madamanchi, A.; Boyd, K.L.; O’Rear, L.D.; Nashabi, A.; Li, Z.; Dupont, W.D.; Zijlstra, A.; Zutter, M.M. The alpha(2)beta(1) integrin is a metastasis suppressor in mouse models and human cancer. J. Clin. Invest. 2011, 121, 226–237. [Google Scholar] [CrossRef]
- Huck, L.; Pontier, S.M.; Zuo, D.M.; Muller, W.J. beta1-integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc. Nat. Acad. Sci. USA 2010, 107, 15559–15564. [Google Scholar] [CrossRef]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef]
- Leng, X.; Ding, T.; Lin, H.; Wang, Y.; Hu, L.; Hu, J.; Feig, B.; Zhang, W.; Pusztai, L.; Symmans, W.F.; et al. Inhibition of lipocalin 2 impairs breast tumorigenesis and metastasis. Cancer Res. 2009, 69, 8579–8584. [Google Scholar] [CrossRef]
- Davis, V.L.; Shaikh, F.; Gallagher, K.M.; Villegas, M.; Rea, S.L.; Cline, J.M.; Hughes, C.L. Inhibition of Neu-induced mammary carcinogenesis in transgenic mice expressing ERDelta3, a dominant negative estrogen receptor alpha variant. Hormones Cancer 2012, 3, 227–239. [Google Scholar] [CrossRef]
- Heckman-Stoddard, B.M.; Vargo-Gogola, T.; McHenry, P.R.; Jiang, V.; Herrick, M.P.; Hilsenbeck, S.G.; Settleman, J.; Rosen, J.M. Haploinsufficiency for p190B RhoGAP inhibits MMTV-Neu tumor progression. Breast Cancer Res. 2009, 11, R61. [Google Scholar] [CrossRef]
- Worzfeld, T.; Swiercz, J.M.; Looso, M.; Straub, B.K.; Sivaraj, K.K.; Offermanns, S. ErbB-2 signals through Plexin-B1 to promote breast cancer metastasis. J. Clin. Invest. 2012, 122, 1296–1305. [Google Scholar] [CrossRef]
- Laurin, M.; Huber, J.; Pelletier, A.; Houalla, T.; Park, M.; Fukui, Y.; Haibe-Kains, B.; Muller, W.J.; Cote, J.F. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc. Nat. Acad. Sci. USA 2013, 110, 7434–7439. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef]
- Julien, S.G.; Dube, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nature Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.M.; Zhuang, G.; Hicks, D.; Fang, W.B.; Hwang, Y.; Cates, J.M.; Coffman, K.; Jackson, D.; Bruckheimer, E.; Muraoka-Cook, R.S.; et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Invest. 2008, 118, 64–78. [Google Scholar] [CrossRef]
- Ke, Y.; Wu, D.; Princen, F.; Nguyen, T.; Pang, Y.; Lesperance, J.; Muller, W.J.; Oshima, R.G.; Feng, G.S. Role of Gab2 in mammary tumorigenesis and metastasis. Oncogene 2007, 26, 4951–4960. [Google Scholar] [CrossRef]
- Freudenberg, J.A.; Wang, Q.; Katsumata, M.; Drebin, J.; Nagatomo, I.; Greene, M.I. The role of HER2 in early breast cancer metastasis and the origins of resistance to HER2-targeted therapies. Exp. Mol. Pathol. 2009, 87, 1–11. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J. Clin. Invest. 2009, 119, 1438–1449. [Google Scholar] [CrossRef]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Invest. 2009, 119, 1417–1419. [Google Scholar] [CrossRef]
- Roxanis, I. Occurrence and significance of epithelial-mesenchymal transition in breast cancer. J. Clin. Pathol. 2013, 66, 517–521. [Google Scholar] [CrossRef]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef]
- Bastid, J. EMT in carcinoma progression and dissemination: facts, unanswered questions, and clinical considerations. Cancer Metast Rev. 2012, 31, 277–283. [Google Scholar] [CrossRef]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metast Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Wendt, M.K.; Schiemann, W.P. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009, 11, R68. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef]
- Ponzo, M.G.; Lesurf, R.; Petkiewicz, S.; O’Malley, F.P.; Pinnaduwage, D.; Andrulis, I.L.; Bull, S.B.; Chughtai, N.; Zuo, D.; Souleimanova, M.; et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc. Nat. Acad. Sci. USA 2009, 106, 12903–12908. [Google Scholar] [CrossRef]
- Leontovich, A.A.; Zhang, S.; Quatraro, C.; Iankov, I.; Veroux, P.F.; Gambino, M.W.; Degnim, A.; McCubrey, J.; Ingle, J.; Galanis, E.; et al. Raf-1 oncogenic signaling is linked to activation of mesenchymal to epithelial transition pathway in metastatic breast cancer cells. Int. J. Oncol. 2012, 40, 1858–1864. [Google Scholar]
- Soini, Y.; Tuhkanen, H.; Sironen, R.; Virtanen, I.; Kataja, V.; Auvinen, P.; Mannermaa, A.; Kosma, V.M. Transcription factors zeb1, twist and snai1 in breast carcinoma. BMC Cancer 2011, 11, 73. [Google Scholar] [CrossRef]
- Al Saleh, S.; Sharaf, L.H.; Luqmani, Y.A. Signalling pathways involved in endocrine resistance in breast cancer and associations with epithelial to mesenchymal transition (Review). Int. J. Oncol. 2011, 38, 1197–1217. [Google Scholar]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Giordano, A.; Gao, H.; Anfossi, S.; Cohen, E.; Mego, M.; Lee, B.N.; Tin, S.; de Laurentiis, M.; Parker, C.A.; Alvarez, R.H.; et al. Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer . Mol. Cancer Ther. 2012, 11, 2526–2534. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brix, D.M.; Clemmensen, K.K.B.; Kallunki, T. When Good Turns Bad: Regulation of Invasion and Metastasis by ErbB2 Receptor Tyrosine Kinase. Cells 2014, 3, 53-78. https://doi.org/10.3390/cells3010053
Brix DM, Clemmensen KKB, Kallunki T. When Good Turns Bad: Regulation of Invasion and Metastasis by ErbB2 Receptor Tyrosine Kinase. Cells. 2014; 3(1):53-78. https://doi.org/10.3390/cells3010053
Chicago/Turabian StyleBrix, Ditte Marie, Knut Kristoffer Bundgaard Clemmensen, and Tuula Kallunki. 2014. "When Good Turns Bad: Regulation of Invasion and Metastasis by ErbB2 Receptor Tyrosine Kinase" Cells 3, no. 1: 53-78. https://doi.org/10.3390/cells3010053