Glucocorticoid Induced Cerebellar Toxicity in the Developing Neonate: Implications for Glucocorticoid Therapy during Bronchopulmonary Dysplasia

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
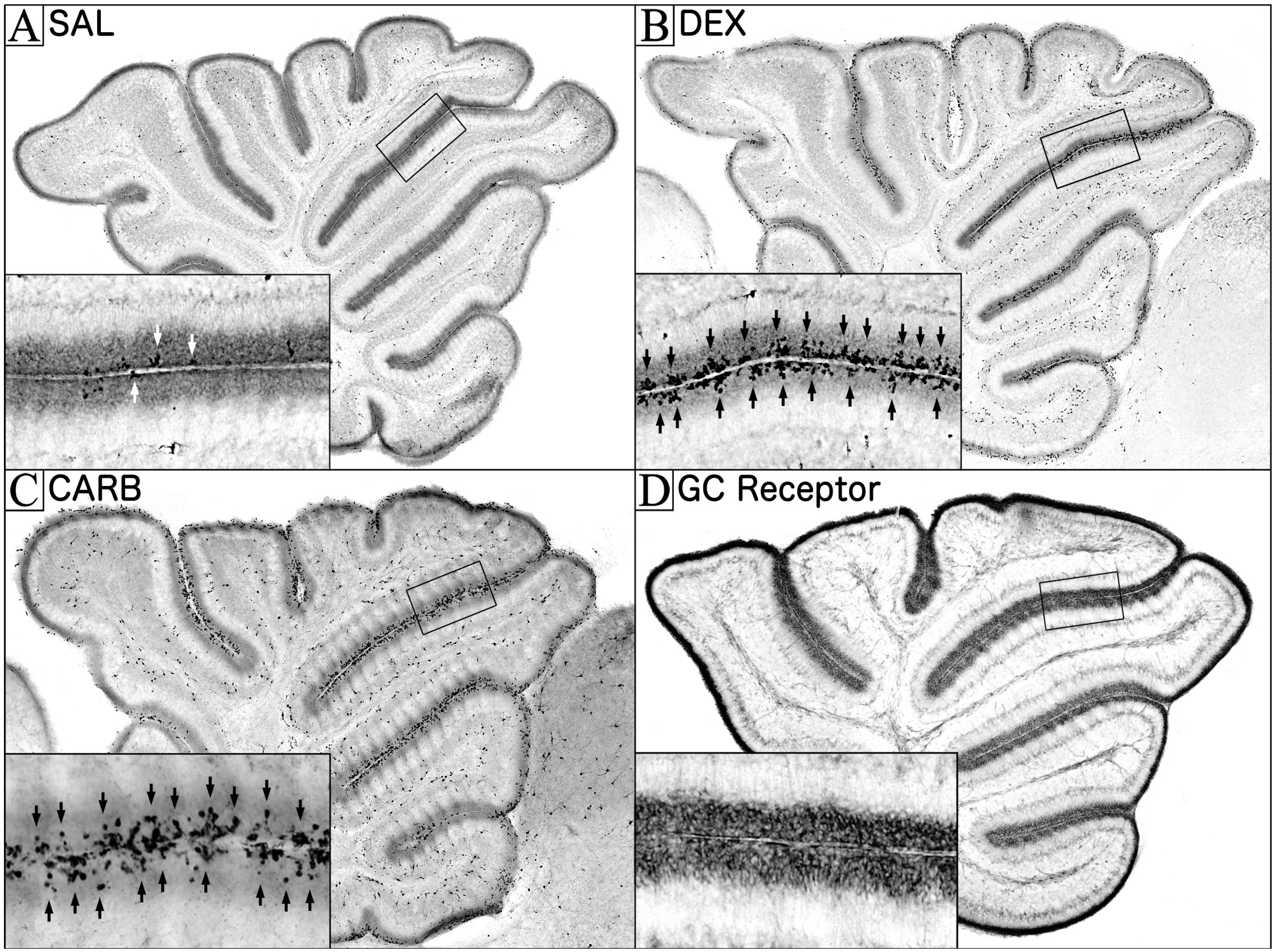
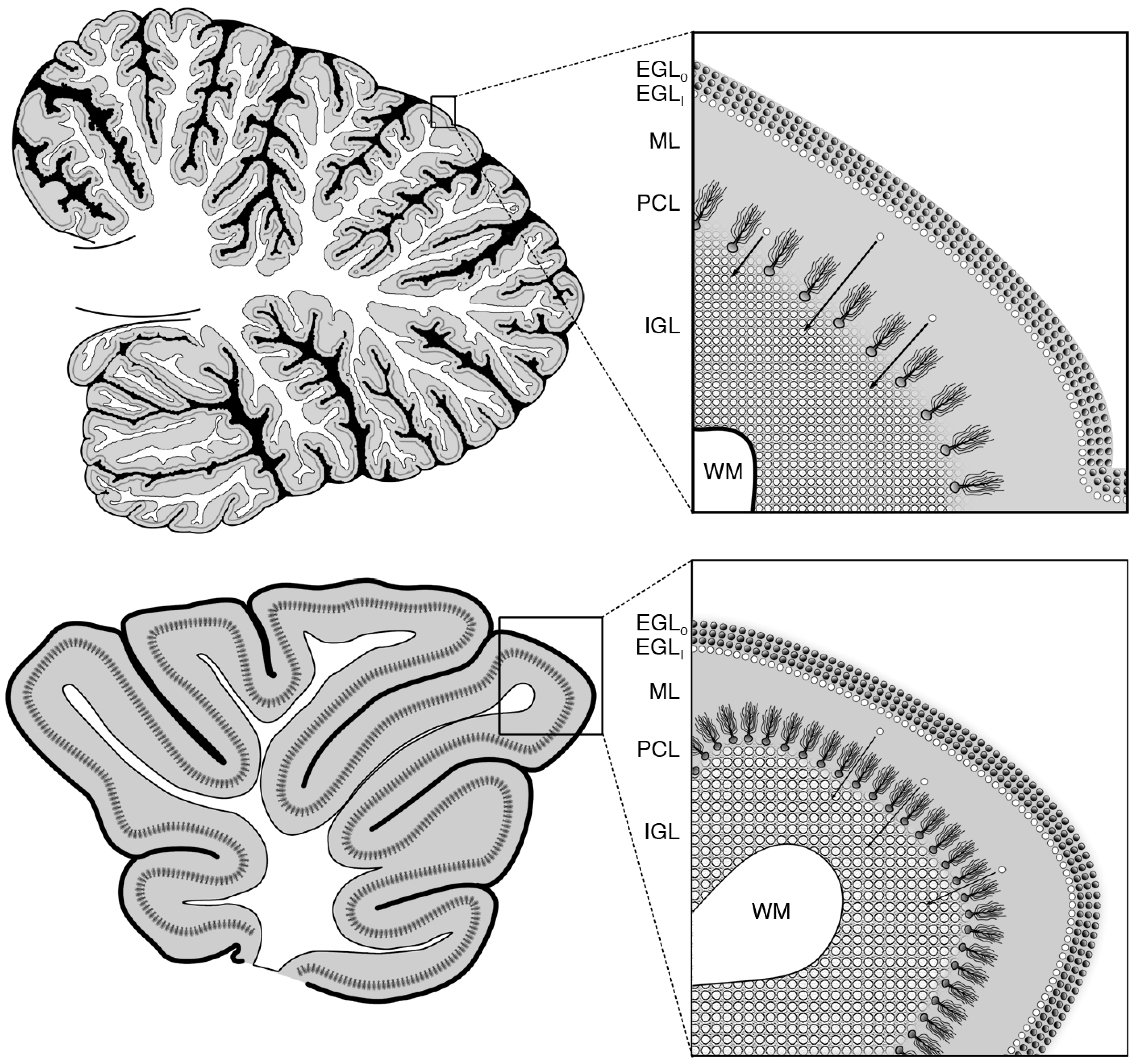
2. Glucocorticoid Induced Neural Progenitor Cell Death in the Cerebellum
2.1. The EGL and Its Role in Cerebellar Development

2.2. Apoptosis and Development
3. The Role of Glucocorticoids in Cerebellar Neurodevelopment
3.1. GC Stimulation in the EGL is Carefully Regulated during Its Disappearance
3.2. Interactions between GC Stimulation and the Sonic Hedgehog Pathway
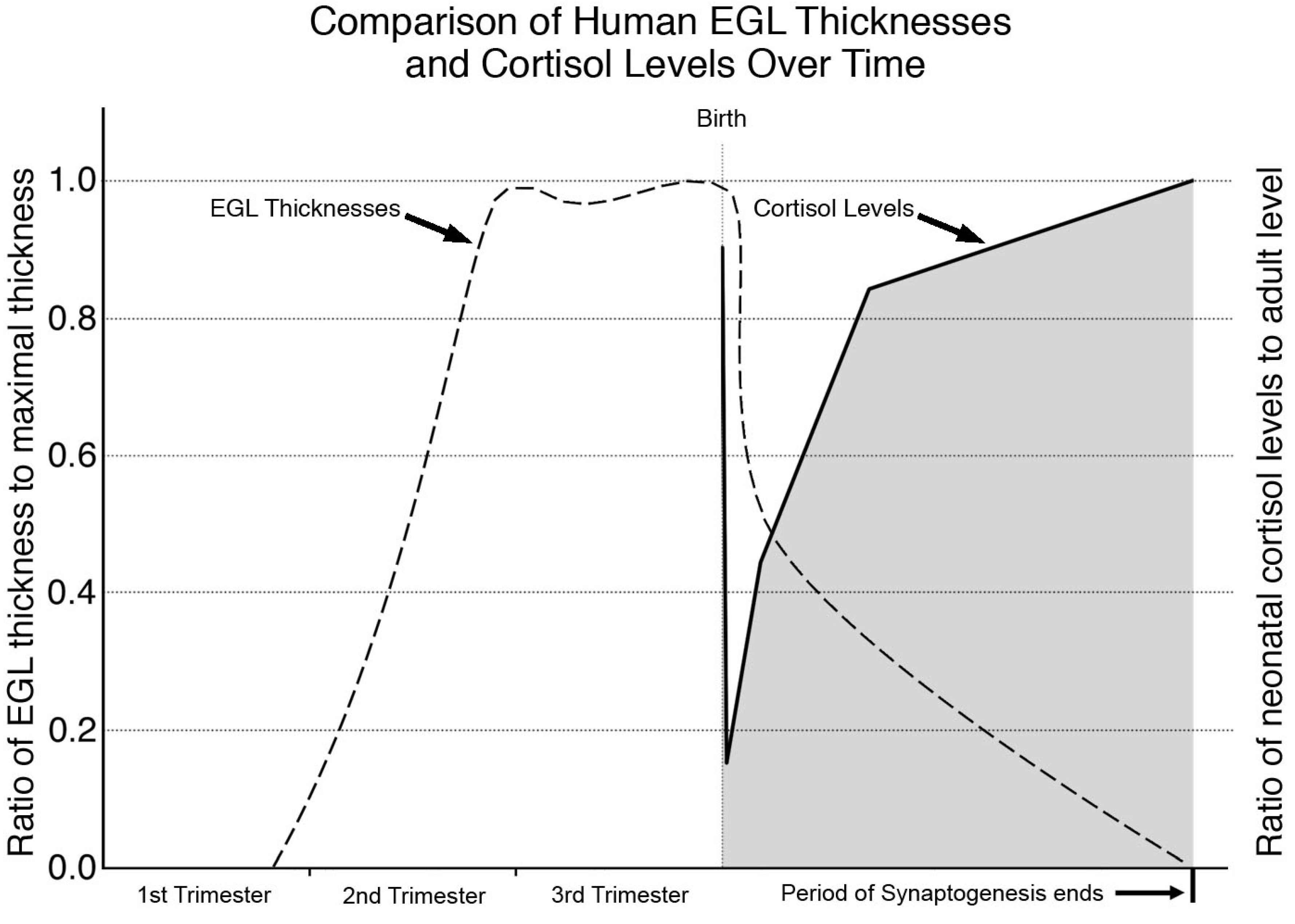
4. Does GC Stimulation Have Similar Cerebellar Effects in Humans?

Human Neurodevelopment and the Cerebellum
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ehrenkranz, R.A.; Walsh, M.C.; Vohr, B.R.; Jobe, A.H.; Wright, L.L.; Fanaroff, A.A.; Wrage, L.A.; Poole, K.; National Institutes of Child Health and Human Development Neonatal Research Network. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics 2005, 116, 1353–1360. [Google Scholar] [CrossRef]
- National Institutes of Health Consensus Development Panel. Effect of corticosteroids for fetal maturation on perinatal outcomes. NIH Consens Statement 1994, 12, 1–24. [Google Scholar]
- Yeh, T.F.; Lin, Y.J.; Huang, C.C.; Chen, Y.J.; Lin, C.H.; Lin, H.C.; Hsieh, W.S.; Lien, Y.J. Early dexamethasone therapy in preterm infants: A follow-up study. Pediatrics 1998, 101, E7. [Google Scholar]
- Yeh, T.F.; Lin, Y.J.; Lin, H.C.; Huang, C.C.; Hsieh, W.S.; Lin, C.H.; Tsai, C.H. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. N. Engl. J. Med. 2004, 350, 1304–1313. [Google Scholar] [CrossRef]
- Shinwell, E.S.; Karplus, M.; Reich, D.; Weintraub, Z.; Blazer, S.; Bader, D.; Yurman, S.; Dolfin, T.; Kogan, A.; Dollberg, S.; et al. Early postnatal dexamethasone treatment and increased incidence of cerebral palsy. Arch. Dis. Child Fetal. Neonatal. Ed. 2000, 83, F177–F181. [Google Scholar] [CrossRef]
- Parikh, N.A.; Lasky, R.E.; Kennedy, K.A.; Moya, F.R.; Hochhauser, L.; Romo, S.; Tyson, J.E. Postnatal dexamethasone therapy and cerebral tissue volumes in extremely low birth weight infants. Pediatrics 2007, 119, 265–272. [Google Scholar] [CrossRef]
- Tam, E.W.; Chau, V.; Ferriero, D.M.; Barkovich, A.J.; Poskitt, K.J.; Studholme, C.; Fok, E.D.; Grunau, R.E.; Glidden, D.V.; Miller, S.P. Preterm cerebellar growth impairment after postnatal exposure to glucocorticoids. Sci. Transl. Med. 2011, 3, 105ra105. [Google Scholar] [CrossRef]
- National Institutes of Health Consensus Development Panel. Antenatal corticosteroids revisited: Repeat courses. NIH Consens Statement 2000, 17, 1–18. [Google Scholar]
- Committee on Fetus and Newborn. Postnatal corticosteroids to treat or prevent chronic lung disease in preterm infants. Pediatrics 2002, 109, 330–338. [Google Scholar] [CrossRef]
- Watterberg, K.L. Policy statement--postnatal corticosteroids to prevent or treat bronchopulmonary dysplasia. Pediatrics 2010, 126, 800–808. [Google Scholar] [CrossRef]
- Jobe, A.H. Postnatal corticosteroids for bronchopulmonary dysplasia. Clin. Perinatol. 2009, 36, 177–188. [Google Scholar] [CrossRef]
- Tin, W.; Wiswell, T.E. Drug therapies in bronchopulmonary dysplasia: Debunking the myths. Semin. Fetal. Neonatal Med. 2009, 14, 383–390. [Google Scholar] [CrossRef]
- Rodier, P.M. Environmental causes of central nervous system maldevelopment. Pediatrics 2004, 113, 1076–1083. [Google Scholar]
- Field, E.J. Effect of cortisone on the neonatal rat. Nature 1954, 174, 182. [Google Scholar] [CrossRef]
- Buckingham, S. Is lung an analog of Moog’s developing intestine? 1. Phosphotases and pulmonary differentiation in fetal rabbits. Fed. Am. Soc. Exp. Biol. 1968, 27, 328. [Google Scholar]
- Weichsel, M.E., Jr. Glucocorticoid effect upon thymidine kinase in the developing cerebellum. Pediatr. Res. 1974, 8, 843–847. [Google Scholar] [CrossRef]
- Ferguson, S.A.; Holson, R.R. Neonatal dexamethasone on day 7 causes mild hyperactivity and cerebellar stunting. Neurotoxicol. Teratol. 1999, 21, 71–76. [Google Scholar] [CrossRef]
- Bohn, M.C.; Lauder, J.M. Cerebellar granule cell genesis in the hydrocortisone-treated rats. Dev. Neurosci. 1980, 3, 81–89. [Google Scholar] [CrossRef]
- Noguchi, K.K.; Lau, K.; Smith, D.J.; Swiney, B.S.; Farber, N.B. Glucocorticoid receptor stimulation and the regulation of neonatal cerebellar neural progenitor cell apoptosis. Neurobiol. Dis. 2011, 43, 356–363. [Google Scholar] [CrossRef]
- Noguchi, K.K.; Walls, K.C.; Wozniak, D.F.; Olney, J.W.; Roth, K.A.; Farber, N.B. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ. 2008, 15, 1582–1592. [Google Scholar] [CrossRef]
- Maloney, S.E.; Noguchi, K.K.; Wozniak, D.F.; Fowler, S.C.; Farber, N.B. Long-term effects of multiple glucocorticoid exposures in neonatal mice. Behav. Sci. (Basel). 2001, 1, 4–30. [Google Scholar]
- Pedraz, J.L.; Lanao, J.M.; Dominguez-Gil, A. Interspecies pharmacokinetics of ketamine. In Status of Ketamine in Anesthesiology; Domino, E.F., Ed.; NPP Books: Ann Arbor, MI, USA, 1990; pp. 285–295. [Google Scholar]
- Keil, D.E.; Luebke, R.W.; Pruett, S.B. Differences in the effects of dexamethasone on macrophage nitrite production: Dependence on exposure regimen (in vivo or in vitro) and activation stimuli. Int. J. Immunopharmacol. 1995, 17, 157–166. [Google Scholar] [CrossRef]
- Carletti, B.; Rossi, F. Neurogenesis in the cerebellum. Neuroscientist 2008, 14, 91–100. [Google Scholar] [CrossRef]
- Andersen, B.B.; Korbo, L.; Pakkenberg, B. A quantitative study of the human cerebellum with unbiased stereological techniques. J. Comp. Neurol. 1992, 326, 549–560. [Google Scholar] [CrossRef]
- Harvey, R.J.; Napper, R.M. Quantitative study of granule and Purkinje cells in the cerebellar cortex of the rat. J. Comp. Neurol. 1988, 274, 151–157. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The human brain in numbers: A linearly scaled-up primate brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef]
- Sajan, S.A.; Waimey, K.E.; Millen, K.J. Novel approaches to studying the genetic basis of cerebellar development. Cerebellum 2010, 9, 272–283. [Google Scholar]
- Alder, J.; Cho, N.K.; Hatten, M.E. Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron 1996, 17, 389–399. [Google Scholar] [CrossRef]
- Sotelo, C. Cellular and genetic regulation of the development of the cerebellar system. Prog. Neurobiol. 2004, 72, 295–339. [Google Scholar] [CrossRef]
- Rakic, P.; Sidman, R.L. Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. J. Comp. Neurol. 1970, 139, 473–500. [Google Scholar]
- Porterfield, S.P. Vulnerability of the developing brain to thyroid abnormalities: Environmental insults to the thyroid system. Environ. Health Perspect. 1994, 102 Suppl, 125–130. [Google Scholar]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Kuida, K.; Zheng, T.S.; Na, S.; Kuan, C.; Yang, D.; Karasuyama, H.; Rakic, P.; Flavell, R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 1996, 384, 368–372. [Google Scholar] [CrossRef]
- Kuida, K.; Haydar, T.F.; Kuan, C.Y.; Gu, Y.; Taya, C.; Karasuyama, H.; Su, M.S.; Rakic, P.; Flavell, R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94, 325–337. [Google Scholar] [CrossRef]
- Cecconi, F.; Alvarez-Bolado, G.; Meyer, B.I.; Roth, K.A.; Gruss, P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998, 94, 727–737. [Google Scholar] [CrossRef]
- Altman, J.; Bayer, S.A. Development of the Cerebellar System: In Relation to Its Evolution, Structure, and Functions; CRC Press: Boca Raton, USA, 1997. [Google Scholar]
- Yang, X.; Klein, R.; Tian, X.; Cheng, H.T.; Kopan, R.; Shen, J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev. Biol. 2004, 269, 81–94. [Google Scholar] [CrossRef]
- Depaepe, V.; Suarez-Gonzalez, N.; Dufour, A.; Passante, L.; Gorski, J.A.; Jones, K.R.; Ledent, C.; Vanderhaeghen, P. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature 2005, 435, 1244–1250. [Google Scholar] [CrossRef]
- McFarland, K.N.; Wilkes, S.R.; Koss, S.E.; Ravichandran, K.S.; Mandell, J.W. Neural-specific inactivation of ShcA results in increased embryonic neural progenitor apoptosis and microencephaly. J. Neurosci. 2006, 26, 7885–7897. [Google Scholar] [CrossRef]
- La Rosa, E.J.; de Pablo, F. Cell death in early neural development: Beyond the neurotrophic theory. Trends Neurosci. 2000, 23, 454–458. [Google Scholar] [CrossRef]
- Sommer, L.; Rao, M. Neural stem cells and regulation of cell number. Prog. Neurobiol. 2002, 66, 1–18. [Google Scholar] [CrossRef]
- Akhtar, R.S.; Geng, Y.; Klocke, B.J.; Roth, K.A. Neural precursor cells possess multiple p53-dependent apoptotic pathways. Cell Death Differ 2006, 13, 1727–1739. [Google Scholar] [CrossRef]
- De Zio, D.; Giunta, L.; Corvaro, M.; Ferraro, E.; Cecconi, F. Expanding roles of programmed cell death in mammalian neurodevelopment. Semin. Cell Dev. Biol. 2005, 16, 281–294. [Google Scholar] [CrossRef]
- Haydar, T.F.; Kuan, C.Y.; Flavell, R.A.; Rakic, P. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb. Cortex 1999, 9, 621–626. [Google Scholar] [CrossRef]
- Medh, R.D.; Thompson, E.B. Hormonal regulation of physiological cell turnover and apoptosis. Cell Tissue Res. 2000, 301, 101–124. [Google Scholar] [CrossRef]
- Gruver-Yates, A.; Cidlowski, J. Tissue-specific actions of glucocorticoids on apoptosis: A double-edged sword. Cells 2013, 2, 202–223. [Google Scholar] [CrossRef]
- Liggins, G.C. The role of cortisol in preparing the fetus for birth. Reprod. Fertil. Dev. 1994, 6, 141–150. [Google Scholar] [CrossRef]
- Pavlík, A.; Buresová, M. The neonatal cerebellum: The highest level of glucocorticoid receptors in the brain. Brain Res. 1984, 314, 13–20. [Google Scholar]
- Schmidt, M.V.; Oitzl, M.S.; Levine, S.; de Kloet, E.R. The HPA system during the postnatal development of CD1 mice and the effects of maternal deprivation. Brain Res. Dev. Brain Res. 2002, 139, 39–49. [Google Scholar] [CrossRef]
- Dakine, N.; Oliver, C.; Grino, M. Effects of experimental hypothyroidism on the development of the hypothalamo-pituitary-adrenal axis in the rat. Life Sci. 2000, 67, 2827–2844. [Google Scholar] [CrossRef]
- Holmes, M.C.; Sangra, M.; French, K.L.; Whittle, I.R.; Paterson, J.; Mullins, J.J.; Seckl, J.R. 11beta-Hydroxysteroid dehydrogenase type 2 protects the neonatal cerebellum from deleterious effects of glucocorticoids. Neuroscience 2006, 137, 865–873. [Google Scholar] [CrossRef]
- Robson, A.C.; Leckie, C.M.; Seckl, J.R.; Holmes, M.C. 11 Beta-hydroxysteroid dehydrogenase type 2 in the postnatal and adult rat brain. Brain Res. Mol. Brain Res. 1998, 61, 1–10. [Google Scholar] [CrossRef]
- Schmidt, M.V.; Enthoven, L.; van der Mark, M.; Levine, S.; de Kloet, E.R.; Oitzl, M.S. The postnatal development of the hypothalamic-pituitary-adrenal axis in the mouse. Int. J. Dev. Neurosci. 2003, 21, 125–132. [Google Scholar] [CrossRef]
- Seckl, J.R. Prenatal glucocorticoids and long-term programming. Eur. J. Endocrinol. 2004, 151 Suppl, U49–U62. [Google Scholar] [CrossRef]
- Suzuki, T.; Sasano, H.; Suzuki, S.; Hirasawa, G.; Takeyama, J.; Muramatsu, Y.; Date, F.; Nagura, H.; Krozowski, Z.S. 11Beta-hydroxysteroid dehydrogenase type 2 in human lung: Possible regulator of mineralocorticoid action. J. Clin. Endocrinol. Metab. 1998, 83, 4022–4025. [Google Scholar] [CrossRef]
- Heine, V.M.; Griveau, A.; Chapin, C.; Ballard, P.L.; Chen, J.K.; Rowitch, D.H. A small-molecule smoothened agonist prevents glucocorticoid-induced neonatal cerebellar injury. Sci. Transl. Med. 2011, 3, 105ra104. [Google Scholar] [CrossRef]
- Benesová, O.; Pavlík, A. Brain glucocorticoid receptors and their role in behavioural teratogenicity of synthetic glucocorticoids. Arch. Toxicol. Suppl. 1985, 8, 73–76. [Google Scholar] [CrossRef]
- Benesová, O.; Pavlík, A. Perinatal treatment with glucocorticoids and the risk of maldevelopment of the brain. Neuropharmacology 1989, 28, 89–97. [Google Scholar] [CrossRef]
- Meyer, J.S. Early adrenalectomy stimulates subsequent growth and development of the rat brain. Exp. Neurol. 1983, 82, 432–446. [Google Scholar] [CrossRef]
- Yehuda, R.; Fairman, K.R.; Meyer, J.S. Enhanced brain cell proliferation following early adrenalectomy in rats. J. Neurochem. 1989, 53, 241–248. [Google Scholar] [CrossRef]
- Yehuda, R.; Meyer, J.S. Regional patterns of brain growth during the first three weeks following early adrenalectomy. Physiol. Behav. 1991, 49, 233–237. [Google Scholar] [CrossRef]
- Goldowitz, D.; Hamre, K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998, 21, 375–382. [Google Scholar] [CrossRef]
- Vaillant, C.; Monard, D. SHH pathway and cerebellar development. Cerebellum 2009, 8, 291–301. [Google Scholar] [CrossRef]
- Heine, V.M.; Rowitch, D.H. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism. J. Clin. Invest. 2009, 119, 267–277. [Google Scholar]
- Heine, V.M.; Priller, M.; Ling, J.; Rowitch, D.H.; Schüller, U. Dexamethasone destabilizes Nmyc to inhibit the growth of hedgehog-associated medulloblastoma. Cancer Res. 2010, 70, 5220–5225. [Google Scholar] [CrossRef]
- McCune, J.S.; Hawke, R.L.; LeCluyse, E.L.; Gillenwater, H.H.; Hamilton, G.; Ritchie, J.; Lindley, C. In vivo and in vitro induction of human cytochrome P4503A4 by dexamethasone. Clin. Pharmacol. Ther. 2000, 68, 356–366. [Google Scholar] [CrossRef]
- Liberman, A.C.; Druker, J.; Refojo, D.; Holsboer, F.; Arzt, E. Glucocorticoids inhibit GATA-3 phosphorylation and activity in T cells. FASEB J. 2009, 23, 1558–1571. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Bond, M.C.; Chen, M.; Ren, X.R.; Lyerly, H.K.; Barak, L.S.; Chen, W. Identification of select glucocorticoids as Smoothened agonists: Potential utility for regenerative medicine. Proc. Natl. Acad. Sci. USA 2010, 107, 9323–9328. [Google Scholar]
- Wang, J.; Barak, L.S.; Mook, R.A.; Chen, W. Glucocorticoid hedgehog agonists in neurogenesis. Vitam. Horm. 2011, 87, 207–215. [Google Scholar]
- Wang, Y.; Davidow, L.; Arvanites, A.C.; Blanchard, J.; Lam, K.; Xu, K.; Oza, V.; Yoo, J.W.; Ng, J.M.; Curran, T.; et al. Glucocorticoid compounds modify smoothened localization and hedgehog pathway activity. Chem. Biol. 2012, 19, 972–982. [Google Scholar] [CrossRef]
- Schmahmann, J.D. Disorders of the cerebellum: Ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 367–378. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Soul, J.S.; Gauvreau, K.; Huppi, P.S.; Warfield, S.K.; Bassan, H.; Robertson, R.L.; Volpe, J.J.; du Plessis, A.J. Late gestation cerebellar growth is rapid and impeded by premature birth. Pediatrics 2005, 115, 688–695. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Bassan, H.; Gauvreau, K.; Robertson, R.L., Jr.; Sullivan, N.R.; Benson, C.B.; Avery, L.; Stewart, J.; Soul, J.S.; Ringer, S.A.; et al. Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, Learning, and behavioral disability in survivors? Pediatrics 2007, 120, 584–593. [Google Scholar] [CrossRef]
- Benders, M.J.; Groenendaal, F.; van Bel, F.; Ha Vinh, R.; Dubois, J.; Lazeyras, F.; Warfield, S.K.; Hüppi, P.S.; de Vries, L.S. Brain development of the preterm neonate after neonatal hydrocortisone treatment for chronic lung disease. Pediatr. Res. 2009, 66, 555–559. [Google Scholar] [CrossRef]
- Lodygensky, G.A.; Rademaker, K.; Zimine, S.; Gex-Fabry, M.; Lieftink, A.F.; Lazeyras, F.; Groenendaal, F.; de Vries, L.S.; Huppi, P.S. Structural and functional brain development after hydrocortisone treatment for neonatal chronic lung disease. Pediatrics 2005, 116, 1–7. [Google Scholar] [CrossRef]
- Peltoniemi, O.M.; Lano, A.; Puosi, R.; Yliherva, A.; Bonsante, F.; Kari, M.A.; Hallman, M.; Neonatal Hydrocortisone Working Group. Trial of early neonatal hydrocortisone: Two-year follow-up. Neonatology 2009, 95, 240–247. [Google Scholar] [CrossRef]
- Rademaker, K.J.; Uiterwaal, C.S.; Groenendaal, F.; Venema, M.M.; van Bel, F.; Beek, F.J.; van Haastert, I.C.; Grobbee, D.E.; de Vries, L.S. Neonatal hydrocortisone treatment: Neurodevelopmental outcome and MRI at school age in preterm-born children. J. Pediatr. 2007, 150, 351–357. [Google Scholar] [CrossRef]
- Rademaker, K.J.; de Vries, W.B. Long-term effects of neonatal hydrocortisone treatment for chronic lung disease on the developing brain and heart. Semin. Fetal. Neonatal. Med. 2009, 14, 171–177. [Google Scholar] [CrossRef]
- Watterberg, K.L. Adrenocortical function and dysfunction in the fetus and neonate. Semin. Neonatol. 2004, 9, 13–21. [Google Scholar] [CrossRef]
- Doyle, L.W.; Ehrenkranz, R.A.; Halliday, H.L. Postnatal hydrocortisone for preventing or treating bronchopulmonary dysplasia in preterm infants: A systematic review. Neonatology 2010, 98, 111–117. [Google Scholar] [CrossRef]
- Sloboda, D.M.; Challis, J.R.G.; Moss, T.J.M.; Newnham, J.P. Synthetic glucocorticoids: Antenatal administration and long-term implications. Curr. Pharm. Des. 2005, 11, 1459–1472. [Google Scholar] [CrossRef]
- Scott, S.M.; Wells, L. Corticosteroid-binding globulin in preterm infants in an intensive care unit. Horm. Res. 1995, 44, 218–221. [Google Scholar] [CrossRef]
- Sippell, W.G.; Dörr, H.G.; Bidlingmaier, F.; Knorr, D. Plasma levels of aldosterone, corticosterone, 11-deoxycorticosterone, progesterone, 17-hydroxyprogesterone, cortisol, and cortisone during infancy and childhood. Pediatr. Res. 1980, 14, 39–46. [Google Scholar] [CrossRef]
- Hanna, C.E.; Jett, P.L.; Laird, M.R.; Mandel, S.H.; LaFranchi, S.H.; Reynolds, J.W. Corticosteroid binding globulin, Total serum cortisol, and stress in extremely low-birth-weight infants. Am. J. Perinatol. 1997, 14, 201–204. [Google Scholar]
- Lavezzi, A.M.; Ottaviani, G.; Terni, L.; Matturri, L. Histological and biological developmental characterization of the human cerebellar cortex. Int. J. Dev. Neurosci. 2006, 24, 365–371. [Google Scholar] [CrossRef]
- Mastorakos, G.; Ilias, I. Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann. N. Y. Acad. Sci. 2003, 997, 136–149. [Google Scholar] [CrossRef]
- Seckl, J.R.; Miller, W.L. How safe is long-term prenatal glucocorticoid treatment? JAMA 1997, 277, 1077–1079. [Google Scholar] [CrossRef]
- Matthews, S.G.; Owen, D.; Kalabis, G.; Banjanin, S.; Setiawan, E.B.; Dunn, E.A.; Andrews, M.H. Fetal glucocorticoid exposure and hypothalamo-pituitary-adrenal (HPA) function after birth. Endocr. Res. 2004, 30, 827–836. [Google Scholar] [CrossRef]
- St John, E.B.; Carlo, W.A. Respiratory distress syndrome in VLBW infants: Changes in management and outcomes observed by the NICHD Neonatal Research Network. Semin. Perinatol. 2003, 27, 288–292. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Noguchi, K.K. Glucocorticoid Induced Cerebellar Toxicity in the Developing Neonate: Implications for Glucocorticoid Therapy during Bronchopulmonary Dysplasia. Cells 2014, 3, 36-52. https://doi.org/10.3390/cells3010036
Noguchi KK. Glucocorticoid Induced Cerebellar Toxicity in the Developing Neonate: Implications for Glucocorticoid Therapy during Bronchopulmonary Dysplasia. Cells. 2014; 3(1):36-52. https://doi.org/10.3390/cells3010036
Chicago/Turabian StyleNoguchi, Kevin K. 2014. "Glucocorticoid Induced Cerebellar Toxicity in the Developing Neonate: Implications for Glucocorticoid Therapy during Bronchopulmonary Dysplasia" Cells 3, no. 1: 36-52. https://doi.org/10.3390/cells3010036