Abstract
Damage to DNA is common and can arise from numerous environmental and endogenous sources. In response to ubiquitous DNA damage, Y-family DNA polymerases are induced by the SOS response and are capable of bypassing DNA lesions. In Escherichia coli, these Y-family polymerases are DinB and UmuC, whose activities are modulated by their interaction with the polymerase manager protein UmuD. Many, but not all, bacteria utilize DinB and UmuC homologs. Recently, a C-family polymerase named ImuC, which is similar in primary structure to the replicative DNA polymerase DnaE, was found to be able to copy damaged DNA and either carry out or suppress mutagenesis. ImuC is often found with proteins ImuA and ImuB, the latter of which is similar to Y‑family polymerases, but seems to lack the catalytic residues necessary for polymerase activity. This imuAimuBimuC mutagenesis cassette represents a widespread alternative strategy for translesion synthesis and mutagenesis in bacteria. Bacterial Y‑family and ImuC DNA polymerases contribute to replication past DNA damage and the acquisition of antibiotic resistance.
Keywords:
DNA damage; mutagenesis; SOS response; DNA pol IV (DinB); DNA pol V (UmuD'2C); dnaE; dnaE2; imuA; imuB; imuC 1. Introduction
Various endogenous and exogenous agents can cause damage to DNA, creating lesions and leading to mutagenesis [1]. Replicative DNA polymerases, enzymes that catalyze DNA replication, are incapable of replicating damaged DNA [1], although this inability is not absolute [2,3,4,5]. Therefore, cells across all domains of life are equipped with specialized DNA polymerases that have the ability to replicate damaged DNA in a process known as translesion synthesis (TLS), which was first proposed over 30 years ago [6,7,8,9]. This family of specialized DNA polymerases is known as the Y family of DNA polymerases, after the X family, because Y-family DNA polymerases play a greater role in DNA damage tolerance than in chromosomal replication [8,10]. Despite this specialized function to bypass damaged DNA, Y-family polymerases may cause mutations, because they are unable to replicate undamaged DNA with the same high fidelity as typical replicative polymerases [6,7]. These two functions have important implications for human health, as the mutagenic functions of Y family DNA polymerases contribute to antibiotic resistance, whereas the damage bypass functions can both prevent damage-induced mutations that can lead to cancer as well as decrease the effectiveness of DNA‑damaging cancer chemotherapy drugs by allowing cells to tolerate such damage [11]. This review will focus on the two E. coli Y-family polymerases as well as Y-family and other specialized DNA polymerases found in other species of eubacteria.
The mechanism governing Y-family polymerases and the regulation of TLS in E. coli is known as the SOS response [9,11]. Under normal cellular conditions (i.e., non-stress conditions), a repressor protein inhibits the expression of the Y-family polymerase genes, an idea first proposed by Witkin in 1967 [12]. This mechanism has since been clarified; namely, the repressor protein LexA was identified and shown to bind to operator sites, repress gene expression, and to become inactivated upon DNA damage [11]. When DNA damage is present and replicative polymerases are inhibited, a region of single-stranded DNA (ssDNA) is formed. RecA protein binds to and polymerizes on the newly formed ssDNA, forming a nucleoprotein filament, which is stabilized by the presence of ATP. The LexA repressor protein then binds to the RecA/ssDNA nucleoprotein complex, which induces the autocatalytic cleavage of LexA at the Ala84-Gly85 bond, thereby upregulating the Y-family polymerase genes and others [13,14,15].
Y-family polymerases from E. coli and other species of bacteria will be the focus of this review. However, Y-family polymerases are also found in all domains of life, such as archaeal Sulfolobus solfataricus Dpo4 [16], Saccharomyces cerevisiae Pol η and Rev1 [8], and Homo sapiens Pol η, Pol ι, Pol κ, and Rev1 [8,17]. Y-family polymerases share common characteristic structural features such as the palm, finger, and thumb domains [18,19,20,21,22]. Also characteristic of Y-family polymerases is the presence of the little finger domain [23]. The overall size of the finger and thumb domains of Y-family polymerases is smaller than those of their replicative counterparts, which results in an open, solvent‑accessible DNA-binding region to allow for large, bulky lesions to enter the active site [8,24]. In addition, the Y-family polymerases lack intrinsic 3' to 5' exonucleolytic proofreading and lack the characteristic α-helix, known as the ‘O-helix’ in E. coli Pol I, which is used in replicative polymerases to improve their fidelity. The lack of this α-helix presumably contributes to the ability of Y-family polymerases to accommodate damaged DNA templates and to their lower fidelity on undamaged DNA [23,25,26,27].
2. Translesion Synthesis in E. coli
2.1. UmuD
Prior to discussing E. coli Y-family polymerases DinB (Pol IV) and UmuD'2C (Pol V), the functions of the E. coli umuD gene products will be briefly described, as the umuD gene products play critical roles in regulating the activities of both DinB and UmuC. UmuD is the product of one of the genes whose expression is coordinately upregulated along with dinB and umuC [13,28,29]. The umuDC genes are organized in an operon; however, the levels of UmuD appear to correlate more closely with those of DinB than UmuC [30]. Notably, UmuD is not found in all species that have UmuC present, indicating that UmuD is not generally required to regulate UmuC or DinB, although other proteins may fulfill this function in organisms that lack UmuD [8]. UmuD2 is the predominant form of the protein for the first 20 to 40 minutes after induction by the SOS response [31]. UmuD, in conjunction with UmuC, acts in a DNA damage checkpoint [32]. When E. coli cells are grown at 30 °C and UmuD and UmuC are present at elevated levels, they inhibit DNA replication in a role distinct from their function in TLS [31,33]. They also inhibit the replication process after the cell has been exposed to UV light [31]. When UmuD2 interacts with the RecA/ssDNA nucleoprotein filament, the filament facilitates UmuD autocatalytic cleavage, thereby removing the 24 N-terminal amino acids of UmuD to form UmuD' [34,35,36]. UmuD cleavage is similar to the autocatalytic cleavage of LexA, also facilitated by the RecA/ssDNA nucleoprotein filament [37,38,39]. However, the catalytic efficiency of cleavage is much greater for LexA than it is for UmuD2[34]. UmuD2 cleavage typically occurs about 20 to 40 minutes after the initiation of the SOS response [11,31]. The cleaved form of UmuD2, UmuD'2, then interacts with UmuC to form UmuD'2C (Pol V), which is capable of performing TLS [11,40,41,42]. UmuD' and UmuC prevent RecA-dependent homologous recombination as a result of the interaction between UmuD'2C and the RecA/ssDNA nucleoprotein filament [43,44,45]. Full-length UmuD2 is involved in prevention of mutagenesis by UmuC or DinB, whereas UmuD'2 is involved in facilitation of mutagenesis via Pol V; thus, cleavage of UmuD represents a switch from a non‑mutagenic state to a mutagenic state of a cell [46].
2.2. DinB (DNA Pol IV)
DinB, initially identified as the product of the dinP gene [47], was discovered in 1980 as one of the DNA damage-inducible genes and was named dinB [48]; both names are used in the literature. The dinB gene encodes one of the two E. coli Y-family DNA polymerases (DinB, or Pol IV) capable of bypassing lesions in DNA via translesion synthesis [49]. DinB is the only Y-family DNA polymerase that is conserved throughout all domains of life, although S. cerevisiae apparently lacks a DinB ortholog [8,50]. In non-SOS conditions, DinB is expressed at approximately 250 molecules per cell; however, the number of DinB molecules increases by approximately 10-fold under SOS-induced conditions [51]. During the SOS response, DinB is expressed at the highest level of all five DNA polymerases in E. coli [51]. Like other Y-family polymerases, DinB is a DNA-dependent DNA polymerase that is capable of copying damaged primer-template structures and lacks 3'-5' exonuclease proofreading abilities [49]. The fidelity of the dinB gene product is lower than that of the replicative polymerase in E. coli, namely the Pol III holoenzyme [52]. The presence of DinB during TLS can increase mutagenesis as a result of its relatively low fidelity and the lack of 3'-5' exonuclease activity. [49,51,53,54,55,56].
In addition to being upregulated by the SOS response, DinB is involved in a process known as adaptive mutagenesis [57,58]. In an experiment using Lac reporter strains of E. coli, there was a seven‑fold decrease in mutations in strains lacking the dinB gene compared to in the wild-type strain, suggesting that DinB can induce mutations [57]. These mutations, which result from the adaptive mutagenesis process, can cause cells to have a selective advantage during stressful conditions [59]. The precise mechanism for adaptive mutagenesis and how it leads to high levels of DinB expression is not fully understood [60,61,62], although one mechanism may be through DinB involvement in error‑prone double-strand break repair [63]. Overall, DinB expression can be considered a general stress response mechanism that can lead to high rates of mutagenesis and could ultimately result in antibiotic resistance, as described below [57,64,65,66]. Moreover, DinB contributes to cellular fitness and long‑term survival in stationary phase [67]. DinB also has a non-catalytic function, in that elevated levels of DinB slow the replication fork in a checkpoint-like phenomenon [68,69,70].
Currently, no crystal structure of DinB has been solved; however, homology models [71,72,73] have been constructed using the crystal structures of Dpo4 from Sulfolobus solfataricus [23] and Dbh from Sulfolobus acidocaldaricus [16] as templates. These models allow for the prediction of specific residues involved in DinB function. For example, the steric gate residue, which is the amino acid residue of a DNA polymerase that prevents ribonucleotides from entering the active site [74,75], of DinB is F13 [73]. Changing the steric gate residue of DinB increases the frequency of ribonucleotide incorporation from less than 10−5 to 10−3, as well as increases the ability of DinB to replicate undamaged DNA [73].
DinB is known to bypass certain dG adducts (Table 1). For example, DinB bypasses N2-furfuryl dG [73], N2-benzo[a]pyrene-dG [72,76], N2-(-1-carboxyethyl)-dG [77], N2-N2-dG interstrand crosslinks [78], and γ-hydroxypropano-dG [79]. DinB is effective in bypass of N2-dG adducts formed from benzo[a]pyrene (B[a]P), a bulky polycyclic carcinogen which is metabolically activated to form 7R,8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene [72,76]. In the presence of DinB, N2-B[a]P-dG adducts are bypassed with relatively high fidelity and efficiency with a misincorporation frequency of 10−2 to 10−4 [76]. In addition, DinB has been shown to accurately and efficiently bypass N2-(1-carboxyethyl)-2'-deoxyguanosine (N2-CEdG) adducts [77]. N2-CEdG minor groove adducts are formed endogenously from methylglyoxal, which is a byproduct of glycolysis [80,81], and are detected at one lesion per 107 nucleotides in human melanoma cells [80].
An E. coli strain containing a deletion of the dinB gene is sensitive to both nitrofurazone (NFZ) and 4-nitroquinoline-1-oxide (4NQO) [73]. Both of these DNA-damaging agents are thought to form N2‑dG adducts in vivo [82,83]. DinB shows greater accuracy and 15-fold increased proficiency of dCTP insertion opposite N2-furfuryl-dG, an N2-dG adduct likely formed from NFZ, than opposite undamaged DNA [73]. DinB has also been shown to accurately bypass N2-N2-dG interstrand crosslinks (ICLs), which can disrupt DNA replication [78]. Interstrand crosslinks are typically repaired by cooperation between homologous recombination and nucleotide excision repair [84], but recent work has shown that TLS by DinB may also play a role in repair of ICLs [78]. The γ-hydroxypropano-dG adduct, as well as other adducts formed from α,β-unsaturated aldehydes, can form DNA-peptide crosslinks [79]. DinB has been shown to bypass these acrolein-mediated adducts as well as the interstrand crosslinks and the peptide conjugates that form from these adducts [79,85].

Table 1.
Comparison of adducts bypassed in vitro and in vivo by DinB or UmuD'2C. Relevant references and abbreviations are listed in the text.
| DNA polymerase | in vivo | in vitro |
|---|---|---|
| DinB (Pol IV) | N2-furfuryl-dG (the presumed adduct formed from nitrofurazone); N2-B[a]P-dG; N2-CE-dG; ICLs; adducts formed from reactive oxygen species | N2-furfuryl-dG; N2-B[a]P-dG; N2-CE-dG; ICLs; 8oxodG, 2oxodA, 5-fodU, hmdU (adducts formed from reactive oxygen species); abasic sites |
| UmuD'2C (Pol V) | Abasic site; T-T CPD; T-T (6-4) photoproduct; C8-AAF; adducts formed from oxidized dG | Abasic sites; T-T CPD; T-T (6-4) photoproduct; C8-AAF; N2-B[a]P-dG; N6-B[a]P-dA |
DinB confers resistance to the alkylating agent methyl methanesulfonate (MMS) [86]. A cluster of DinB residues referred to as the ‘aromatic triad’, F12, F13, and Y79, is important for survival of E. coli cells in the presence of MMS [87]. E. coli strains that contain single-point mutations in the ‘aromatic triad’ residues within DinB show fewer MMS-induced mutations than nitrofurazone-induced mutations, which suggests that these residues not only play a significant role in TLS, but also are involved in modulating the accuracy of DinB in bypassing specific lesions [87].
DinB is also involved in bypass of lesions that result from various reactive oxygen species leading to A:T → G:C transitions [88]. In studies with defined lesions, DinB was shown to preferentially insert dATP opposite 5-formyluracil (5-fodU) and 5-hydroxymethyluracil (5-hmdU); both dCTP and dATP opposite 7,8-dihydro-8-oxoguanine (8-oxo-dG) with low efficiency; and both dCTP and dTTP opposite 1,2-dihydro-2-oxoadenine (2-oxo-dA) with dCTP inserted more efficiently [88]. In addition, DinB was found to incorporate 8-hydroxy-dGTP opposite both adenine and cytosine and 2-hydroxy-dATP opposite both guanine and thymine in vitro [89,90]. Oxidation of dGTP to 8-oxo-dGTP is the cause of cell death that results from treatment with antibiotics and from elevated levels of DinB, due to increased incorporation of 8-oxo-dGTP by DinB [89]. The evidence supports a model in which cytotoxicity results from double strand breaks caused by incomplete repair of 8-oxo-dG lesions that are closely spaced [89].
It has been further shown that DinB is capable of adding dGTP opposite the modified pyrimidine 1,3-diaza-2-oxophenothiazine (tC) specifically but is incapable of continuing TLS beyond the modified base [91]. This is intriguing since DinB binds slightly more strongly to DNA primer/template constructs that contain the tC analog than it binds to an undamaged DNA primer/template, which may indicate a specific inability to bypass major-groove modified bases in DNA [91]. However, it was also found that DinB inserts tC opposite G in the DNA template and is capable of extending from the newly-incorporated tC, suggesting that DinB shows asymmetric discrimination against the modified DNA template and the incoming nucleotide [91].
The error frequency of DinB on undamaged DNA is approximately 2.1 × 10−4 for generating frameshift mutations and about 5.1 × 10−5 for generating base substitution mutations [52]. DinB is also known to bypass abasic sites, causing −1 frameshift mutations [52,92]. One model for this is a ‘dNTP‑stabilized’ misalignment mechanism in which dNTP is placed correctly opposite a template base downstream rather than placing an incorrect nucleotide opposite the next available template base [52]. More recently however, evidence for another mechanism involving template slippage has been observed, which provides another possible explanation for the generation of −1 frameshift mutations [93]. The template slippage mechanism causes single base deletions on DNA containing homopolymeric runs [93]. When UmuD2 is bound to DinB, a non-slipped conformation is preferred which prevents the generation of frameshift mutations [93].
It is known that the following proteins interact directly with DinB and affect its replication efficiency: UmuD2, RecA, NusA, Rep helicase, single-stranded DNA binding protein (SSB), and the β-processivity clamp subunit [30,94,95,96,97,98]. The presence of UmuD and RecA improves the catalytic efficiency of DinB and also reduces the number of −1 frameshift mutations generated by DinB in vitro [30]. Deletion of the umuD gene leads to an increase in the frequency of −1 frameshift mutations; however, the resistance of such cells to nitrofurazone was not affected, suggesting that the ability of DinB to perform TLS and the mechanism for generating −1 frameshift mutations are separable functions [30].
The C-terminus of DinB (residues 347–351) binds to the β-clamp subunit of the Pol III holoenzyme; this interaction contributes substantially to both enzyme processivity in TLS and dNTP-binding affinity [94,99,100,101]. A second binding site has also been observed between residues 303–305 of the DinB little finger domain and residues located near the dimer interface of the β-clamp subunit [100]. Binding of DinB to the β-clamp subunit increases the processivity of the polymerase, helps to position DinB correctly at the replication fork, and coordinates polymerase usage [100,102,103,104,105,106,107].
NusA, which has roles in elongation, termination, and anti-termination of transcription [108,109,110], has also been shown to interact with DinB. NusA recruits DinB to gaps in the DNA template strand during transcription-coupled TLS when RNA polymerase is stalled by a lesion in DNA [95,96]. NusA has been shown to be necessary for stress-induced mutagenesis by DinB [111]. The exact binding site of DinB on NusA is unknown, but it is believed to be located in the C-terminal domain of NusA [95,112]. The NusA-DinB interaction is predicted to bridge the gap between replication-coupled TLS and transcription-coupled TLS [96]; therefore, this work demonstrates a crucial connection between replication and transcription, especially in the presence of DNA damage.
2.3. UmuD'2C (DNA Pol V)
E. coli Pol V is the second of the two Y-family polymerases found in E. coli. Pol V consists of two different protein subunits, one UmuD'2 dimer and UmuC, which interact to form UmuD'2C [42,113,114,115]. The umuC gene was discovered in the late 1970s to be required for E. coli cells exposed to UV light to mutate [114]. The ability of UmuC to bypass UV-induced DNA adducts via TLS was not discovered until well after UmuC was characterized as being involved in SOS mutagenesis. Several models were proposed over the course of the next few decades to explain the role of UmuC in mutagenesis [116,117,118,119]; however in the late 1990s, it was determined that UmuC was in fact a DNA polymerase [40,41,42,120]. This was confirmed when it was shown that UmuC exhibited low fidelity lesion bypass on its own, but its fidelity and efficiency increased in the presence of RecA, SSB, and UmuD' [40,120,121]. In a key finding, UmuC maintained its ability to function even in the absence of the Pol III holoenzyme [40,120].
Similar to the SOS regulation of dinB expression, the genes encoding the protein constituents of Pol V, umuC and umuD, are both regulated by the SOS response but the umuD and umuC genes are located within the same operon [28,29,122]. Also similar to DinB, upon induction of the SOS response, the expression of the Umu proteins increases 10-fold, with UmuC increasing from approximately 15 to 200 molecules and UmuD increasing from approximately 180 to 2,400 molecules [123]. DNA repair processes such as nucleotide excision repair typically remove a lesion once it has formed in DNA [124,125]; however, in the event that nucleotide excision repair does not take place, replication will recover upon induction of Pol V [125]. It has been shown that when the umuC gene is deleted, there is moderate DNA synthesis recovery and when the recJ gene is deleted, there is poor recovery of DNA synthesis after damage [126]. When both the umuC and recJ genes are deleted, there is no recovery of DNA synthesis after damage, indicating that recJ and umuC both act to restore stalled replication forks [126].
The ability of Pol V to perform TLS is dependent on the formation of the UmuD'2C complex and the presence of RecA [40,127]. Full-length UmuD is involved in preventing UmuC from engaging in TLS and therefore preventing mutagenesis by UmuC. Changing the active site Ser60 residue to Ala in full-length UmuD prevents autocatalytic cleavage of UmuD2[35]. Full-length UmuD is involved in prevention of mutagenesis; thus it was found that UmuD2 harboring the S60A mutation results in a large reduction of UV-induced mutagenesis [31,128,129]. Cells that contain UmuD2-S60A with UmuC experience greater sensitivity to UV light relative to cells that contain wild-type UmuD and UmuC [35,128,129]. Molecular interactions between UmuC and UmuD have been difficult to determine; however, through immunoprecipitation, glycerol gradient analysis, and yeast two hybrid assays, the physical interaction of UmuD' and UmuC was confirmed [115,130]. The interaction of full-length UmuD and UmuC was also confirmed using affinity chromatography and velocity sedimentation analysis [115]. The physical interaction between UmuD' and UmuC consists of one UmuC protein bound to a dimeric UmuD' [115], with UmuD' interacting with the 25-amino acid C‑terminal end of UmuC [32,115].
To date, there is no experimentally-determined structure of UmuC. Homology models have been constructed of the polymerase and little finger domains [24,128,131], but as the C-terminal domain possesses little homology to proteins of known structure, a model of the entire protein cannot be constructed at this time. Still, the model has allowed specific predictions of the functions of particular residues to be tested. In particular, the steric gate residue (Y11) [131,132,133], hydrophobic residues (I38, A39) near the nascent base pair [134], and a cluster of residues (N32, N33, D34) near the incoming nucleotides have all been shown to contribute to UmuC function [135].
In general, Pol V is capable of bypassing lesions formed in DNA that DinB is incapable of bypassing. Pol V can bypass lesions caused by exposure to UV light such as thymine-thymine (T-T) cis-syn cyclobutane pyrimidine dimers (CPD) and T-T (6-4) photoproducts (Table 1) [101,105,106,136]. In addition to bypassing lesions from UV light, Pol V is capable of bypassing abasic sites, C8-dG lesions such as N-2-acetylaminofluorine (C8-AAF), as well as being required for replicating DNA containing 5'S-8,5'-cyclo-2'dG [92,101,105,106,136,137]. Pol V bypasses lesions caused by UV light with greater efficiency when in the presence of the β-clamp subunit, the RecA/ssDNA nucleoprotein filament, and SSB [136]. Pol V can be mutagenic when carrying out TLS. For example, Pol V inserts dGTP opposite the 3'T in T-T (6-4) photoproducts instead of dATP with a six-fold greater frequency [136]. However, Pol V is also known to bypass certain lesions with high accuracy. For instance, Pol V faithfully inserts dATP opposite both Ts of T-T CPDs [136] as well as dCTP opposite C8-AAF-dG [106]. Pol V also bypasses N2-benzo[a]pyrene-dG adducts, N6-benzo[a]pyrene-dA adducts and adducts resulting from oxidation with varying accuracy [72,76,138]. Pol V replicates undamaged DNA with error frequencies of 10−3 to 10−4, which is a much lower fidelity than the Pol III holoenzyme and a lower fidelity than DinB [136]. It should be noted that Y-family DNA polymerases can be accurate or error-prone depending on the cellular and DNA context in which they are acting.
In addition to facilitating the autocatalytic cleavage of the LexA repressor protein as well as facilitating the cleavage of full-length UmuD2 to UmuD'2, RecA/ssDNA nucleoprotein filaments also play a direct role in TLS [40,41,139,140,141]. RecA has been determined to stimulate both nucleotide insertion and extension [141]. One model suggests that Pol V and two RecA molecules form a complex that activates Pol V for TLS in the presence of ATP [127,142]. There is evidence that a distinct RecA/ssDNA nucleoprotein filament transfers RecA and an ATP from the 3' end of this trans filament to Pol V, which activates Pol V for TLS [143,144]. However, other work suggests that the RecA/ssDNA nucleoprotein filament acts in cis on DNA directly downstream of pol V rather than in trans to facilitate the activation of Pol V and TLS [107]. These differences are likely the result of differences in experimental details. The complex of activated RecA with UmuD'2C is termed the Pol V mutasome [127].
In addition to RecA, the β-processivity clamp and the γ clamp loader increase the processivity of Pol V by allowing the enzyme to remain bound to the DNA and by providing additional stability [145]. The β clamp increases processivity approximately three-fold to five-fold in one study [146] and about 100-fold in another study [107]. Clearly, the β clamp stimulates processivity of Pol V, but the extent of the increase in processivity depends on the specifics of the experimental system [19].
2.4. E. coli Translesion Polymerases in Antibiotic Resistance
As both DinB and Pol V can be mutagenic depending on the cellular context and the nature of DNA lesions encountered, it was speculated that they could contribute to antibiotic resistance [64,65,66]. Indeed, in E. coli cells grown under stress conditions, DinB and to a lesser extent Pol V are responsible for base substitution mutations in the ampD gene that result in resistance to the β-lactam antibiotic ampicillin [147]. Moreover, DNA Pols II, IV, and V all contribute to E. coli resistance to the antibiotic ciprofloxacin, as well [148]. While the contribution of each polymerase was examined in bacterial cultures, in a mouse model of infection with pathogenic E. coli, LexA cleavage was required for resistance to ciprofloxacin or rifampicin [148]. Thus, SOS-regulated DNA polymerases and possibly other genes under LexA control contribute to the evolution of antibiotic resistance in bacteria both when they are grown in the free-living state and during infection within a mammalian host.
2.5. E. coli Translesion Polymerases in Pathogenesis
A recent study has shown that DinB of uropathogenic E. coli (UPEC) is required for virulence of UPEC strains in bladder infections in mice [149]. Deletion of dinB in all UPEC isolates tested results in a reduced ability to colonize host bladders. No reduction in virulence of the dinB-deletion mutant is observed in mice that have a reduced Toll-Like Receptor-4 (TLR4)-dependent inflammatory response, indicating that DinB is important in helping UPEC cope with the stresses produced by host inflammation. In contrast, deletion of umuDC does not reduce the virulence of these UPEC strains. Surprisingly, cells of the dinB-deletion mutant recovered from the host have a mutation frequency similar to that of the wild-type parent. This is in contrast to the phenotype observed in culture, in which the dinB-deletion mutant of the UPEC strain UTI89 has a reduced rate of spontaneous mutation when grown in either rich medium or in human urine. This study demonstrated a clear role for DinB in UPEC pathogenesis and virulence. However, DinB does not appear to influence the acquisition of mutations by UPEC in the host environment.
3. DinB and UmuC Orthologs in Other Bacteria
Although the Y-family polymerases of E. coli have been well studied, Y-family polymerases are present throughout all domains of life [8]. The following will discuss recent developments in the study of Y-family polymerases in other species of eubacteria. The close relative of E. coli, Salmonella typhimurium, possesses homologs of DinB, UmuD, UmuC, and a second pair of UmuD and UmuC homologs, known as SamA and SamB [150]. A S. typhimurium strain lacking DinB and both Pol V homologs had a sharp reduction in the frequency of spontaneous deletion formation [150]. Conversely, a strain overproducing Pol IV (but not the Pol V homologs) has an increased frequency of spontaneous deletions [150]. Acinetobacter species have a range of configurations of umuD, umuC, and dinB genes [151]. For example, Acinetobacter baylyi possesses a umuD gene with a 5' extension but only fragments of umuC [152], whereas Acinetobacter ursingii harbors the extended version of umuD in an operon with dinB [151]. Moreover, despite the presence of umuD-, umuC-, and dinB-like genes in Acinetobacter species, UV-induced mutagenesis was observed in only a few of the species tested, including in the opportunistic pathogens Acinetobacter baumanii and Acinetobacter ursingii [151].
Y-family polymerases in the bacterium Bacillus subtilis have been demonstrated to be involved in mutagenesis [153]. The Y-family polymerases encoded in the B. subtilis genome are UvrX, YqjH, and YqjW, which have significant homology to the E. coli Y-family polymerases DinB and UmuC [8]. The B. subtilis genome sequence data indicates that UvrX is encoded in the prophage known as SPβ [154]. UvrX has 25% sequence identity to E. coli DinB and is involved in repair of UV damage [154]. The constitutive YqjH protein has 36% sequence identity to E. coli DinB and the SOS-inducible YqjW protein has 26% sequence identity to E. coli UmuC [155]. Inactivation of the yqjH and yqjW genes results in increased UV sensitivity and decreases the frequency of UV-induced mutagenesis [156]. The lack of a UmuD homolog in B. subtilis could indicate that another protein is fulfilling its function of regulating mutagenesis. Recently, it has been found that YqjH and YqjW are involved in protecting sporulating cells of B. subtilis [157]. Deletion of yqjH and yqjW genes decreases sporulation efficiency as well as increases sensitivity to chemical mutagens such as hydrogen peroxide, tert‑butylhydroperoxide, mitomycin C (MMC), and UV-C radiation [157]. It was concluded that YqjH and YqjW proteins are involved in TLS in sporulating B. subtilis cells and cause spontaneous mutations [157].
Mycobacterium tuberculosis contains two Y-family DNA polymerases, both of which are homologous to E. coli DinB. They are identified as DinB1 (or DinX), which is encoded by the gene Rv1537, and DinB2 (or DinP), which is encoded by the gene Rv3056 [158]. These proteins possess sequence similarity to their homologs in E. coli [49] as well as to those in Pseudomonas aeruginosa [159], leading to the presumption that DinB1 and DinB2 both have DNA polymerase activity. Unlike Y-family polymerases from E. coli and most other eubacteria, expression of DinB1 and DinB2 does not depend on the RecA protein, the SOS response, or even the existence of damaged DNA [160,161,162,163]. In contrast, DinB1 and DinB2 are regulated by separate mechanisms whereby DinB1 is expressed in pulmonary tuberculosis [164] and DinB2 is expressed upon exposure to novobiocin [160]. While this work determined that the DinB homologs in M. tuberculosis are not induced upon DNA damage as in other organisms, the C-family DNA polymerase DnaE2 was induced by the presence of DNA damage (see also Sections 4 and Sections 7.1) [165]. The C-family DNA polymerases were previously considered high‑fidelity, replicative DNA polymerases in bacteria; however, the C family includes a subfamily of polymerases, including DnaE2, the members of which are capable of carrying out TLS [166]. DnaE2, rather than the DinB homologs, was therefore predicted to play the primary role in adaptive mutagenesis in M. tuberculosis [165].
The bacterium Mycobacterium smegmatis also contains homologs of E. coli Y-family polymerases. It was found that the genome of M. smegmatis contains three DinB homologs encoded by the genes msmeg_1014, msmeg_3172, and msmeg_6443 according to the KEGG PATHWAY Database [167]. Interestingly, the key residues necessary for functional polymerase activity are conserved in msmeg_1014 (also known as MsDpo4) [168]. MsDpo4 is capable of performing template-dependent nucleotide insertion and can promote mismatches on undamaged DNA templates [168]. In addition, MsDpo4 has been shown to preferentially promote G:T and T:G mismatches, indicating that it has the ability to increase the frequency of untargeted mutations [168].
Y-family DNA polymerase homologs are also present in species of the bacterial genus Pseudomonas [8]. Pseudomonas aeruginosa contains a homolog of E. coli DinB (PaDinB), which is lacking in intrinsic proofreading capabilities [159]. PaDinB promotes C to A transversions as well as induces −1 frameshift mutations [159]. Strains lacking the dinB gene are sensitive to the DNA‑damaging agents nitrofurazone (NFZ) and 4-nitroquinoline oxide (4NQO) showing that PaDinB most likely plays a role in TLS similar to that of E. coli DinB [159]. On the other hand, PaDinB accurately copies TT CPDs but contributes to H2O2-induced mutagenesis [169]. A DinB homolog was also found in Pseudomonas putida and was shown to be involved in 1-base pair (bp) deletions in starving cells, yet was also reported to be expressed in a RecA-independent process [170]. P. putida also possesses a plasmid‑borne homolog of Pol V that confers resistance to DNA damage, increases fitness, and whose expression is regulated in a RecA-dependent manner [171].
4. The Function of dnaE and Discovery of a Second dnaE Gene
In most bacteria the main replicative polymerase is the C-family DNA polymerase DnaE (α subunit) or PolC [172,173], which is the polymerase subunit of the DNA Pol III holoenzyme, a complex of 10 different subunits [166]. In some organisms such as E. coli, there are two or three copies of DnaE in the Pol III holoenzyme, which is encoded by a single gene, dnaE [166,174,175]. In other organisms such as B. subtilis [176], one α subunit is encoded by dnaE and another α subunit is encoded by polC, each of which has a distinct role corresponding to DNA synthesis on the leading and lagging strands in the replication process [166]. Notably, B. subtilis DnaE is SOS-inducible and is capable of TLS [177]; similarly Streptococcus pyogenes DnaE is error-prone and can carry out TLS [178].
As early as 1995, it was noted that various Mycobacterium and Mycoplasma species contained an extra dnaE gene [158,179,180,181], which due to the lack of an identifiable 3'-5' exonuclease domain were characterized as another α subunit gene. This extra dnaE gene resulted in the primary, replicative gene to now be designated dnaE1 (for example, Rv1547c in Mycobacterium tuberculosis) and the extra copy designated as dnaE2 (Rv3370c in M. tuberculosis).
In some cyanobacteria the dnaE gene products are DnaE1 and DnaE2, which are split by inteins and combine to form the intact PolC [182], compounding the number of different “dnaE2”s in the literature. In order to deal with the proliferation of dnaE relatives, a new system of nomenclature has been proposed [166]. The dnaE2 gene, referring to a homolog of E. coli dnaE that is not essential for replication, is often found following or accompanied by the two genes imuA and imuB, and thus dnaE2 is now referred to as imuC (Table 2)[166].
Table 2.
Summary of DNA polymerases and their accessory factors. For each protein, a species that contains the most studied or most representative protein is listed. Especially for newly described mutagenesis cassettes, the roles of these proteins are still uncertain or incomplete.
| Protein | Role | Representative species |
|---|---|---|
| DinB | Bypass of N2-dG adducts, −1 frameshift mutagenesis | E. coli |
| UmuD'2C | Bypass of UV-induced lesions, induced mutagenesis | E. coli |
| SamAB | Plasmid-borne UmuDC homologs | S. typhimurium |
| UvrX | UV damage repair, sporulation | B. subtilis |
| YqjH | UV damage repair, sporulation | B. subtilis |
| YqjW | UV damage repair, sporulation | B. subtilis |
| ImuC | Induced mutagenesis from UV/MMC | M. tuberculosis |
| ImuB | Binds processivity factor; role in polymerase switching? | M. tuberculosis |
| ImuY | Same pathway as D. deserti ImuC, analogous to ImuB? | D. deserti |
| ImuA | Mostly unknown, found in species with ImuB/ImuC | C. crescentus |
| ImuA' | Mostly unknown, interacts with ImuB | M. tuberculosis |
5. Discovery of Associated Genes imuA, imuA', imuB, imuY
In Pseudomonas putida, reverse transcriptase PCR (RT-PCR) showed multiple genes in a cassette annotated as lexA2, sulA, dinP (by analogy to E. coli dinP or dinB), and dnaE are expressed under the direct control of the lexA2 gene as a single transcription unit [183]. Phylogenetic analysis showed a widespread occurrence of this mutagenic cassette. The cassette is unlikely to have been acquired recently by these genomes, given the similar GC content of each cassette with that of its genomic environment [183]. The sulA, dinP, and dnaE genes were all determined to be involved in DNA replication and mutagenesis [183]. Subsequently, the original annotations of sulA, dinP, and dnaE were changed to imuA, imuB, and imuC, respectively, to reflect the re-classification of these genes as encoding novel proteins [183,184,185]. The names of imuA and imuB are derived from “inducible mutagenesis” [186]. As the third gene in the cassette, dnaE2 was proposed to be renamed imuC as the logical extension of the names of the genes in the operon [166,187].
While the ImuB amino acid sequence is closely related to those of Y-family polymerases, the amino acids that correspond to catalytic aspartic acids in most other Y-family polymerases are missing, and thus this protein is thought to be biochemically inactive in translesion synthesis [187,188]. ImuA and its homolog in M. tuberculosis, ImuA', share some sequence similarity with LexA, RecA, and SulA, but little is known about the function of the ImuA' proteins. Bdellovibrio bacteriovorus imuA is not able to complement an E. coli recA− mutant [189]. In some organisms, prominently in the Actinomycetales, such as Mycobacterium tuberculosis, the imuA candidate gene is so dissimilar from the proteobacterial imuA that the M. tuberculosis gene has been named imuA' to mark its notable difference [190]. Similarly, imuY in Deinoccocus deserti [191,192] lacks similarity to known imuB genes, and since it was implicated in translesion synthesis with a Y-family polymerase-like sequence, was termed imuY [193].
6. imuABC Operon Regulation and Organization
Expression of the imuA, imuB, and imuC genes is almost exclusively controlled by LexA-SOS systems. Many of the genes in the imuABC family were discovered during searches for SOS-box-containing LexA binding motifs. The imuABC genes were found following the SOS boxes, closely linking the discovery of LexA binding motifs and imuABC genes. Elucidating the evolutionary history of LexA binding motifs can be difficult due to the short sequence of the SOS box [184]. With the increasing number of imuABC gene sequences now known, the identification of putative imuABC genes can be utilized to augment phylogenetic analysis of the LexA binding motifs [190]. In organisms with a recognizable SOS box motif, imuABC open reading frame(s) provide an opportunity to track the SOS-box with much higher precision than the SOS box alone.
In P. putida, there are two different LexA regulons controlled by LexA1 and LexA2 [184]. It was shown that LexA2 directly controls the lexA2-imuA-imuB-imuC operon as a single transcriptional unit when induced by MMC in P. putida [183]. There seems to be evolutionary pressure to include LexA regulation of the imuABC mutagenic cassette once an organism has acquired at least imuB and imuC [190].
The imuABC genes are not found in cyanobacteria or gram-positive bacteria. A complete mutagenesis cassette is often found in the form of a single operon imuA-imuB-imuC such as in many Alphaproteobacteria (Figure 1). For some Alphaproteobacteria such as Sinorhizobium meliloti and Agrobacterium tumefaciens, not only are their cassettes a single uninterrupted operon, cassettes can be found on both the chromosome as well as on plasmids [190]. In Ralstonia solanacearum,the cassette exists only on its plasmid [183,194].
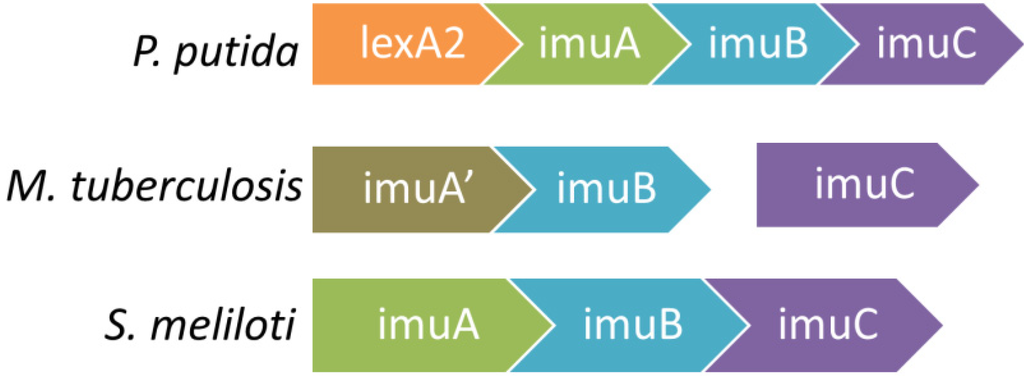
Figure 1.
Some characteristic configurations of the imuABC mutagenesis cassette are shown below. The plethora of imuABC operons has been characterized extensively [183,190,195]. While P. putida and S. meliloti have imuA, imuB, and imuC together as one operon, M. tuberculosis contains imuC separated from imuA and imuB, as well as imuA' instead of imuA.
Members of the imuABC cassette can be organized as an uninterrupted imuABC operon, or in other configurations where various members are found in different loci or with members missing. A lone imuC gene is found in some bacteria, such as Kineococcus radiotolerans, Symbiobacterium thermophilum, and Actinomyces naeslundii, while an imuBC cassette is found in Streptomyces coelicolor [190]. M. tuberculosis, which has one of the most thoroughly studied imuABC systems, has a configuration with imuA' and imuB together at one locus and imuC at another locus, both of which are controlled by LexA [190]. Almost all configurations exist in different bacteria, including each of the imuA, imuB, and imuC genes located at a distinct locus each with their own SOS box [190,195].
7. Known Functions of the Mutagenesis Cassette Gene Products
7.1. Mycobacterium
When identifying the SOS boxes of Mycobacterium tuberculosis and the induction levels of the genes proposed to be lexA regulated, Davis et al. found a gene annotated as dnaE2 (i.e., imuC) with a preceding M. tuberculosis LexA-binding SOS box [196]. On average, the Rv3370c (imuC) gene was up-regulated more than 10-fold following induction by MMC [196]. It has been shown that MMC induces imuC, recA, and lexA in strains containing a functional RecA, but that imuA'B and imuC are not induced by MMC in a recA-deletion mutant [162,197]. An imuC null mutant of M. tuberculosis has a reduced virulence relative to that of the wild type and is deficient in UV-induced mutagenesis [165]. This experiment showed that imuC-mediated mutagenesis is the sole source of UV-induced mutagenesis in M. tuberculosis, with a mutational spectrum that resembles that of a signature for translesion synthesis [165]. Strains with imuC reproducibly generated CC to TT mutations, consistent with bypass of a UV damage-induced pyrimidine dimer, whereas in strains without imuC, this mutation was not observed [165]. Overexpression of imuC in non-UV-treated cells does not increase the mutation frequency, suggesting that imuC requires additional subunits to function [165].
In M. tuberculosis, recA controls expression of imuA', imuB, and imuC [188,198]. MMC also induces the SOS response in M. smegmatis. Loss of imuA', imuB, or imuC individually or in combination results in the same level of hypersensitivity to MMC, suggesting that the products of these genes function as part of a single pathway for resistance to MMC [188]. M. tuberculosis imuC has three aspartic acids that correspond to the known active site acidic residues of C-family Pol III polymerase catalytic subunits. The M. smegmatis441DID443 to 441AIA443 mutation, which changes two of the three conserved active site residues, eliminates UV-induced mutagenesis and confers on M. smegmatis hypersensitivity to MMC, mimicking the imuC deletion phenotype [188]. These experiments established strong evidence that imuC is responsible for induced mutagenesis and survival under DNA-damage stress conditions via translesion synthesis [188].
An extensive study by Warner et al. elucidated many of the interactions between the imuA, imuB, and imuC gene products [188]. Whereas ImuC lacks a β-binding motif to interact with the processivity factor at the replication fork, ImuB does contain a β-clamp-binding motif. Only ImuB interacts with DnaE1 or with the β-processivity clamp. ImuA' and ImuC showed interactions with only ImuB and not with each other or with the β clamp [188]. The ImuB-β interaction can be disrupted by mutation of the β-binding motif or by truncation of the C-terminal end of ImuB including the β-binding motif. Truncations up to, but not including, the β-binding motif of ImuB did not disrupt the interactions with ImuA', ImuC, or β [188]. Truncation of the C-terminal 44 residues of ImuB disrupted the ImuA'-ImuB interaction [188]. Thus, each of the three proteins expressed from the imuABC operon interact in a pairwise fashion with ImuB, which leaves open the possibility of ternary complex formation with ImuB occupying a central position in such a ‘mutasome’.
7.2. Deinoccocus
In Deinococcus deserti, which was isolated in the Sahara desert [191,192], the genome contains a mutagenesis cassette in the form of lexA-(imuB-like protein)-imuC, as a single transcriptional unit [193]. This operon has unusual characteristics compared to other imuABC cassettes. The imuB-like gene is different enough from other imuBs that the gene in D. deserti was termed imuY, to recognize its homology to Y-family DNA polymerases, rather than imuB [193]. In addition there is a hypothetical protein of 243 base pairs between imuY and imuC [193]. D. deserti has three recA genes encoding two RecA products: recAC, encoding chromosomal RecAC, and recAP1 and recAP3, which both encode the same plasmid-derived RecAP product [193]. The mutagenesis cassette was induced by RecAC but not by RecAP [193]. Interestingly, while transcriptional regulation of the cassette was dependent on RecAC, recAC mutants did not show a loss in UV or gamma radiation survival [193]. Deletion of imuY, imuC, or both imuY-imuC showed the same 10-fold decrease in UV-induced mutagenesis, and deletion of imuY could be complemented by a plasmid carrying the imuY gene [193]. To explain the lack of decreased survival upon imuY or imuC deletion, Dulermo et al. note that the conditions in the native environment of the Sahara desert starkly differ from the mild conditions in the laboratory; under the combined stress of dessication, starvation, and other environmental conditions, D. deserti may depend more heavily on imuY and imuC for survival [193].
In Deinococcus ficus, a lexA-imuB-imuC gene cassette and a dinB2 gene are carried on an accessory plasmid [199]. Disruption of either imuB or imuC showed equal loss of survival and loss of mutagenesis following UV exposure, suggesting the same pathway of action for both survival and induced mutagenesis [199]. Deinoccocus ficus naturally possesses keratinolytic activity to break down feathers, which could be used in agricultural and industrial applications [199]. Using the inherent mutator properties of ImuC, UV exposure was utilized as a mutagen to create improved keratinolytic activity [199]. Induced mutagenesis by UV light led to at least one mutant strain with a two-fold higher activity [199]. Zeng et al. suggest that increased keratinolytic activity after UV exposure could come from imuBC-dependent induced mutations, but note the possibility of the two dinB genes to contribute to this process [199]. In addition, D. grandis contains a putative imuB showing high similarity to the D. ficus imuB [199].
The two most thoroughly studied Deinoccocus species, D. radiodurans and D. geothermalis, do not contain mutagenic or translesion synthesis polymerases [193]. It has been hypothesized that it is advantageous for Deinoccocus species not to have error-prone translesion synthesis or mutagenic polymerases in order for them to accomplish their striking feats of DNA repair [200]. The discovery of a mutagenic cassette in D. deserti and D. ficus provide an interesting example of how different species within the same genus can develop different survival strategies [193,199].
7.3. Caulobacter
In Caulobacter crescentus, imuABC genes are responsible for UV- and MMC-induced mutagenesis [186] and are strongly repressed by LexA, with increased expression in a lexA mutant strain by 15-fold over that of the wild type [201]. While deletion of a single gene in the mutagenic cassette results in slight sensitivity to UV exposure and abolishes induced mutagenesis, a double imuB-imuC deletion mutant has no further increase in sensitivity or reduction of mutagenesis, strengthening evidence that these genes function in the same pathway in this bacterium [186].
In wild type C. crescentus, UV-induced mutagenesis results in a mixture of G:C to A:T transitions, G:C transversions, A:T to G:C or C:G mutations, and tandem substitutions [186]. Under conditions with either imuB or imuC absent, the mutation spectrum drastically shifts to become dominated by G:C to A:T transitions, with the remainder of mutations being A:T to G:C transitions [186]. The dependence on imuB and imuC for G:C to C:G transversions, A:T to C:G transitions, and tandem substitutions presents a unique mutation spectrum compared to that of E. coli umuDC [56,202,203] or M. tuberculosis imuA'BC [165,186].
7.4. Streptomyces and Streptococcus
In Streptomyces coelicolor, the dinB2 and imuC genes overlap by 4 bp, an organization found in most Streptomyces and some other bacteria such as Sinorhizobium [204]. In S. coelicolor, imuC deletion strains have no defect in end patching of telomeres, conjugal transfer, UV survival, or UV‑induced mutagenesis, even though imuC is induced by UV exposure and MMC. The authors argue that imuC is a rapidly evolving gene and that it may be still developing a new/optimal function [204]. In Streptococcus uberis, a mutagenesis cassette has been reported that is induced by UV light and that induces mutagenesis after UV exposure; the genes composing this cassette seem to be present throughout Streptococcacaea [205].
7.5. Pseudomonas
P. putida contains an imuABC operon [185,206]; it has been shown that during stationary phase mutagenesis imuC reduces the frequency of base substitution mutations, whereas imuB increases base substitution mutations as well as 1-bp deletions [185]. When imuC was deleted, the number of base substitution mutations increased with no change in 1-bp deletions [185]. When imuB was deleted, 1-bp frameshifts were decreased with no change seen in base substitution mutations [185].
Pol I, an A-family DNA polymerase, can act as a translesion synthesis polymerase [155]. In a P. putida Pol I-deficient background, the spontaneous mutation frequency was similar in the presence or absence of imuC [206]. However, after UV exposure, A to T and A to G mutations decreased in an imuC− strain [206]. Frequency of UV-induced mutations increased two-fold in imuC− compared to wild type, but not in a Pol I-deficient background [206]. It was concluded that in an unstressed Pol I deficient background, ImuC does not meaningfully contribute to DNA synthesis, but in the presence of DNA damage ImuC becomes involved in DNA synthesis [206].
In P. aeruginosa in response to ciprofloxacin, imuC and dnaE1 are upregulated two- and six-fold, respectively [207]. Also in this species, imuC has been shown to be responsible for induced mutagenesis [159]. It must be noted that while P. aeruginosa imuC and P. putida imuC share 73% identity, they have phenotypically opposite effects [185], where imuC (also referred to as polC in P. aeruginosa) is an anti-mutator in P. putida [185,206] and a mutator in P. aeruginosa [159].
8. Prevalence and Diversity of imuABC Genes
The imuABC genes have only recently garnered attention compared to their relatives, umuDC and dinB. While the model organism E. coli utilizes the umuDC family for induced mutagenesis and translesion synthesis, it is now becoming clear that this may be the exception rather than the rule in bacteria [184,208]. The UmuD'2C and ImuABC systems are observed to be exclusive; that is, organisms with UmuD and UmuC do not have ImuABC [190]. For example, the shared set of SOS response genes between E. coli and M. tuberculosis are lexA, recA, uvrA, and a set of inducible polymerase genes: imuABC in M. tuberculosis and umuDC in E. coli [184]. The imuABC and umuDC genes seem to fulfill the same role of induced mutagenesis in the SOS response [198,208].
There is considerable diversity in the gene products of this operon (see Figure 2, Figure 3 and Figure 4). For example C. crescentus ImuA and M. tuberculosis ImuA' show very little identity. Additionally, various ImuB proteins, such as S. meliloti and P. putida ImuB (and D. deserti ImuY) have few highly conserved residues [190,193]. Even ImuC variants, which are more highly conserved across different organisms, can cause quite different phenotypes. Here P. aeruginosa and P. putida serve as a prominent example: despite 72% sequence similarity, P. aeruginosa ImuC acts as a mutator and P. putida ImuC acts as an anti-mutator [159,185,206]. Some species, such as S. coelicolor, have an ImuC to which no function has been assigned, although end patching of telomeres, conjugal transfer, UV survival, and UV‑induced mutagenesis have been investigated [204].
Transient mutators have the ability to turn on their mutator activity only under stressed conditions, then to turn off their mutator ability to maintain their fitness level once resistance to a selective pressure has been achieved [10,209]. Maintenance of a high mutation frequency under non-stressed conditions would be deleterious to the survival of the organism. SOS-induced mutagenesis is a transient mutator system and in most organisms this response is carried out by the lexA-imuABC mutagenesis cassette [11,209]. For example, some rifampicin resistant clinical strains of M. tuberculosis have high levels of imuC expression as a consequence of the same mutation that confers antibiotic resistance [210]. Since these strains have lower fitness in the absence of antibiotic selection, Bergval et al. suggest that the reduction in fitness may be due to the inappropriate expression of imuC, which is known to have mutator activity in M. tuberculosis [210]. The imuC-deficient strains of M. tuberculosis are less virulent than wild-type strains, and mice infected with these strains experience lower mortality than those infected with the wild type [165]. Elucidation of how hypermutation is associated with infection [211], but not with antibiotic resistance [212] in P. aeruginosa may shed more light on the molecular mechanisms of imuABC-mediated mutagenesis and its role in bacterial adaptation.

Figure 2.
Percent identities by ClustalW2 [213,214] for the ImuA protein and related proteins. “(p)” represents the plasmid gene product, “(c)” represents the chromosomal gene product. The accessions are from Uniprot [215], from left to right: P0A7C2, P0A154, P0AFZ5, P0A7G6, Q07447, P42443, C1D2C5, C1CXY5, C1D2K8, Q9A3J1, Q6MQS4, Q50730, Q88I84, Q9I5Q0, Q92ZJ8, Q92LA5.
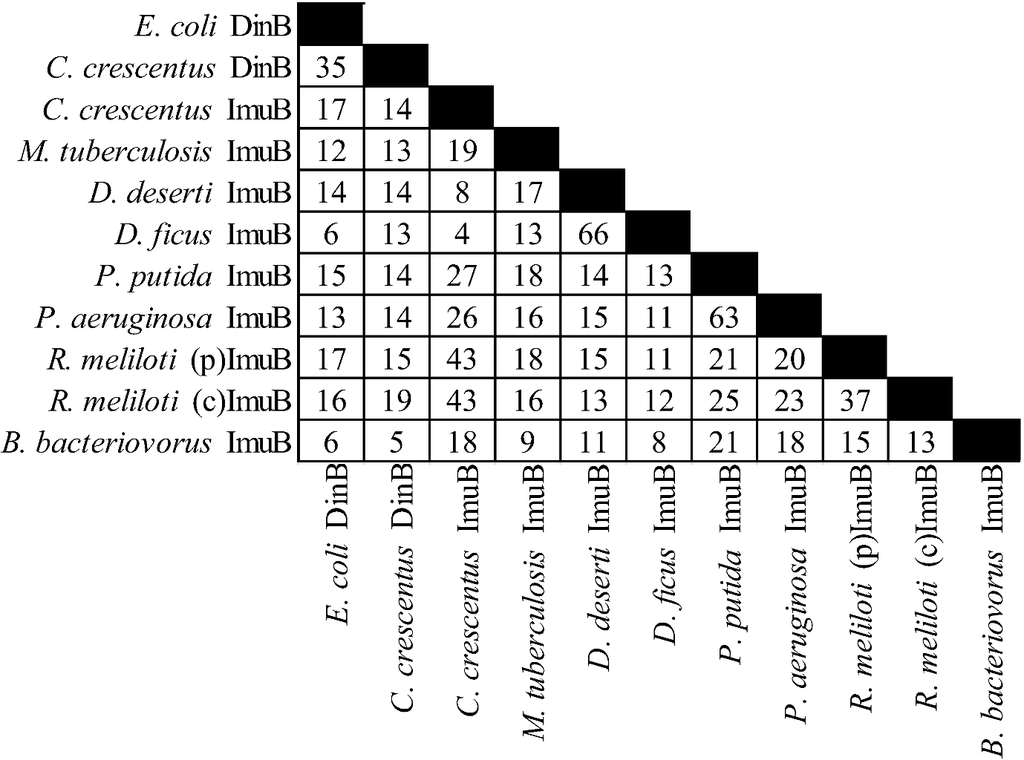
Figure 3.
Percent identities by ClustalW2 [213,214] for ImuB protein and related proteins. “(p)” represents the plasmid gene product, “(c)” represents the chromosomal gene product. The accessions are from Uniprot [215] with the exception of D. ficus ImuB from RefSeq [216], from left to right: Q47155, Q9A5I1, B8H428, O50419, C1D2K9, ADO33718, Q88I83, Q9I5Q1, Q92ZJ7, Q92JS7, Q6MQS5.

Figure 4.
Percent identities by ClustalW2 [213,214] for ImuC proteins and related proteins. “(p)” represents the plasmid gene product, “(c)” represents the chromosomal gene product. The accessions are from Uniprot [215] with the exception of D. ficus ImuC from RefSeq [216], from left to right: P10443, B8GWS6, Q9XDH5, B8H427, Q6MQS6, O50399, C1D2L1, ADO33730, Q9S291, Q88I82, Q9I5Q2, Q92ZJ6, Q92LA6.
9. Questions and Conclusions
E. coli Y-family DNA polymerases are critical in conferring resistance to various DNA damaging agents including UV light and chemical mutagens. The two Y-family polymerases in E. coli are capable of bypassing certain lesions and are also involved in the regulation of DNA replication. Y-family polymerases are important in facilitating mutagenesis, contributing to their involvement in antibiotic resistance. The discovery of the imuABC mutagenesis cassette indicates another strategy for bacterial mutagenesis and translesion synthesis. Indeed, ImuC facilitates induced mutagenesis and DNA damage tolerance, and possibly provides a missing link between replicative C-family polymerases and the mutagenic Y-family polymerases. The wide phylogenetic and phenotypic diversity of the imuABC cassette makes it a prime case study for how mutagenic cassettes appear, evolve, or disappear, and their effects on survival, adaptation, and resistance.
The discovery of the mutagenesis cassettes that include imuA, imuB,and imuC genes (Table 2) raises a number of questions. One key question is whether imuC will demonstrate specificity for certain types of damage, as has been observed for Y-family DNA polymerases. As the number of DNA polymerases and the apparent complexity of DNA damage responses in bacteria continue to increase, a key question is how these polymerases are managed and how access to the replication fork is controlled in response to DNA damage. The possible functions of accessory factors in DNA damage recognition as well as access to the replication fork also remain to be elucidated.
Acknowledgments
This work was supported by the National Science Foundation (Career Award MCB-0845033 to P.J.B), the American Cancer Society (Research Scholar Grant RSG-12-161-01-DMC to P.J.B.), and the NU Office of the Provost. P.J.B. is a Cottrell Scholar of the Research Corporation for Science Advancement.
Conflict of Interest
The authors declare no conflict of interest.
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Higuchi, K.; Katayama, T.; Iwai, S.; Hidaka, M.; Horiuchi, T.; Maki, H. Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells 2003, 8, 437–449. [Google Scholar] [CrossRef]
- Pages, V.; Fuchs, R.P. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 2003, 300, 1300–1303. [Google Scholar] [CrossRef]
- Yeeles, J.T.; Marians, K.J. The Escherichia coli replisome is inherently DNA damage tolerant. Science 2011, 334, 235–238. [Google Scholar]
- Gon, S.; Napolitano, R.; Rocha, W.; Coulon, S.; Fuchs, R.P. Increase in dNTP pool size during the DNA damage response plays a key role in spontaneous and induced-mutagenesis in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 19311–19316. [Google Scholar] [CrossRef]
- Bridges, B.A. Error-prone DNA repair and translesion DNA synthesis. II: The inducible SOS hypothesis. DNA Repair (Amst) 2005, 4, 725–726, 739. [Google Scholar] [CrossRef]
- Bridges, B.A. Error-prone DNA repair and translesion synthesis: Focus on the replication fork. DNA Repair (Amst) 2005, 4, 618–619, 634. [Google Scholar] [CrossRef]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Radman, M. SOS repair hypothesis: Phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 1975, 5A, 355–367. [Google Scholar]
- Nohmi, T. Environmental stress and lesion-bypass DNA polymerases. Annu. Rev. Microbiol. 2006, 60, 231–253. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Witkin, E.M. The radiation sensitivity of Escherichia coli B: A hypothesis relating filament formation and prophage induction. Proc. Natl. Acad. Sci. U. S. A. 1967, 57, 1275–1279. [Google Scholar] [CrossRef]
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64. [Google Scholar]
- Horii, T.; Ogawa, T.; Nakatani, T.; Hase, T.; Matsubara, H.; Ogawa, H. Regulation of SOS functions: Purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein. Cell 1981, 27, 515–522. [Google Scholar] [CrossRef]
- Little, J.W.; Kim, B.; Roland, K.L.; Smith, M.H.; Lin, L.L.; Slilaty, S.N. Cleavage of LexA repressor. Methods Enzymol. 1994, 244, 266–284. [Google Scholar] [CrossRef]
- Silvian, L.F.; Toth, E.A.; Pham, P.; Goodman, M.F.; Ellenberger, T. Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat. Struct. Biol. 2001, 8, 984–989. [Google Scholar] [CrossRef]
- Goodman, M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002, 71, 17–50. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Fischhaber, P.L.; Kisker, C. Error-prone DNA polymerases: Novel structures and the benefits of infidelity. Cell 2001, 107, 9–12. [Google Scholar] [CrossRef]
- Walsh, J.M.; Hawver, L.A.; Beuning, P.J. Escherichia coli Y family DNA polymerases. Front. Biosci. 2012, 17, 3164–3182. [Google Scholar]
- Yang, W.; Woodgate, R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15591–15598. [Google Scholar] [CrossRef]
- Pata, J.D. Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta. 2010, 1804, 1124–1135. [Google Scholar] [CrossRef]
- Washington, M.T.; Carlson, K.D.; Freudenthal, B.D.; Pryor, J.M. Variations on a theme: Eukaryotic Y-family DNA polymerases. Biochim. Biophys. Acta. 2010, 1804, 1113–1123. [Google Scholar] [CrossRef]
- Ling, H.; Boudsocq, F.; Woodgate, R.; Yang, W. Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell 2001, 107, 91–102. [Google Scholar] [CrossRef]
- Chandani, S.; Jacobs, C.; Loechler, E.L. Architecture of y-family DNA polymerases relevant to translesion DNA synthesis as revealed in structural and molecular modeling studies. J. Nucleic Acids 2010, 2010, 784081. [Google Scholar]
- Beard, W.A.; Wilson, S.H. Structural insights into the origins of DNA polymerase fidelity. Structure 2003, 11, 489–496. [Google Scholar] [CrossRef]
- Kaushik, N.; Pandey, V.N.; Modak, M.J. Significance of the O-helix residues of Escherichia coli DNA polymerase I in DNA synthesis: Dynamics of the dNTP binding pocket. Biochemistry 1996, 35, 7256–7266. [Google Scholar] [CrossRef]
- Ogawa, M.; Tosaka, A.; Ito, Y.; Yoshida, S.; Suzuki, M. Enhanced ribonucleotide incorporation by an O-helix mutant of Thermus aquaticus DNA polymerase I. Mutat. Res. 2001, 485, 197–207. [Google Scholar] [CrossRef]
- Shinagawa, H.; Kato, T.; Ise, T.; Makino, K.; Nakata, A. Cloning and characterization of the umu operon responsible for inducible mutagenesis in Escherichia coli. Gene 1983, 23, 167–174. [Google Scholar] [CrossRef]
- Elledge, S.J.; Walker, G.C. Proteins required for ultraviolet light and chemical mutagenesis. Identification of the products of the umuC locus of Escherichia coli. J. Mol. Biol. 1983, 164, 175–192. [Google Scholar] [CrossRef]
- Godoy, V.G.; Jarosz, D.F.; Simon, S.M.; Abyzov, A.; Ilyin, V.; Walker, G.C. UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol. Cell 2007, 28, 1058–1070. [Google Scholar] [CrossRef]
- Opperman, T.; Murli, S.; Smith, B.T.; Walker, G.C. A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9218–9223. [Google Scholar] [CrossRef]
- Sutton, M.D.; Walker, G.C. umuDC-mediated cold sensitivity is a manifestation of functions of the UmuD(2)C complex involved in a DNA damage checkpoint control. J. Bacteriol. 2001, 183, 1215–1224. [Google Scholar] [CrossRef]
- Marsh, L.; Walker, G.C. Cold sensitivity induced by overproduction of UmuDC in Escherichia coli. J. Bacteriol. 1985, 162, 155–161. [Google Scholar]
- Burckhardt, S.E.; Woodgate, R.; Scheuermann, R.H.; Echols, H. UmuD mutagenesis protein of Escherichia coli: Overproduction, purification, and cleavage by RecA. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 1811–1815. [Google Scholar] [CrossRef]
- Nohmi, T.; Battista, J.R.; Dodson, L.A.; Walker, G.C. RecA-mediated cleavage activates UmuD for mutagenesis: Mechanistic relationship between transcriptional derepression and posttranslational activation. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 1816–1820. [Google Scholar] [CrossRef]
- Shinagawa, H.; Iwasaki, H.; Kato, T.; Nakata, A. RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 1806–1810. [Google Scholar] [CrossRef]
- Paetzel, M.; Strynadka, N.C. Common protein architecture and binding sites in proteases utilizing a Ser/Lys dyad mechanism. Protein Sci. 1999, 8, 2533–2536. [Google Scholar]
- Brent, R.; Ptashne, M. Mechanism of action of the lexA gene product. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4204–4208. [Google Scholar] [CrossRef]
- Little, J.W.; Mount, D.W.; Yanisch-Perron, C.R. Purified lexA protein is a repressor of the recA and lexA genes. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4199–4203. [Google Scholar] [CrossRef]
- Reuven, N.B.; Arad, G.; Maor-Shoshani, A.; Livneh, Z. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD', RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 1999, 274, 31763–31766. [Google Scholar] [CrossRef]
- Tang, M.; Shen, X.; Frank, E.G.; O'Donnell, M.; Woodgate, R.; Goodman, M.F. UmuD'(2)C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 8919–8924. [Google Scholar]
- Bruck, I.; Woodgate, R.; McEntee, K.; Goodman, M.F. Purification of a soluble UmuD'C complex from Escherichia coli. Cooperative binding of UmuD'C to single-stranded DNA. J. Biol. Chem. 1996, 271, 10767–10774. [Google Scholar] [CrossRef]
- Rehrauer, W.M.; Lavery, P.E.; Palmer, E.L.; Singh, R.N.; Kowalczykowski, S.C. Interaction of Escherichia coli RecA protein with LexA repressor. I. LexA repressor cleavage is competitive with binding of a secondary DNA molecule. J. Biol. Chem. 1996, 271, 23865–23873. [Google Scholar]
- Sommer, S.; Bailone, A.; Devoret, R. The appearance of the UmuD'C protein complex in Escherichia coli switches repair from homologous recombination to SOS mutagenesis. Mol. Microbiol. 1993, 10, 963–971. [Google Scholar] [CrossRef]
- Szpilewska, H.; Bertrand, P.; Bailone, A.; Dutreix, M. In vitro inhibition of RecA-mediated homologous pairing by UmuD'C proteins. Biochimie 1995, 77, 848–853. [Google Scholar] [CrossRef]
- Ollivierre, J.N.; Fang, J.; Beuning, P.J. The Roles of UmuD in Regulating Mutagenesis. J. Nucleic Acids 2010, 2010, 947680. [Google Scholar]
- Ohmori, H.; Hatada, E.; Qiao, Y.; Tsuji, M.; Fukuda, R. dinP, a new gene in Escherichia coli, whose product shows similarities to UmuC and its homologues. Mutat. Res. 1995, 347, 1–7. [Google Scholar] [CrossRef]
- Kenyon, C.J.; Walker, G.C. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 2819–2823. [Google Scholar] [CrossRef]
- Wagner, J.; Gruz, P.; Kim, S.R.; Yamada, M.; Matsui, K.; Fuchs, R.P.; Nohmi, T. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell 1999, 4, 281–286. [Google Scholar] [CrossRef]
- Fuchs, R.P.; Fujii, S.; Wagner, J. Properties and functions of Escherichia coli: Pol IV and Pol V. Adv. Protein Chem. 2004, 69, 229–264. [Google Scholar] [CrossRef]
- Kim, S.R.; Matsui, K.; Yamada, M.; Gruz, P.; Nohmi, T. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol. Genet. Genomics. 2001, 266, 207–215. [Google Scholar] [CrossRef]
- Kobayashi, S.; Valentine, M.R.; Pham, P.; O'Donnell, M.; Goodman, M.F. Fidelity of Escherichia coli DNA polymerase IV. Preferential generation of small deletion mutations by dNTP-stabilized misalignment. J. Biol. Chem. 2002, 277, 34198–34207. [Google Scholar]
- Kuban, W.; Jonczyk, P.; Gawel, D.; Malanowska, K.; Schaaper, R.M.; Fijalkowska, I.J. Role of Escherichia coli DNA polymerase IV in in vivo replication fidelity. J. Bacteriol. 2004, 186, 4802–4807. [Google Scholar] [CrossRef]
- Satou, K.; Yamada, M.; Nohmi, T.; Harashima, H.; Kamiya, H. Mutagenesis induced by oxidized DNA precursors: Roles of Y family DNA polymerases in Escherichia coli. Chem. Res. Toxicol. 2005, 18, 1271–1278. [Google Scholar] [CrossRef]
- Strauss, B.S.; Roberts, R.; Francis, L.; Pouryazdanparast, P. Role of the dinB gene product in spontaneous mutation in Escherichia coli with an impaired replicative polymerase. J. Bacteriol. 2000, 182, 6742–6750. [Google Scholar] [CrossRef]
- Wolff, E.; Kim, M.; Hu, K.; Yang, H.; Miller, J.H. Polymerases leave fingerprints: Analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J. Bacteriol. 2004, 186, 2900–2905. [Google Scholar] [CrossRef]
- Tompkins, J.D.; Nelson, J.L.; Hazel, J.C.; Leugers, S.L.; Stumpf, J.D.; Foster, P.L. Error-prone polymerase, DNA polymerase IV, is responsible for transient hypermutation during adaptive mutation in Escherichia coli. J. Bacteriol. 2003, 185, 3469–3472. [Google Scholar] [CrossRef]
- McKenzie, G.J.; Lee, P.L.; Lombardo, M.J.; Hastings, P.J.; Rosenberg, S.M. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol. Cell 2001, 7, 571–579. [Google Scholar] [CrossRef]
- Foster, P.L. Stress-induced mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 373–397. [Google Scholar] [CrossRef]
- Hersh, M.N.; Ponder, R.G.; Hastings, P.J.; Rosenberg, S.M. Adaptive mutation and amplification in Escherichia coli: Two pathways of genome adaptation under stress. Res. Microbiol. 2004, 155, 352–359. [Google Scholar] [CrossRef]
- McKenzie, G.J.; Magner, D.B.; Lee, P.L.; Rosenberg, S.M. The dinB operon and spontaneous mutation in Escherichia coli. J. Bacteriol. 2003, 185, 3972–3977. [Google Scholar] [CrossRef]
- Slechta, E.S.; Bunny, K.L.; Kugelberg, E.; Kofoid, E.; Andersson, D.I.; Roth, J.R. Adaptive mutation: General mutagenesis is not a programmed response to stress but results from rare coamplification of dinB with lac. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12847–12852. [Google Scholar] [CrossRef]
- Ponder, R.G.; Fonville, N.C.; Rosenberg, S.M. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol. Cell 2005, 19, 791–804. [Google Scholar] [CrossRef]
- Cirz, R.T.; Romesberg, F.E. Controlling mutation: Intervening in evolution as a therapeutic strategy. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 341–354. [Google Scholar] [CrossRef]
- McKenzie, G.J.; Rosenberg, S.M. Adaptive mutations, mutator DNA polymerases and genetic change strategies of pathogens. Curr. Opin. Microbiol. 2001, 4, 586–594. [Google Scholar] [CrossRef]
- Smith, P.A.; Romesberg, F.E. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat. Chem. Biol. 2007, 3, 549–556. [Google Scholar] [CrossRef]
- Yeiser, B.; Pepper, E.D.; Goodman, M.F.; Finkel, S.E. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 8737–8741. [Google Scholar]
- Indiani, C.; Langston, L.D.; Yurieva, O.; Goodman, M.F.; O'Donnell, M. Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6031–6038. [Google Scholar]
- Mori, T.; Nakamura, T.; Okazaki, N.; Furukohri, A.; Maki, H.; Akiyama, M.T. Escherichia coli DinB inhibits replication fork progression without significantly inducing the SOS response. Genes Genet. Syst. 2012, 87, 75–87. [Google Scholar] [CrossRef]
- Uchida, K.; Furukohri, A.; Shinozaki, Y.; Mori, T.; Ogawara, D.; Kanaya, S.; Nohmi, T.; Maki, H.; Akiyama, M. Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol. Microbiol. 2008, 70, 608–622. [Google Scholar] [CrossRef]
- Jarosz, D.F.; Cohen, S.E.; Delaney, J.C.; Essigmann, J.M.; Walker, G.C. A DinB variant reveals diverse physiological consequences of incomplete TLS extension by a Y-family DNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21137–21142. [Google Scholar] [CrossRef]
- Seo, K.Y.; Nagalingam, A.; Miri, S.; Yin, J.; Chandani, S.; Kolbanovskiy, A.; Shastry, A.; Loechler, E.L. Mirror image stereoisomers of the major benzo[a]pyrene N-2-dG adduct are bypassed by different lesion-bypass DNA polymerases in E. coli. DNA Repair (Amst) 2006, 5, 515–522. [Google Scholar] [CrossRef]
- Jarosz, D.F.; Godoy, V.G.; Delaney, J.C.; Essigmann, J.M.; Walker, G.C. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 2006, 439, 225–228. [Google Scholar] [CrossRef]
- Astatke, M.; Ng, K.; Grindley, N.D.; Joyce, C.M. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 3402–3407. [Google Scholar] [CrossRef]
- Brown, J.A.; Suo, Z. Unlocking the sugar "steric gate" of DNA polymerases. Biochemistry 2011, 50, 1135–1142. [Google Scholar] [CrossRef]
- Shen, X.; Sayer, J.M.; Kroth, H.; Ponten, I.; O'Donnell, M.; Woodgate, R.; Jerina, D.M.; Goodman, M.F. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J. Biol. Chem. 2002, 277, 5265–5274. [Google Scholar]
- Yuan, B.F.; Cao, H.C.; Jiang, Y.; Hong, H.Z.; Wang, Y.S. Efficient and accurate bypass of N-2-(1-carboxyethyl)-2 '-deoxyguanosine by DinB DNA polyrnerase in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8679–8684. [Google Scholar] [CrossRef]
- Kumari, A.; Minko, I.G.; Harbut, M.B.; Finkel, S.E.; Goodman, M.F.; Lloyd, R.S. Replication bypass of interstrand cross-link intermediates by Escherichia coli DNA polymerase IV. J. Biol. Chem. 2008, 283, 27433–27437. [Google Scholar] [CrossRef]
- Minko, I.G.; Yamanaka, K.; Kozekov, I.D.; Kozekova, A.; Indiani, C.; O'Donnell, M.E.; Jiang, Q.; Goodman, M.F.; Rizzo, C.J.; Lloyd, R.S. Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 2008, 21, 1983–1990. [Google Scholar] [CrossRef]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11. [Google Scholar]
- Thornalley, P.J. Pharmacology of methylglyoxal: Formation, modification of proteins and nucleic acids, and enzymatic detoxification—A role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Whiteway, J.; Koziarz, P.; Veall, J.; Sandhu, N.; Kumar, P.; Hoecher, B.; Lambert, I.B. Oxygen-insensitive nitroreductases: Analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli. J. Bacteriol. 1998, 180, 5529–5539. [Google Scholar]
- Panigrahi, G.B.; Walker, I.G. The N2-guanine adduct but not the C8-guanine or N6-adenine adducts formed by 4-nitroquinoline 1-oxide blocks the 3'-5' exonuclease action of T4 DNA polymerase. Biochemistry 1990, 29, 2122–2126. [Google Scholar] [CrossRef]
- Cole, R.S. Repair of DNA containing interstrand crosslinks in Escherichia coli: Sequential excision and recombination. Proc. Natl. Acad. Sci. U. S. A. 1973, 70, 1064–1068. [Google Scholar] [CrossRef]
- Minko, I.G.; Kozekov, I.D.; Harris, T.M.; Rizzo, C.J.; Lloyd, R.S.; Stone, M.P. Chemistry and biology of DNA containing 1,N(2)-deoxyguanosine adducts of the alpha, beta-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem. Res. Toxicol. 2009, 22, 759–778. [Google Scholar] [CrossRef]
- Bjedov, I.; Dasgupta, C.N.; Slade, D.; Le Blastier, S.; Selva, M.; Matic, I. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics 2007, 176, 1431–1440. [Google Scholar] [CrossRef]
- Benson, R.W.; Norton, M.D.; Lin, I.; Du Comb, W.S.; Godoy, V.G. An active site aromatic triad in Escherichia coli DNA Pol IV coordinates cell survival and mutagenesis in different DNA damaging agents. PLoS One 2011, 6, e19944. [Google Scholar]
- Hori, M.; Yonekura, S.; Nohmi, T.; Gruz, P.; Sugiyama, H.; Yonei, S.; Zhang-Akiyama, Q.M. Error-Prone Translesion DNA Synthesis by Escherichia coli DNA Polymerase IV (DinB) on Templates Containing 1,2-dihydro-2-oxoadenine. J. Nucleic Acids 2010, 2010, 807579. [Google Scholar]
- Foti, J.J.; Devadoss, B.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 2012, 336, 315–319. [Google Scholar]
- Yamada, M.; Nunoshiba, T.; Shimizu, M.; Gruz, P.; Kamiya, H.; Harashima, H.; Nohmi, T. Involvement of Y-family DNA polymerases in mutagenesis caused by oxidized nucleotides in Escherichia coli. J. Bacteriol. 2006, 188, 4992–4995. [Google Scholar] [CrossRef]
- Walsh, J.M.; Bouamaied, I.; Brown, T.; Wilhelmsson, L.M.; Beuning, P.J. Discrimination against the Cytosine Analog tC by Escherichia coli DNA Polymerase IV DinB. J. Mol. Biol. 2011, 409, 89–100. [Google Scholar] [CrossRef]
- Maor-Shoshani, A.; Hayashi, K.; Ohmori, H.; Livneh, Z. Analysis of translesion replication across an abasic site by DNA polymerase IV of Escherichia coli. DNA Repair (Amst) 2003, 2, 1227–1238. [Google Scholar] [CrossRef]
- Foti, J.J.; Delucia, A.M.; Joyce, C.M.; Walker, G.C. UmuD(2) inhibits a non-covalent step during DinB-mediated template slippage on homopolymeric nucleotide runs. J. Biol. Chem. 2010, 285, 23086–23095. [Google Scholar]
- Wagner, J.; Fujii, S.; Gruz, P.; Nohmi, T.; Fuchs, R.P.P. The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 2000, 1, 484–488. [Google Scholar]
- Cohen, S.E.; Godoy, V.G.; Walker, G.C. Transcriptional modulator NusA interacts with translesion DNA polymerases in Escherichia coli. J. Bacteriol. 2009, 191, 665–672. [Google Scholar] [CrossRef]
- Cohen, S.E.; Lewis, C.A.; Mooney, R.A.; Kohanski, M.A.; Collins, J.J.; Landick, R.; Walker, G.C. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15517–15522. [Google Scholar]
- Sladewski, T.E.; Hetrick, K.M.; Foster, P.L. Escherichia coli Rep DNA helicase and error-prone DNA polymerase IV interact physically and functionally. Mol. Microbiol. 2011, 80, 524–541. [Google Scholar] [CrossRef]
- Furukohri, A.; Nishikawa, Y.; Tatsumi Akiyama, M.; Maki, H. Interaction between Escherichia coli DNA polymerase IV and single-stranded DNA-binding protein is required for DNA synthesis on SSB-coated DNA. Nucleic Acids Res. 2012, 40, 6039–6048. [Google Scholar] [CrossRef]
- Lenne-Samuel, N.; Wagner, J.; Etienne, H.; Fuchs, R.P. The processivity factor beta controls DNA polymerase IV traffic during spontaneous mutagenesis and translesion synthesis in vivo. EMBO Rep. 2002, 3, 45–49. [Google Scholar] [CrossRef]
- Bunting, K.A.; Roe, S.M.; Pearl, L.H. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J. 2003, 22, 5883–5892. [Google Scholar] [CrossRef]
- Becherel, O.J.; Fuchs, R.P.P. SOS mutagenesis results from up-regulation of translesion synthesis. J. Mol. Biol. 1999, 294, 299–306. [Google Scholar] [CrossRef]
- Wagner, J.; Etienne, H.; Fuchs, R.P.; Cordonnier, A.; Burnouf, D. Distinct beta-clamp interactions govern the activities of the Y family PolIV DNA polymerase. Mol. Microbiol. 2009, 74, 1143–1151. [Google Scholar] [CrossRef]
- Heltzel, J.M.; Maul, R.W.; Wolff, D.W.; Sutton, M.D. Escherichia coli DNA Polymerase IV (Pol IV), but Not Pol II, Dynamically Switches with a Stalled Pol III* Replicase. J. Bacteriol. 2012, 194, 3589–3600. [Google Scholar] [CrossRef]
- Heltzel, J.M.; Maul, R.W.; Scouten Ponticelli, S.K.; Sutton, M.D. A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12664–12669. [Google Scholar]
- Fujii, S.; Fuchs, R.P. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004, 23, 4342–4352. [Google Scholar] [CrossRef]
- Fujii, S.; Gasser, W.; Fuchs, R.P. The biochemical requirements of DNA polymerase V-mediated translesion synthesis revisited. J. Mol. Biol. 2004, 341, 405–417. [Google Scholar] [CrossRef]
- Fujii, S.; Fuchs, R.P. Biochemical basis for the essential genetic requirements of RecA and the beta-clamp in Pol V activation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14825–14830. [Google Scholar] [CrossRef]
- Farnham, P.J.; Greenblatt, J.; Platt, T. Effects of NusA protein on transcription termination in the tryptophan operon of Escherichia coli. Cell 1982, 29, 945–951. [Google Scholar] [CrossRef]
- Greenblatt, J.; Li, J. Interaction of the sigma factor and the nusA gene protein of E. coli with RNA polymerase in the initiation-termination cycle of transcription. Cell 1981, 24, 421–428. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, Y.; Severinov, K.; Das, A.; Hanna, M.M. Role of Escherichia coli RNA polymerase alpha subunit in modulation of pausing, termination and anti-termination by the transcription elongation factor NusA. EMBO J. 1996, 15, 150–161. [Google Scholar]
- Cohen, S.E.; Walker, G.C. The transcription elongation factor NusA is required for stress-induced mutagenesis in Escherichia coli. Curr. Biol. 2010, 20, 80–85. [Google Scholar] [CrossRef]
- Nakamura, Y.; Mizusawa, S.; Court, D.L.; Tsugawa, A. Regulatory defects of a conditionally lethal nusAts mutant of Escherichia coli. Positive and negative modulator roles of NusA protein in vivo. J. Mol. Biol. 1986, 189, 103–111. [Google Scholar] [CrossRef]
- Bagg, A.; Kenyon, C.J.; Walker, G.C. Inducibility of a gene product required for UV and chemical mutagenesis in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 5749–5753. [Google Scholar] [CrossRef]
- Kato, T.; Shinoura, Y. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol. Gen. Genet. 1977, 156, 121–131. [Google Scholar]
- Woodgate, R.; Rajagopalan, M.; Lu, C.; Echols, H. UmuC mutagenesis protein of Escherichia coli: Purification and interaction with UmuD and UmuD'. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 7301–7305. [Google Scholar] [CrossRef]
- Bridges, B.A.; Woodgate, R. Mutagenic repair in Escherichia coli: Products of the recA gene and of the umuD and umuC genes act at different steps in UV-induced mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 4193–4197. [Google Scholar] [CrossRef]
- Bridges, B.A.; Woodgate, R. The two-step model of bacterial UV mutagenesis. Mutat. Res. 1985, 150, 133–139. [Google Scholar] [CrossRef]
- Rajagopalan, M.; Lu, C.; Woodgate, R.; O'Donnell, M.; Goodman, M.F.; Echols, H. Activity of the purified mutagenesis proteins UmuC, UmuD', and RecA in replicative bypass of an abasic DNA lesion by DNA polymerase III. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 10777–10781. [Google Scholar] [CrossRef]
- Walker, G.C. Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli. Microbiol. Rev. 1984, 48, 60–93. [Google Scholar]
- Tang, M.; Bruck, I.; Eritja, R.; Turner, J.; Frank, E.G.; Woodgate, R.; O'Donnell, M.; Goodman, M.F. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: Reconstitution of in vitro lesion bypass dependent on the UmuD'2C mutagenic complex and RecA protein. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 9755–9760. [Google Scholar]
- Arad, G.; Hendel, A.; Urbanke, C.; Curth, U.; Livneh, Z. Single-stranded DNA-binding protein recruits DNA polymerase V to primer termini on RecA-coated DNA. J. Biol. Chem. 2008, 283, 8274–8282. [Google Scholar]
- Schlacher, K.; Goodman, M.F. Timeline—Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 587–594. [Google Scholar] [CrossRef]
- Woodgate, R.; Ennis, D.G. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol. Gen. Genet. 1991, 229, 10–16. [Google Scholar]
- Courcelle, J.; Hanawalt, P.C. Participation of recombination proteins in rescue of arrested replication forks in UV-irradiated Escherichia coli need not involve recombination. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8196–8202. [Google Scholar] [CrossRef]
- Courcelle, C.T.; Belle, J.J.; Courcelle, J. Nucleotide excision repair or polymerase V-mediated lesion bypass can act to restore UV-arrested replication forks in Escherichia coli. J. Bacteriol. 2005, 187, 6953–6961. [Google Scholar] [CrossRef]
- Courcelle, C.T.; Chow, K.H.; Casey, A.; Courcelle, J. Nascent DNA processing by RecJ favors lesion repair over translesion synthesis at arrested replication forks in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9154–9159. [Google Scholar]
- Schlacher, K.; Leslie, K.; Wyman, C.; Woodgate, R.; Cox, M.M.; Goodman, M.F. DNA polymerase V and RecA protein, a minimal mutasome. Mol. Cell 2005, 17, 561–572. [Google Scholar] [CrossRef]
- Beuning, P.J.; Simon, S.M.; Zemla, A.; Barsky, D.; Walker, G.C. A non-cleavable UmuD variant that acts as a UmuD' mimic. J. Biol. Chem. 2006, 281, 23296–23296. [Google Scholar]
- Battista, J.R.; Ohta, T.; Nohmi, T.; Sun, W.; Walker, G.C. Dominant negative umuD mutations decreasing RecA-mediated cleavage suggest roles for intact UmuD in modulation of SOS mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 7190–7194. [Google Scholar] [CrossRef]
- Jonczyk, P.; Nowicka, A. Specific in vivo protein-protein interactions between Escherichia coli SOS mutagenesis proteins. J. Bacteriol. 1996, 178, 2580–2585. [Google Scholar]
- Vaisman, A.; Kuban, W.; McDonald, J.P.; Karata, K.; Yang, W.; Goodman, M.F.; Woodgate, R. Critical amino acids in Escherichia coli UmuC responsible for sugar discrimination and base-substitution fidelity. Nucleic Acids Res. 2012, 40, 6144–6157. [Google Scholar] [CrossRef]
- Shurtleff, B.W.; Ollivierre, J.N.; Tehrani, M.; Walker, G.C.; Beuning, P.J. Steric gate variants of UmuC confer UV hypersensitivity on Escherichia coli. J. Bacteriol. 2009, 191, 4815–4823. [Google Scholar] [CrossRef]
- Kuban, W.; Vaisman, A.; McDonald, J.P.; Karata, K.; Yang, W.; Goodman, M.F.; Woodgate, R. Escherichia coli UmuC active site mutants: Effects on translesion DNA synthesis, mutagenesis and cell survival. DNA Repair (Amst) 2012, 11, 726–732. [Google Scholar] [CrossRef]
- Seo, K.Y.; Yin, J.; Donthamsetti, P.; Chandani, S.; Lee, C.H.; Loechler, E.L. Amino acid architecture that influences dNTP insertion efficiency in Y-family DNA polymerase V of E. coli. J. Mol. Biol. 2009, 392, 270–282. [Google Scholar] [CrossRef]
- Hawver, L.A.; Gillooly, C.A.; Beuning, P.J. Characterization of Escherichia coli UmuC active-site loops identifies variants that confer UV hypersensitivity. J. Bacteriol. 2011, 193, 5400–5411. [Google Scholar] [CrossRef]
- Tang, M.J.; Pham, P.; Shen, X.; Taylor, J.S.; O'Donnell, M.; Woodgate, R.; Goodman, M.F. Roles of E-coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature 2000, 404, 1014–1018. [Google Scholar] [CrossRef]
- Jasti, V.P.; Das, R.S.; Hilton, B.A.; Weerasooriya, S.; Zou, Y.; Basu, A.K. (5'S)-8,5'-cyclo-2'-deoxyguanosine is a strong block to replication, a potent pol V-dependent mutagenic lesion, and is inefficiently repaired in Escherichia coli. Biochemistry 2011, 50, 3862–3865. [Google Scholar] [CrossRef]
- Neeley, W.L.; Delaney, S.; Alekseyev, Y.O.; Jarosz, D.F.; Delaney, J.C.; Walker, G.C.; Essigmann, J.M. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J. Biol. Chem. 2007, 282, 12741–12748. [Google Scholar]
- Schlacher, K.; Pham, P.; Cox, M.M.; Goodman, M.F. Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation. Chem. Rev. 2006, 106, 406–419. [Google Scholar]
- Patel, M.; Jiang, Q.F.; Woodgate, R.; Cox, M.M.; Goodman, M.F. A new model for SOS-induced mutagenesis: How RecA protein activates DNA polymerase V. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 171–184. [Google Scholar] [CrossRef]
- Pham, P.; Seitz, E.M.; Saveliev, S.; Shen, X.; Woodgate, R.; Cox, M.M.; Goodman, M.F. Two distinct modes of RecA action are required for DNA polymerase V-catalyzed translesion synthesis. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11061–11066. [Google Scholar]
- Sweasy, J.B. RecA kicks Pol V into gear. Nat. Struct. Mol. Biol. 2005, 12, 215–216. [Google Scholar] [CrossRef]
- Jiang, Q.; Karata, K.; Woodgate, R.; Cox, M.M.; Goodman, M.F. The active form of DNA polymerase V is UmuD'(2)C-RecA-ATP. Nature 2009, 460, 359–363. [Google Scholar] [CrossRef]
- Schlacher, K.; Cox, M.M.; Woodgate, R.; Goodman, M.F. RecA acts in trans to allow replication of damaged DNA by DNA polymerase V. Nature 2006, 442, 883–887. [Google Scholar] [CrossRef]
- Pham, P.; Bertram, J.G.; O'Donnell, M.; Woodgate, R.; Goodman, M.F. A model for SOS-lesion-targeted mutations in Escherichia coli. Nature 2001, 409, 366–370. [Google Scholar] [CrossRef]
- Maor-Shoshani, A.; Livneh, Z. Analysis of the stimulation of DNA polymerase V of Escherichia coli by processivity proteins. Biochemistry 2002, 41, 14438–14446. [Google Scholar] [CrossRef]
- Petrosino, J.F.; Galhardo, R.S.; Morales, L.D.; Rosenberg, S.M. Stress-induced beta-lactam antibiotic resistance mutation and sequences of stationary-phase mutations in the Escherichia coli chromosome. J. Bacteriol. 2009, 191, 5881–5889. [Google Scholar] [CrossRef]
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crecy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005, 3, e176. [Google Scholar] [CrossRef]
- Gawel, D.; Seed, P.C. Urinary tract infection drives genome instability in uropathogenic Escherichia coli and necessitates translesion synthesis DNA polymerase IV for virulence. Virulence 2011, 2, 222–232. [Google Scholar] [CrossRef]
- Koskiniemi, S.; Andersson, D.I. Translesion DNA polymerases are required for spontaneous deletion formation in Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10248–10253. [Google Scholar] [CrossRef]
- Hare, J.M.; Bradley, J.A.; Lin, C.L.; Elam, T.J. Diverse responses to UV light exposure in Acinetobacter include the capacity for DNA damage-induced mutagenesis in the opportunistic pathogens Acinetobacter baumannii and Acinetobacter ursingii. Microbiology 2012, 158, 601–611. [Google Scholar] [CrossRef]
- Hare, J.M.; Adhikari, S.; Lambert, K.V.; Hare, A.E.; Grice, A.N. The Acinetobacter regulatory UmuDAb protein cleaves in response to DNA damage with chimeric LexA/UmuD characteristics. FEMS Microbiol. Lett. 2012. [Google Scholar]
- Sung, H.M.; Yasbin, R.E. Adaptive, or stationary-phase, mutagenesis, a component of bacterial differentiation in Bacillus subtilis. J. Bacteriol. 2002, 184, 5641–5653. [Google Scholar] [CrossRef]
- Kunst, F.; Ogasawara, N.; Moszer, I.; Albertini, A.M.; Alloni, G.; Azevedo, V.; Bertero, M.G.; Bessieres, P.; Bolotin, A.; Borchert, S.; et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 1997, 390, 249–256. [Google Scholar] [CrossRef]
- Duigou, S.; Ehrlich, S.D.; Noirot, P.; Noirot-Gros, M.F. DNA polymerase I acts in translesion synthesis mediated by the Y-polymerases in Bacillus subtilis. Mol. Microbiol. 2005, 57, 678–690. [Google Scholar] [CrossRef]
- Sung, H.M.; Yeamans, G.; Ross, C.A.; Yasbin, R.E. Roles of YqjH and YqjW, homologs of the Escherichia coli UmuC/DinB or Y superfamily of DNA polymerases, in stationary-phase mutagenesis and UV-induced mutagenesis of Bacillus subtilis. J. Bacteriol. 2003, 185, 2153–2160. [Google Scholar] [CrossRef]
- Rivas-Castillo, A.M.; Yasbin, R.E.; Robleto, E.; Nicholson, W.L.; Pedraza-Reyes, M. Role of the Y-family DNA polymerases YqjH and YqjW in protecting sporulating Bacillus subtilis cells from DNA damage. Curr. Microbiol. 2010, 60, 263–267. [Google Scholar]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Sanders, L.H.; Rockel, A.; Lu, H.; Wozniak, D.J.; Sutton, M.D. Role of Pseudomonas aeruginosa dinB-encoded DNA polymerase IV in mutagenesis. J. Bacteriol. 2006, 188, 8573–8585. [Google Scholar] [CrossRef]
- Boshoff, H.I.; Myers, T.G.; Copp, B.R.; McNeil, M.R.; Wilson, M.A.; Barry, C.E., 3rd. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: Novel insights into drug mechanisms of action. J. Biol. Chem. 2004, 279, 40174–40184. [Google Scholar]
- Brooks, P.C.; Movahedzadeh, F.; Davis, E.O. Identification of some DNA damage-inducible genes of Mycobacterium tuberculosis: Apparent lack of correlation with LexA binding. J. Bacteriol. 2001, 183, 4459–4467. [Google Scholar] [CrossRef]
- Rand, L.; Hinds, J.; Springer, B.; Sander, P.; Buxton, R.S.; Davis, E.O. The majority of inducible DNA repair genes in Mycobacterium tuberculosis are induced independently of RecA. Mol. Microbiol. 2003, 50, 1031–1042. [Google Scholar] [CrossRef]
- Kana, B.D.; Abrahams, G.L.; Sung, N.; Warner, D.F.; Gordhan, B.G.; Machowski, E.E.; Tsenova, L.; Sacchettini, J.C.; Stoker, N.G.; Kaplan, G.; et al. Role of the DinB homologs Rv1537 and Rv3056 in Mycobacterium tuberculosis. J. Bacteriol. 2010, 192, 2220–2227. [Google Scholar]
- Rachman, H.; Strong, M.; Ulrichs, T.; Grode, L.; Schuchhardt, J.; Mollenkopf, H.; Kosmiadi, G.A.; Eisenberg, D.; Kaufmann, S.H. Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 2006, 74, 1233–1242. [Google Scholar] [CrossRef]
- Boshoff, H.I.M.; Reed, M.B.; Barry Iii, C.E.; Mizrahi, V. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 2003, 113, 183–193. [Google Scholar] [CrossRef]
- McHenry, C.S. Bacterial replicases and related polymerases. Curr. Opin. Chem. Biol. 2011, 15, 587–594. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef]
- Sharma, A.; Nair, D.T. MsDpo4-a DinB Homolog from Mycobacterium smegmatis-Is an Error-Prone DNA Polymerase That Can Promote G:T and T:G Mismatches. J. Nucleic Acids 2012, 2012, 285481. [Google Scholar]
- Sanders, L.H.; Devadoss, B.; Raja, G.V.; O'Connor, J.; Su, S.; Wozniak, D.J.; Hassett, D.J.; Berdis, A.J.; Sutton, M.D. Epistatic roles for Pseudomonas aeruginosa MutS and DinB (DNA Pol IV) in coping with reactive oxygen species-induced DNA damage. PLoS One 2011, 6, e18824. [Google Scholar]
- Tegova, R.; Tover, A.; Tarassova, K.; Tark, M.; Kivisaar, M. Involvement of error-prone DNA polymerase IV in stationary-phase mutagenesis in Pseudomonas putida. J. Bacteriol. 2004, 186, 2735–2744. [Google Scholar] [CrossRef]
- Tark, M.; Tover, A.; Tarassova, K.; Tegova, R.; Kivi, G.; Horak, R.; Kivisaar, M. A DNA polymerase V homologue encoded by TOL plasmid pWW0 confers evolutionary fitness on Pseudomonas putida under conditions of environmental stress. J. Bacteriol. 2005, 187, 5203–5213. [Google Scholar] [CrossRef]
- Braithwaite, D.K.; Ito, J. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 1993, 21, 787–802. [Google Scholar] [CrossRef]
- Ito, J.; Braithwaite, D.K. Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res. 1991, 19, 4045–4057. [Google Scholar] [CrossRef]
- Johnson, A.; O'Donnell, M. Cellular DNA replicases: Components and dynamics at the replication fork. Annu. Rev. Biochem. 2005, 74, 283–315. [Google Scholar] [CrossRef]
- McInerney, P.; Johnson, A.; Katz, F.; O'Donnell, M. Characterization of a triple DNA polymerase replisome. Mol. Cell 2007, 27, 527–538. [Google Scholar] [CrossRef]
- Dervyn, E.; Suski, C.; Daniel, R.; Bruand, C.; Chapuis, J.; Errington, J.; Janniere, L.; Ehrlich, S.D. Two essential DNA polymerases at the bacterial replication fork. Science 2001, 294, 1716–1719. [Google Scholar]
- Le Chatelier, E.; Becherel, O.J.; d'Alencon, E.; Canceill, D.; Ehrlich, S.D.; Fuchs, R.P.; Janniere, L. Involvement of DnaE, the second replicative DNA polymerase from Bacillus subtilis, in DNA mutagenesis. J. Biol. Chem. 2004, 279, 1757–1767. [Google Scholar]
- Bruck, I.; Goodman, M.F.; O'Donnell, M. The essential C family DnaE polymerase is error-prone and efficient at lesion bypass. J. Biol. Chem. 2003, 278, 44361–44368. [Google Scholar]
- Fraser, C.M.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M.; et al. The minimal gene complement of Mycoplasma genitalium. Science 1995, 270, 397–403. [Google Scholar]
- Himmelreich, R.; Hilbert, H.; Plagens, H.; Pirkl, E.; Li, B.C.; Herrmann, R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996, 24, 4420–4449. [Google Scholar] [CrossRef]
- Barnes, M.H.; Tarantino, P.M., Jr.; Spacciapoli, P.; Brown, N.C.; Yu, H.; Dybvig, K. DNA polymerase III of Mycoplasma pulmonis: Isolation and characterization of the enzyme and its structural gene, polC. Mol. Microbiol. 1994, 13, 843–854. [Google Scholar] [CrossRef]
- Caspi, J.; Amitai, G.; Belenkiy, O.; Pietrokovski, S. Distribution of split DnaE inteins in cyanobacteria. Mol. Microbiol. 2003, 50, 1569–1577. [Google Scholar] [CrossRef]
- Abella, M.; Erill, I.; Jara, M.; Mazon, G.; Campoy, S.; Barbe, J. Widespread distribution of a lexA-regulated DNA damage-inducible multiple gene cassette in the Proteobacteria phylum. Mol. Microbiol. 2004, 54, 212–222. [Google Scholar] [CrossRef]
- Abella, M.; Campoy, S.; Erill, I.; Rojo, F.; Barbé, J. Cohabitation of Two Different lexA Regulons in Pseudomonas putida. J. Bacteriol. 2007, 189, 8855–8862. [Google Scholar] [CrossRef]
- Koorits, L.; Tegova, R.; Tark, M.; Tarassova, K.; Tover, A.; Kivisaar, M. Study of involvement of ImuB and DnaE2 in stationary-phase mutagenesis in Pseudomonas putida. DNA Repair (Amst) 2007, 6, 863–868. [Google Scholar] [CrossRef]
- Galhardo, R.S.; Rocha, R.P.; Marques, M.V.; Menck, C.F. An SOS-regulated operon involved in damage-inducible mutagenesis in Caulobacter crescentus. Nucleic Acids Res. 2005, 33, 2603–2614. [Google Scholar] [CrossRef]
- McHenry, C.S. Breaking the rules: Bacteria that use several DNA polymerase IIIs. EMBO Rep. 2011, 12, 408–414. [Google Scholar] [CrossRef]
- Warner, D.F.; Ndwandwe, D.E.; Abrahams, G.L.; Kana, B.D.; Machowski, E.E.; Venclovas, Č.; Mizrahi, V. Essential roles for imuA′- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13093–13098. [Google Scholar]
- Campoy, S.; Salvador, N.; Cortes, P.; Erill, I.; Barbe, J. Expression of canonical SOS genes is not under LexA repression in Bdellovibrio bacteriovorus. J. Bacteriol. 2005, 187, 5367–5375. [Google Scholar] [CrossRef]
- Erill, I.; Campoy, S.; Mazon, G.; Barbe, J. Dispersal and regulation of an adaptive mutagenesis cassette in the bacteria domain. Nucleic Acids Res. 2006, 34, 66–77. [Google Scholar] [CrossRef]
- de Groot, A.; Chapon, V.; Servant, P.; Christen, R.; Saux, M.F.; Sommer, S.; Heulin, T. Deinococcus deserti sp. nov., a gamma-radiation-tolerant bacterium isolated from the Sahara Desert. Int. J. Syst. Evol. Microbiol. 2005, 55, 2441–2446. [Google Scholar] [CrossRef]
- de Groot, A.; Dulermo, R.; Ortet, P.; Blanchard, L.; Guerin, P.; Fernandez, B.; Vacherie, B.; Dossat, C.; Jolivet, E.; Siguier, P.; et al. Alliance of proteomics and genomics to unravel the specificities of Sahara bacterium Deinococcus deserti. PLoS Genet. 2009, 5, e1000434. [Google Scholar] [CrossRef]
- Dulermo, R.; Fochesato, S.; Blanchard, L.; De Groot, A. Mutagenic lesion bypass and two functionally different RecA proteins in Deinococcus deserti. Mol. Microbiol. 2009, 74, 194–208. [Google Scholar] [CrossRef]
- Genin, S.; Boucher, C. Ralstonia solanacearum: Secrets of a major pathogen unveiled by analysis of its genome. Mol. Plant. Pathol. 2002, 3, 111–118. [Google Scholar] [CrossRef]
- Erill, I.; Campoy, S.; Barbe, J. Aeons of distress: An evolutionary perspective on the bacterial SOS response. FEMS Microbiol. Rev. 2007, 31, 637–656. [Google Scholar] [CrossRef]
- Davis, E.O.; Dullaghan, E.M.; Rand, L. Definition of the mycobacterial SOS box and use to identify LexA-regulated genes in Mycobacterium tuberculosis. J. Bacteriol. 2002, 184, 3287–3295. [Google Scholar] [CrossRef]
- O'Sullivan, D.M.; Hinds, J.; Butcher, P.D.; Gillespie, S.H.; McHugh, T.D. Mycobacterium tuberculosis DNA repair in response to subinhibitory concentrations of ciprofloxacin. J. Antimicrob. Chemother. 2008, 62, 1199–1202. [Google Scholar] [CrossRef]
- Smollett, K.L.; Smith, K.M.; Kahramanoglou, C.; Arnvig, K.B.; Buxton, R.S.; Davis, E.O. Global Analysis of the Regulon of the Transcriptional Repressor LexA, a Key Component of SOS Response in Mycobacterium tuberculosis. J. Biol. Chem. 2012, 287, 22004–22014. [Google Scholar]
- Zeng, Y.-H.; Shen, F.-T.; Tan, C.-C.; Huang, C.-C.; Young, C.-C. The flexibility of UV-inducible mutation in Deinococcus ficus as evidenced by the existence of the imuB–dnaE2 gene cassette and generation of superior feather degrading bacteria. Microbiol. Res. 2011, 167, 40–47. [Google Scholar] [CrossRef]
- Sale, J.E. Radiation resistance: Resurrection by recombination. Curr. Biol. 2007, 17, R12–R14. [Google Scholar] [CrossRef]
- da Rocha, R.P.; Paquola, A.C.; Marques Mdo, V.; Menck, C.F.; Galhardo, R.S. Characterization of the SOS regulon of Caulobacter crescentus. J. Bacteriol. 2008, 190, 1209–1218. [Google Scholar] [CrossRef]
- Maor-Shoshani, A.; Reuven, N.B.; Tomer, G.; Livneh, Z. Highly mutagenic replication by DNA polymerase V (UmuC) provides a mechanistic basis for SOS untargeted mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 565–570. [Google Scholar]
- Livneh, Z.; Cohen-Fix, O.; Skaliter, R.; Elizur, T. Replication of damaged DNA and the molecular mechanism of ultraviolet light mutagenesis. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 465–513. [Google Scholar] [CrossRef]
- Tsai, H.-H.; Shu, H.-W.; Yang, C.-C.; Chen, C.W. Translesion-synthesis DNA polymerases participate in replication of the telomeres in Streptomyces. Nucleic Acids Res. 2012, 40, 1118–1130. [Google Scholar] [CrossRef]
- Varhimo, E.; Savijoki, K.; Jalava, J.; Kuipers, O.P.; Varmanen, P. Identification of a novel streptococcal gene cassette mediating SOS mutagenesis in Streptococcus uberis. J. Bacteriol. 2007, 189, 5210–5222. [Google Scholar] [CrossRef]
- Sidorenko, J.; Jatsenko, T.; Saumaa, S.; Teras, R.; Tark-Dame, M.; Hõrak, R.; Kivisaar, M. Involvement of specialized DNA polymerases Pol II, Pol IV and DnaE2 in DNA replication in the absence of Pol I in Pseudomonas putida. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 714, 63–77. [Google Scholar] [CrossRef]
- Cirz, R.T.; O'Neill, B.M.; Hammond, J.A.; Head, S.R.; Romesberg, F.E. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J. Bacteriol. 2006, 188, 7101–7110. [Google Scholar] [CrossRef]
- Sanchez-Alberola, N.; Campoy, S.; Barbe, J.; Erill, I. Analysis of the SOS response of Vibrio and other bacteria with multiple chromosomes. BMC Genomics 2012, 13, 58. [Google Scholar] [CrossRef]
- Blazquez, J. Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin. Infect. Dis. 2003, 37, 1201–1209. [Google Scholar] [CrossRef]
- Bergval, I.L.; Klatser, P.R.; Schuitema, A.R.J.; Oskam, L.; Anthony, R.M. Specific mutations in the Mycobacterium tuberculosis rpoB gene are associated with increased dnaE2 expression. FEMS Microbiol. Lett. 2007, 275, 338–343. [Google Scholar] [CrossRef]
- Oliver, A.; Canton, R.; Campo, P.; Baquero, F.; Blazquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1254. [Google Scholar] [CrossRef]
- Gutierrez, O.; Juan, C.; Perez, J.L.; Oliver, A. Lack of association between hypermutation and antibiotic resistance development in Pseudomonas aeruginosa isolates from intensive care unit patients. Antimicrob. Agents Chemother. 2004, 48, 3573–3575. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research 2010, 38, W695–W699. [Google Scholar] [CrossRef]
- Consortium, T.U. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012, 40, D71–D75. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).