Adaptive and Pathogenic Responses to Stress by Stem Cells during Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction and Summary of Goals for the Review
2. What Is Stress? General Elements of Cellular Stress
2.1. Stressors
2.2. Adaptive Responses
2.3. Stress Outcomes
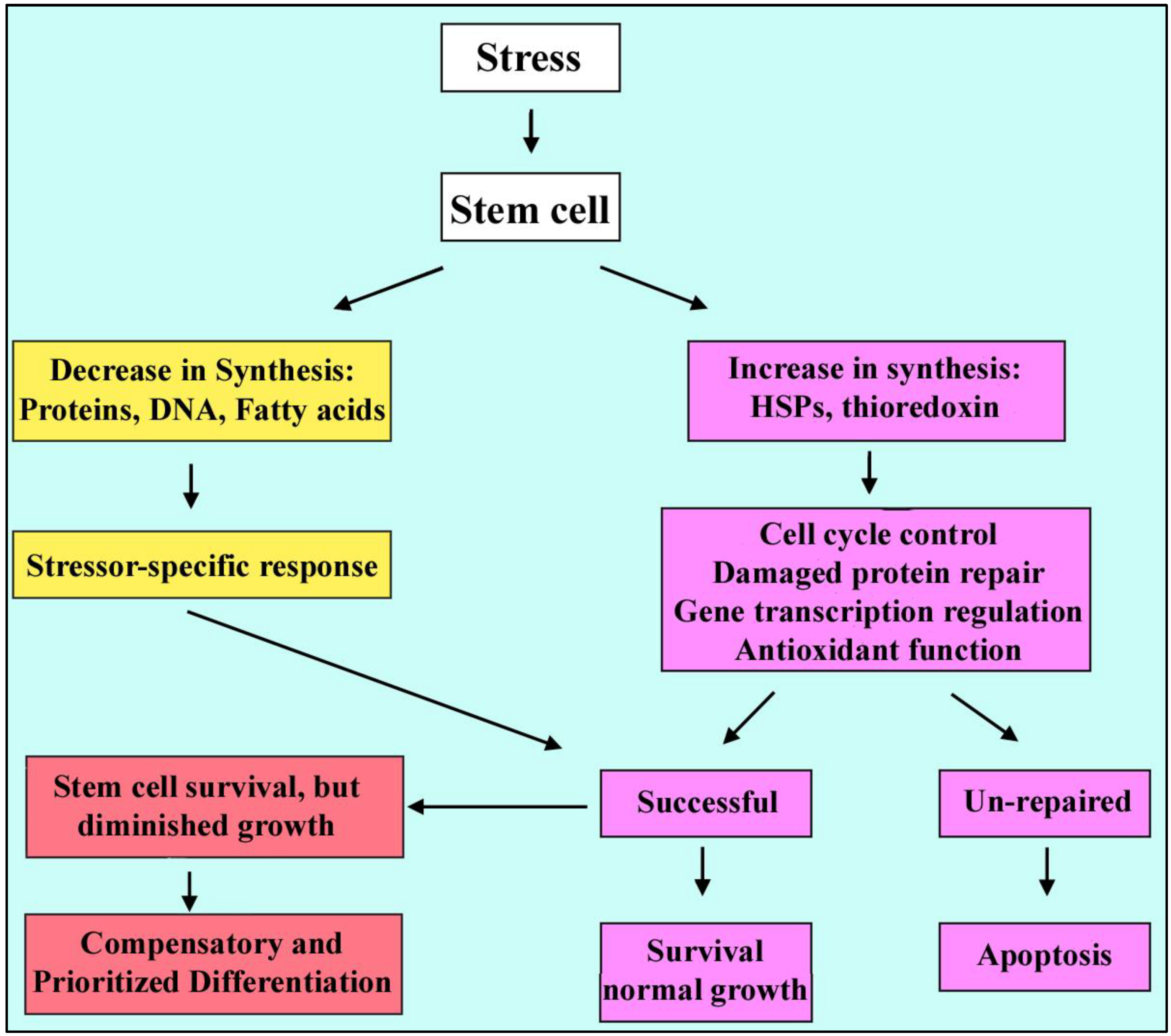
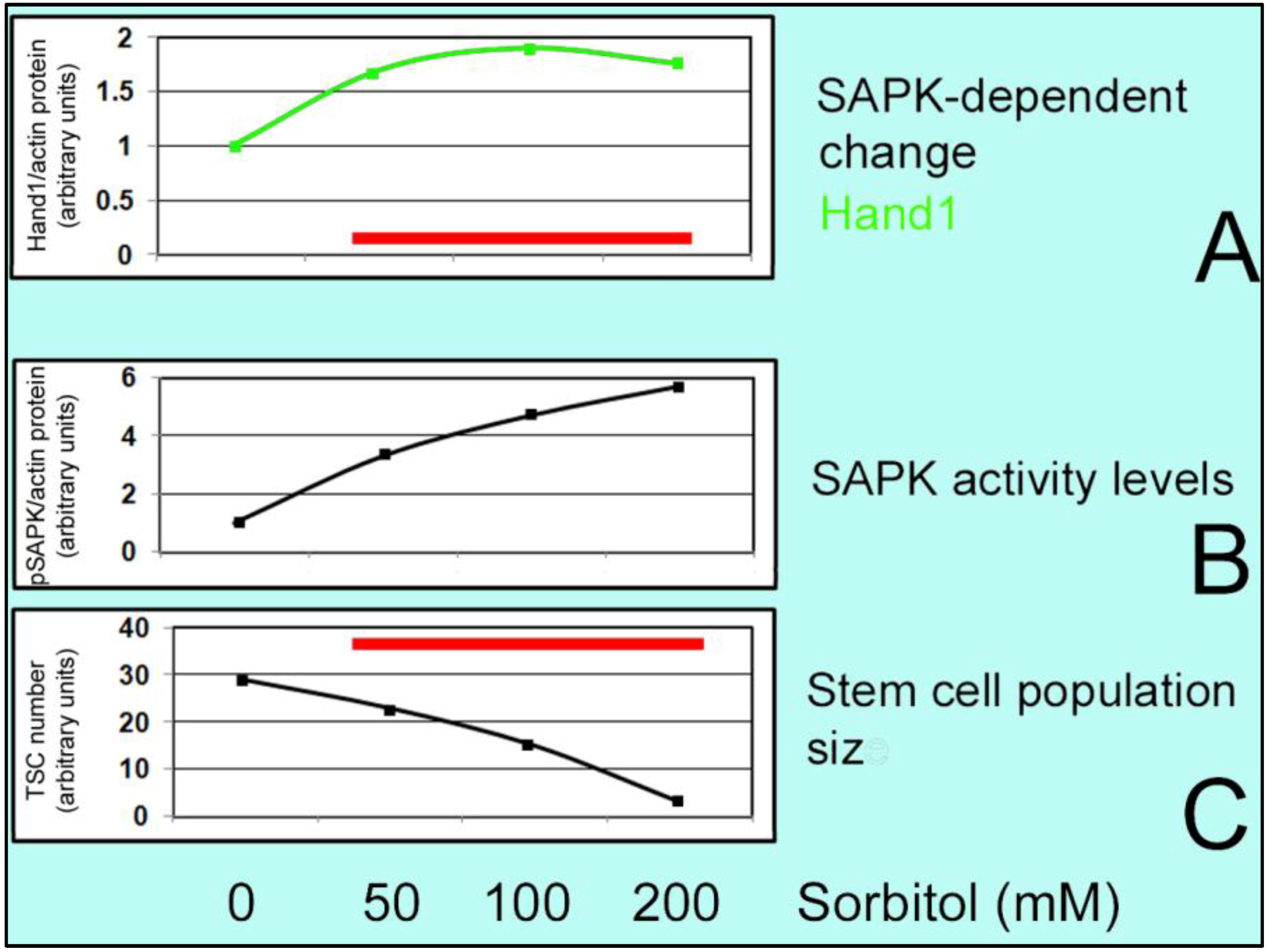
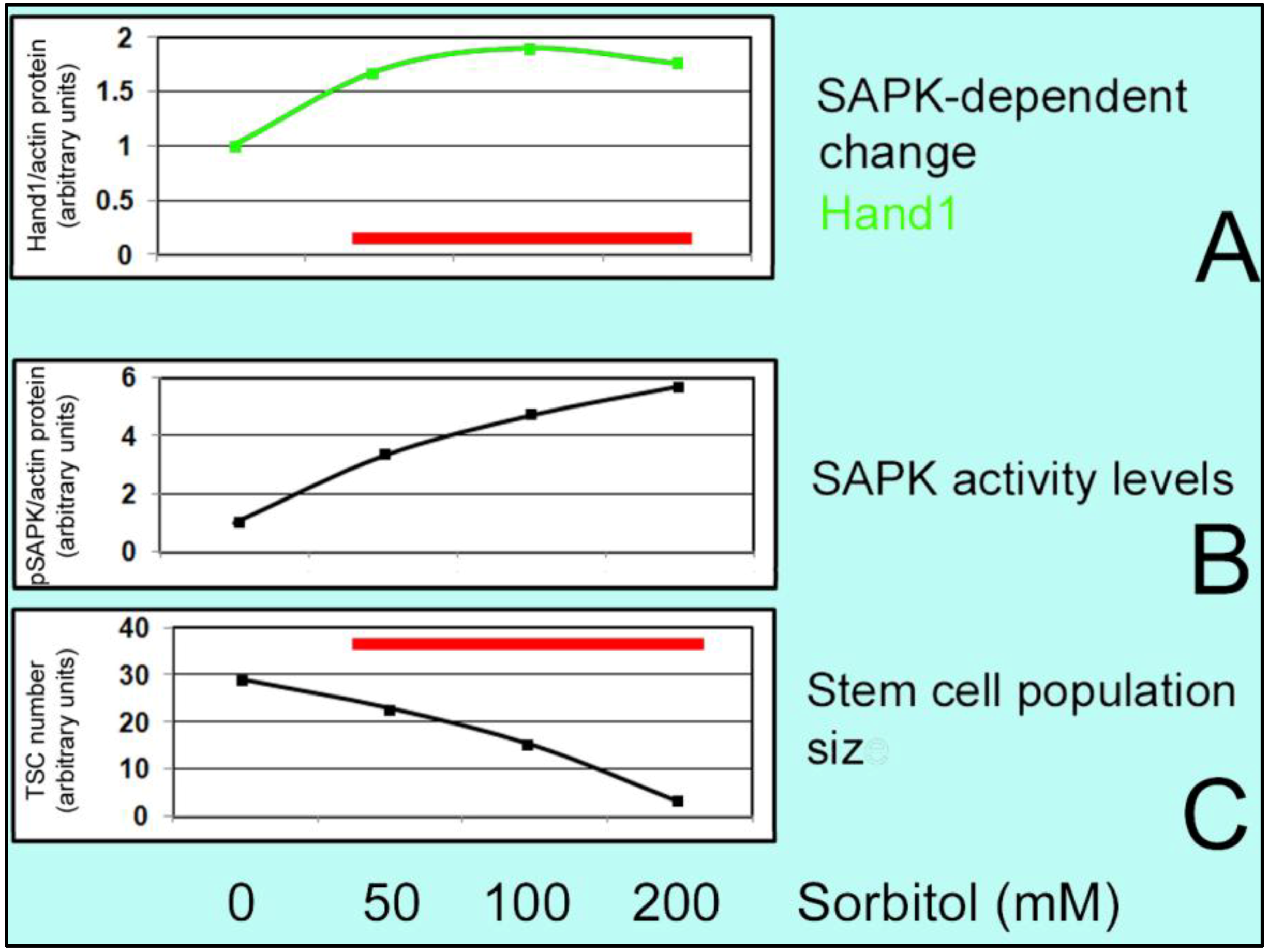
2.4. Stressed Stem Cells

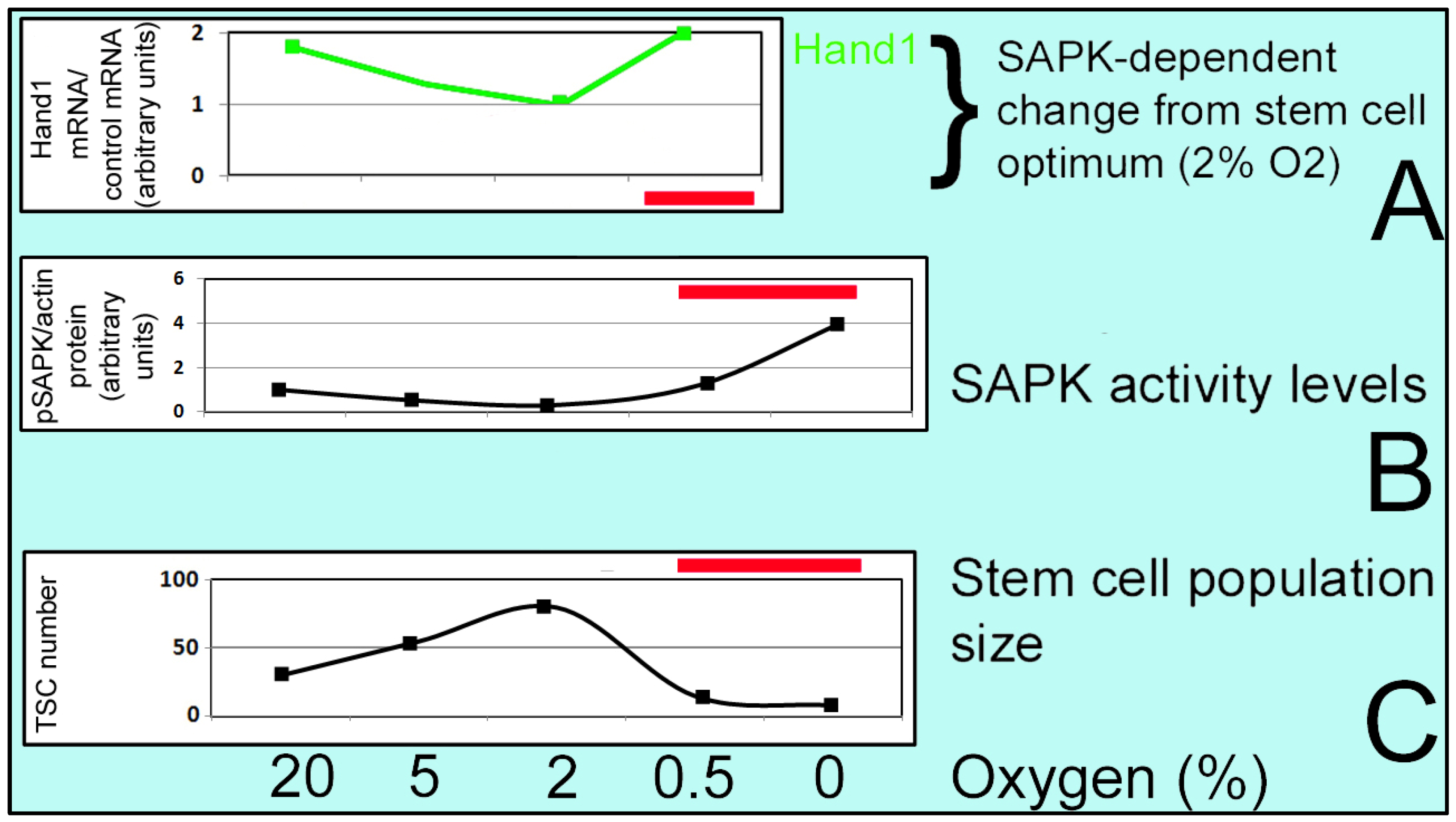
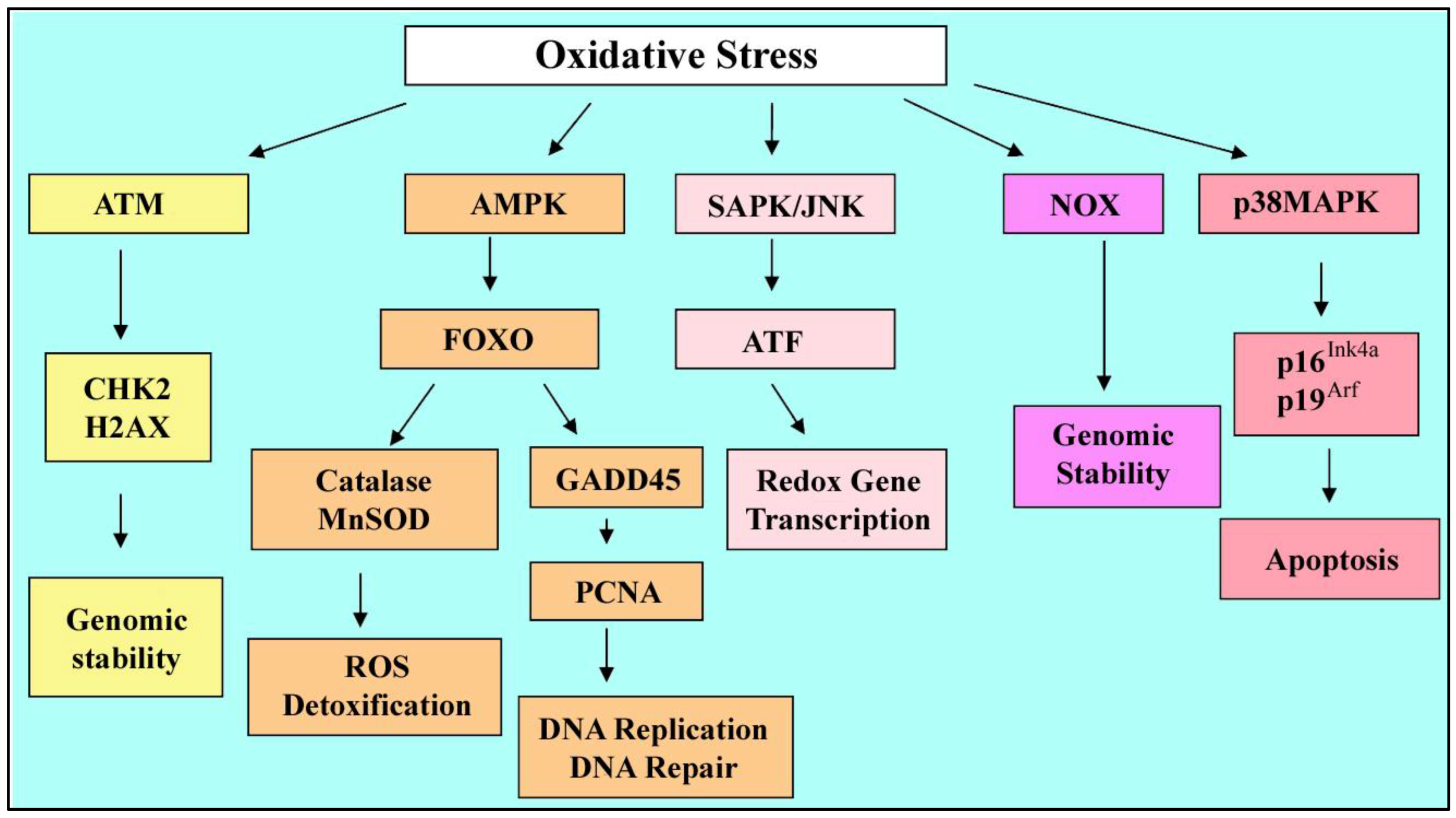
3. Effects of Oxidative Stress on Stem Cells
3.1. Reactive Oxygen Species
3.2. Oxidative Stress Outcomes
3.3. Stressed Stem Cells
3.3.1. Optimal O2 Level in Cultures
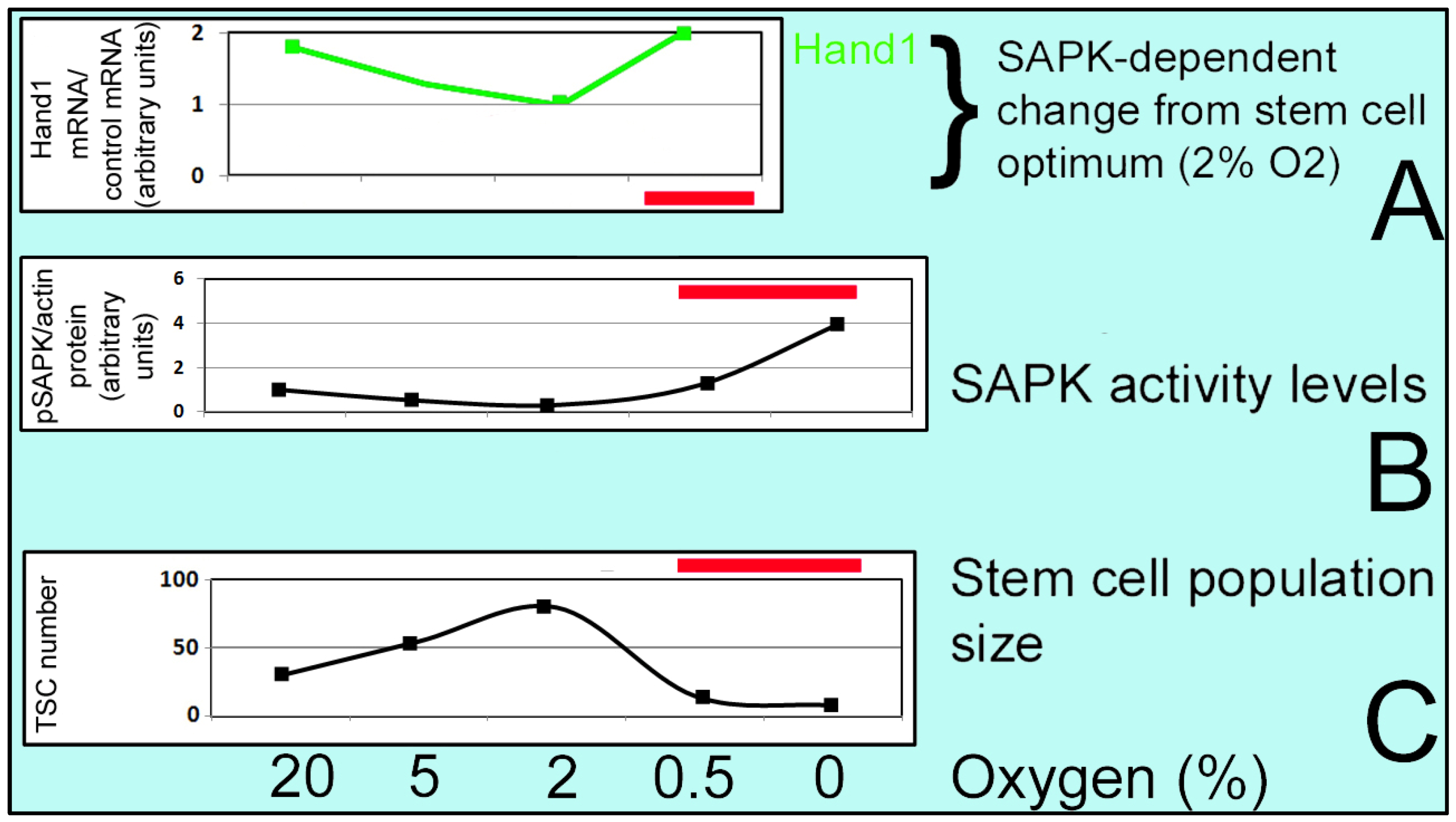
3.3.2. ATP Production in Stem Cells
3.3.3. Diverse Adaptive Responses and Pathways in HSC and ESC
4. Effects of ER Stress on Stem Cells. Prioritized Differentiation is an Adaptive Response
4.1. The Endoplasmic Reticulum and Stress
4.2. ER Stress Response Pathways
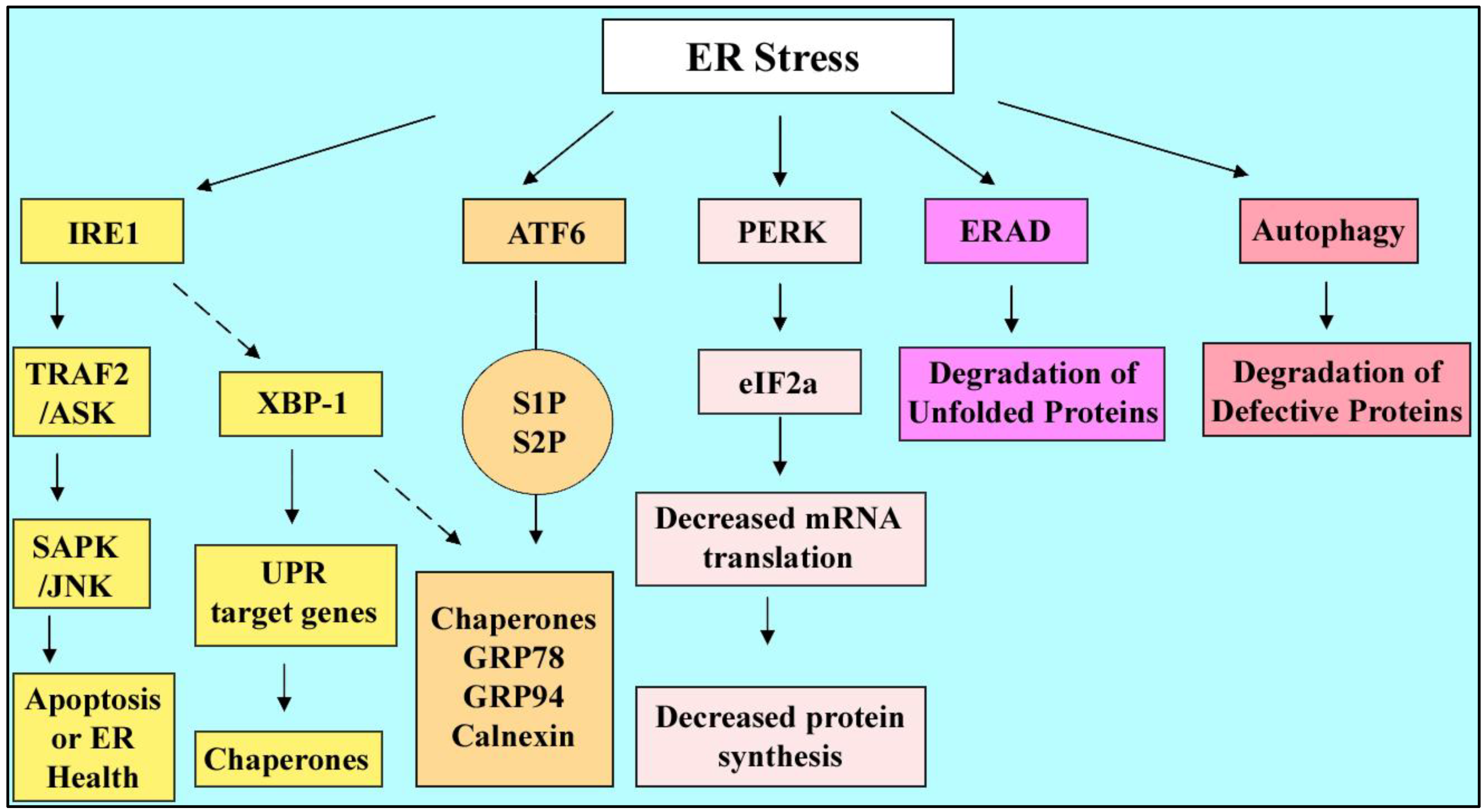
4.3. ER Stress Outcomes: Survival or Death
4.4. ER Stress and Stem Cells
5. Effects of Genotoxic Stress on Stem Cells
6. Effect of Hyperosmolar Stress on Stem Cells
7. Common and Unique Mechanisms and Outcomes of Various Types of Stress
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Woodgett, J.R.; Kyriakis, J.M.; Avruch, J.; Zon, L.I.; Zanke, B.; Templeton, D.J. Reconstitution of novel signalling cascades responding to cellular stresses. Philos Trans. R Soc. Lond B Biol. Sci. 1996, 351, 135–141, discussion 142. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hardie, D.G. Role of the AMP-activated protein kinase in the cellular stress response. Curr. Biol. 1994, 4, 315–324. [Google Scholar] [CrossRef]
- Rappolee, D.A.; Awonuga, A.O.; Puscheck, E.E.; Zhou, S.; Xie, Y. Benzopyrene and experimental stressors cause compensatory differentiation in placental trophoblast stem cells. Syst. Biol. Reprod. Med. 2010, 56, 168–183. [Google Scholar] [CrossRef]
- Zhou, S.; Xie, Y.; Puscheck, E.E.; Rappolee, D.A. Oxygen levels that optimize TSC culture are identified by maximizing growth rates and minimizing stress. Placenta 2011, 32, 475–481. [Google Scholar] [CrossRef]
- Zhong, W.; Xie, Y.; Wang, Y.; Lewis, J.; Trostinskaia, A.; Wang, F.; Puscheck, E.E.; Rappolee, D.A. Use of hyperosmolar stress to measure stress-activated protein kinase activation and function in human HTR cells and mouse trophoblast stem cells. Reprod. Sci. 2007, 14, 534–547. [Google Scholar] [CrossRef]
- Liu, J.; Xu, W.; Sun, T.; Wang, F.; Puscheck, E.; Brigstock, D.; Wang, Q.T.; Davis, R.; Rappolee, D.A. Hyperosmolar Stress Induces Global mRNA Responses in Placental Trophoblast Stem Cells that Emulate Early Post-implantation Differentiation. Placenta 2009, 30, 66–73. [Google Scholar]
- Malhas, A.N.; Vaux, D.J. The nuclear envelope and its involvement in cellular stress responses. Biochem. Soc. Trans. 2011, 39, 1795–1798. [Google Scholar] [CrossRef]
- Cullen, K.E.; Sarge, K.D. Characterization of hypothermia-induced cellular stress response in mouse tissues. J. Biol. Chem. 1997, 272, 1742–1746. [Google Scholar]
- Kasprzak, K.S. Oxidative DNA and protein damage in metal-induced toxicity and carcinogenesis. Free Radic. Biol. Med. 2002, 32, 958–967. [Google Scholar] [CrossRef]
- Farrer, B.T.; Pecoraro, V.L. Heavy-metal complexation by de novo peptide design. Curr. Opin. Drug Discov. Devel. 2002, 5, 937–943. [Google Scholar]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef]
- Cohen, P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol. 1997, 7, 353–361. [Google Scholar] [CrossRef]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest. 2004, 113, 370–378. [Google Scholar]
- Makino, Y.; Okamoto, K.; Yoshikawa, N.; Aoshima, M.; Hirota, K.; Yodoi, J.; Umesono, K.; Makino, I.; Tanaka, H. Thioredoxin: A redox-regulating cellular cofactor for glucocorticoid hormone action. Cross talk between endocrine control of stress response and cellular antioxidant defense system. J. Clin. Invest. 1996, 98, 2469–2477. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Somero, G.N. Biochemical Adaptation: Mechanism and Process in Physiological Evolution; Oxford University Press: Oxford, UK, 2002; p. 466. [Google Scholar] [Green Version]
- van de Water, B.; de Graauw, M.; Le Devedec, S.; Alderliesten, M. Cellular stress responses and molecular mechanisms of nephrotoxicity. Toxicol. Lett. 2006, 162, 83–93. [Google Scholar] [CrossRef]
- Dai, Q.; Zhang, C.; Wu, Y.; McDonough, H.; Whaley, R.A.; Godfrey, V.; Li, H.H.; Madamanchi, N.; Xu, W.; Neckers, L.; Cyr, D.; Patterson, C. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003, 22, 5446–5458. [Google Scholar]
- Nakamura, H. Thioredoxin and its related molecules: update 2005. Antioxid Redox Signal. 2005, 7, 823–828. [Google Scholar] [CrossRef]
- Kultz, D. Molecular and evolutionary basis of the cellular stress response. Annu Rev. Physiol 2005, 67, 225–257. [Google Scholar] [CrossRef]
- Wilmink, G.J.; Roth, C.L.; Ibey, B.L.; Ketchum, N.; Bernhard, J.; Cerna, C.Z.; Roach, W.P. Identification of microRNAs associated with hyperthermia-induced cellular stress response. Cell Stress Chaperones 2010, 15, 1027–1038. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; Rassenti, L.; Alder, H.; Volinia, S.; Liu, C.G.; Kipps, T.J.; Negrini, M.; Croce, C.M. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar]
- Kulshreshtha, R.; Ferracin, M.; Wojcik, S.E.; Garzon, R.; Alder, H.; Agosto-Perez, F.J.; Davuluri, R.; Liu, C.G.; Croce, C.M.; Negrini, M.; Calin, G.A.; Ivan, M. A microRNA signature of hypoxia. Mol. Cell Biol. 2007, 27, 1859–1867. [Google Scholar]
- Calabrese, V.; Mancuso, C.; Sapienza, M.; Puleo, E.; Calafato, S.; Cornelius, C.; Finocchiaro, M.; Mangiameli, A.; Di Mauro, M.; Stella, A.M.; Castellino, P. Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperones 2007, 12, 299–306. [Google Scholar]
- Morano, K.A.; Thiele, D.J. Heat shock factor function and regulation in response to cellular stress, growth, and differentiation signal. Gene Expr. 1999, 7, 271–282. [Google Scholar]
- Kultz, D. Evolution of the cellular stress proteome: From monophyletic origin to ubiquitous function. J. Exp. Biol. 2003, 206, 3119–3124. [Google Scholar] [CrossRef]
- Seetharam, S.; Seidman, M.M. Modulation of ultraviolet light mutational hotspots by cellular stress. J. Mol. Biol. 1992, 228, 1031–1036. [Google Scholar] [CrossRef]
- Ringrose, L.; Paro, R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 2007, 134, 223–232. [Google Scholar] [CrossRef]
- Ringrose, L.; Paro, R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu. Rev. Genet. 2004, 38, 413–443. [Google Scholar] [CrossRef]
- Verbeke, P.; Fonager, J.; Clark, B.F.; Rattan, S.I. Heat shock response and ageing: mechanisms and applications. Cell Biol. Int. 2001, 25, 845–857. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Y.; Liu, X.; Yan, T.; Wu, L.; Alm, E.; Arkin, A.; Thompson, D.K.; Zhou, J. Global transcriptome analysis of the heat shock response of Shewanella oneidensis. J. Bacteriol 2004, 186, 7796–7803. [Google Scholar] [CrossRef]
- Macario, A.J.; Conway de Macario, E. Sick chaperones, cellular stress, and diseas. N Engl. J. Med. 2005, 353, 1489–1501. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: cell survival and cell death. Int J. Cell Biol. 2010, 2010, 214074. [Google Scholar]
- Trosko, J.E. Hierarchical and cybernetic nature of biologic systems and their relevance to homeostatic adaptation to low-level exposures to oxidative stress-inducing agents. Environ. Health Perspect 1998, 106 (Suppl. 1), 331–339. [Google Scholar]
- Tanno, T.; Matsui, W. Development and maintenance of cancer stem cells under chronic inflammation. J. Nihon Med. Sch. 2011, 78, 138–145. [Google Scholar] [CrossRef]
- Saretzki, G.; Armstrong, L.; Leake, A.; Lako, M.; von Zglinicki, T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 2004, 22, 962–971. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Y.; Xiao, Z.; Chen, B.; Wei, Z.; Wang, B.; Zhang, J.; Han, J.; Gao, Y.; Li, L.; Zhao, H.; Zhao, W.; Lin, H.; Dai, J. Alternative translation of OCT4 by an internal ribosome entry site and its novel function in stress response. Stem Cells 2009, 27, 1265–1275. [Google Scholar] [CrossRef]
- Gao, Y.; Wei, J.; Han, J.; Wang, X.; Su, G.; Zhao, Y.; Chen, B.; Xiao, Z.; Cao, J.; Dai, J. The novel function of OCT4B isoform-265 in genotoxic stress. Stem Cells 2012, 30, 665–672. [Google Scholar] [CrossRef]
- Kang, J.; Gemberling, M.; Nakamura, M.; Whitby, F.G.; Handa, H.; Fairbrother, W.G.; Tantin, D. A general mechanism for transcription regulation by Oct1 and Oct4 in response to genotoxic and oxidative stress. Genes Dev. 2009, 23, 208–222. [Google Scholar] [CrossRef]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; von Zglinicki, T.; Lako, M. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef]
- Heyer, B.S.; MacAuley, A.; Behrendtsen, O.; Werb, Z. Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev. 2000, 14, 2072–2084. [Google Scholar]
- Tichy, E.D.; Stambrook, P.J. DNA repair in murine embryonic stem cells and differentiated cells. Exp. Cell Res. 2008, 314, 1929–1936. [Google Scholar] [CrossRef]
- Zhong, W.; Xie, Y.; Abdallah, M.; Awonuga, A.O.; Slater, J.A.; Sipahi, L.; Puscheck, E.E.; Rappolee, D.A. Cellular stress causes reversible, PRKAA1/2-, and proteasome-dependent ID2 protein loss in trophoblast stem cell. Reproduction 2010, 140, 921–930. [Google Scholar] [CrossRef]
- Xie, Y.; Zhong, W.; Wang, Y.; Trostinskaia, A.; Wang, F.; Puscheck, E.E.; Rappolee, D.A. Using hyperosmolar stress to measure biologic and stress-activated protein kinase responses in preimplantation embryos. Mol. Hum. Reprod. 2007, 13, 473–481. [Google Scholar] [CrossRef]
- Xie, Y.; Awonuga, A.O.; Zhou, S.; Puscheck, E.E.; Rappolee, D.A. Interpreting the stress response of early mammalian embryos and their stem cells. Int Rev. Cell Mol. Biol. 2011, 287, 43–95. [Google Scholar] [CrossRef]
- Xie, Y.; Abdallah, M.E.; Awonuga, A.O.; Slater, J.A.; Puscheck, E.E.; Rappolee, D.A. Benzo(a)pyrene causes PRKAA1/2-dependent ID2 loss in trophoblast stem cells. Mol. Reprod. Dev. 2010, 77, 533–539. [Google Scholar] [CrossRef]
- Awonuga, A.O.; Zhong, W.; Abdallah, M.E.; Slater, J.A.; Zhou, S.C.; Xie, Y.F.; Puscheck, E.E.; Rappolee, D.A. Eomesodermin, HAND1, and CSH1 proteins are induced by cellular stress in a stress-activated protein kinase-dependent manner. Mol. Reprod Dev. 2011, 78, 519–528. [Google Scholar] [CrossRef]
- Rappolee, D.A. Impact of transient stress and stress enzymes on development. Dev. Biol 2007, 304, 1–8. [Google Scholar] [CrossRef]
- Rappolee, D.A.; Xie, Y.; Slater, J.A.; Zhou, S.; Puscheck, E.E. Toxic stress prioritizes and imbalances stem cell differentiation: implications for new biomarkers and in vitro toxicology tests. Syst. Biol. Reprod. Med. 2012, 58, 33–40. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 2012, 12, 1–4. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Meth. Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Spasic-Boskovic, O.; Jauniaux, E.; Charnock-Jones, D.S.; Burton, G.J. Nuclear factor-kappa B, p38, and stress-activated protein kinase mitogen-activated protein kinase signaling pathways regulate proinflammatory cytokines and apoptosis in human placental explants in response to oxidative stress: effects of antioxidant vitamin. Am. J. Pathol. 2007, 170, 1511–1520. [Google Scholar] [CrossRef]
- Hool, L.C.; Corry, B. Redox control of calcium channels: From mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2007, 9, 409–435. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Ezashi, T.; Das, P.; Roberts, R.M. Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl Acad Sci. USA 2005, 102, 4783–4788. [Google Scholar]
- Okazaki, K.; Maltepe, E. Oxygen, epigenetics and stem cell fate. Regen Med. 2006, 1, 71–83. [Google Scholar] [CrossRef]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef]
- Eliasson, P.; Jonsson, J.I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J. Cell Physiol. 2010, 222, 17–22. [Google Scholar] [CrossRef]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef]
- Guo, Y.; Einhorn, L.; Kelley, M.; Hirota, K.; Yodoi, J.; Reinbold, R.; Scholer, H.; Ramsey, H.; Hromas, R. Redox regulation of the embryonic stem cell transcription factor oct-4 by thioredoxin. Stem Cells 2004, 22, 259–264. [Google Scholar] [CrossRef]
- Westfall, S.D.; Sachdev, S.; Das, P.; Hearne, L.B.; Hannink, M.; Roberts, R.M.; Ezashi, T. Identification of oxygen-sensitive transcriptional programs in human embryonic stem cells. Stem Cells Dev. 2008, 17, 869–881. [Google Scholar] [CrossRef]
- Matsui, M.; Oshima, M.; Oshima, H.; Takaku, K.; Maruyama, T.; Yodoi, J.; Taketo, M.M. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol. 1996, 178, 179–185. [Google Scholar] [CrossRef]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef]
- Natsuyama, S.; Noda, Y.; Narimoto, K.; Umaoka, Y.; Mori, T. Release of two-cell block by reduction of protein disulfide with thioredoxin from Escherichia coli in mice. J. Reprod. Fertil. 1992, 95, 649–656. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; Ikeda, Y.; Mak, T.W.; Suda, T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar]
- Liu, A.M.; Qu, W.W.; Liu, X.; Qu, C.K. Chromosomal instability inin vitro cultured mouse hematopoietic cells associated with oxidative stress. Am. J. Blood Res. 2012, 2, 71–76. [Google Scholar]
- Maitra, A.; Arking, D.E.; Shivapurkar, N.; Ikeda, M.; Stastny, V.; Kassauei, K.; Sui, G.; Cutler, D.J.; Liu, Y.; Brimble, S.N.; Noaksson, K.; Hyllner, J.; Schulz, T.C.; Zeng, X.; Freed, W.J.; Crook, J.; Abraham, S.; Colman, A.; Sartipy, P.; Matsui, S.; Carpenter, M.; Gazdar, A.F.; Rao, M.; Chakravarti, A. Genomic alterations in cultured human embryonic stem cells. Nat. Genet. 2005, 37, 1099–1103. [Google Scholar]
- Foudah, D.; Redaelli, S.; Donzelli, E.; Bentivegna, A.; Miloso, M.; Dalpra, L.; Tredici, G. Monitoring the genomic stability of in vitro cultured rat bone-marrow-derived mesenchymal stem cells. Chromosome Res. 2009, 17, 1025–1039. [Google Scholar] [CrossRef]
- Rappolee, D.A.; Basilico, C.; Patel, Y.; Werb, Z. Expression and function of FGF-4 in peri-implantation development in mouse embryos. Development 1994, 120, 2259–2269. [Google Scholar]
- Chai, N.; Patel, Y.; Jacobson, K.; McMahon, J.; McMahon, A.; Rappolee, D.A. FGF is an essential regulator of the fifth cell division in preimplantation mouse embryos. Dev. Biol 1998, 198, 105–115. [Google Scholar] [CrossRef]
- Tanaka, S.; Kunath, T.; Hadjantonakis, A.K.; Nagy, A.; Rossant, J. Promotion of trophoblast stem cell proliferation by FGF4. Science 1998, 282, 2072–2075. [Google Scholar] [CrossRef]
- Chung, S.; Arrell, D.K.; Faustino, R.S.; Terzic, A.; Dzeja, P.P. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J. Mol. Cell Cardiol. 2010, 48, 725–734. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; Jung, H.J.; McCaffery, J.M.; Kurland, I.J.; Reue, K.; Lee, W.N.; Koehler, C.M.; Teitell, M.A. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; Suda, T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar]
- Lee, N.; Maurange, C.; Ringrose, L.; Paro, R. Suppression of Polycomb group proteins by JNK signalling induces transdetermination in Drosophila imaginal discs. Nature 2005, 438, 234–237. [Google Scholar]
- de Matos, D.G.; Furnus, C.C. The importance of having high glutathione (GSH) level after bovine in vitro maturation on embryo development effect of beta-mercaptoethanol, cysteine and cystine. Theriogenology 2000, 53, 761–771. [Google Scholar] [CrossRef]
- Naka, K.; Ohmura, M.; Hirao, A. Regulation of the self-renewal ability of tissue stem cells by tumor-related genes. Cancer Biomark 2007, 3, 193–201. [Google Scholar]
- Kang, J.; Shakya, A.; Tantin, D. Stem cells, stress, metabolism and cancer: a drama in two Octs. Trends Biochem Sci. 2009, 34, 491–499. [Google Scholar] [CrossRef]
- Tantin, D.; Schild-Poulter, C.; Wang, V.; Hache, R.J.; Sharp, P.A. The octamer binding transcription factor Oct-1 is a stress sensor. Cancer Res. 2005, 65, 10750–10758. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Tront, J.S.; Sha, X.; Mukherjee, K.; Mohamed-Hadley, A.; Hoffman, B. Gadd45 stress sensors in malignancy and leukemia. Crit. Rev. Oncog. 2011, 16, 129–140. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Yoshida-Araki, K.; Ohta, T.; Ikeda, K.; Motoyama, N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J. Biol. Chem. 2002, 277, 26729–26732. [Google Scholar]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; Chen, C.; Hosokawa, K.; Nakauchi, H.; Nakayama, K.; Nakayama, K.I.; Harada, M.; Motoyama, N.; Suda, T.; Hirao, A. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar]
- Tothova, Z.; Gilliland, D.G. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 2007, 1, 140–152. [Google Scholar]
- Lorimore, S.A.; Chrystal, J.A.; Robinson, J.I.; Coates, P.J.; Wright, E.G. Chromosomal instability in unirradiated hemaopoietic cells induced by macrophages exposed in vivo to ionizing radiation. Cancer Res. 2008, 68, 8122–8126. [Google Scholar] [CrossRef]
- Kim, G.J.; Chandrasekaran, K.; Morgan, W.F. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: A review. Mutagenesis 2006, 21, 361–367. [Google Scholar] [CrossRef]
- Pazhanisamy, S.K.; Li, H.; Wang, Y.; Batinic-Haberle, I.; Zhou, D. NADPH oxidase inhibition attenuates total body irradiation-induced haematopoietic genomic instability. Mutagenesis 2011, 26, 431–435. [Google Scholar] [CrossRef]
- Sabapathy, K.; Klemm, M.; Jaenisch, R.; Wagner, E.F. Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 1997, 16, 6217–6229. [Google Scholar] [CrossRef]
- Coe, J.P.; Rahman, I.; Sphyris, N.; Clarke, A.R.; Harrison, D.J. Glutathione and p53 independently mediate responses against oxidative stress in ES cells. Free Radic. Biol. Med. 2002, 32, 187–196. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar]
- Kim, I.; Shu, C.W.; Xu, W.; Shiau, C.W.; Grant, D.; Vasile, S.; Cosford, N.D.; Reed, J.C. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 2009, 284, 1593–1603. [Google Scholar]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; Liu, Z.; Malyankar, U.M.; Chiovitti, D.; Canny, M.; Durocher, D.; Sicheri, F.; Patterson, J.B. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Winnay, J.N.; Kahn, C.R. PI 3-kinase regulatory subunits as regulators of the unfolded protein response. Meth. Enzymol. 2011, 490, 147–158. [Google Scholar]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 2002, 110, 1383–1388. [Google Scholar]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar]
- Pincus, D.; Chevalier, M.W.; Aragon, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef]
- Merksamer, P.I.; Papa, F.R. The UPR and cell fate at a glance. J. Cell Sci. 2010, 123, 1003–1006. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef]
- Carter, A.M. Placental oxygen consumption. Part I: in vivo studies—A review. Placenta 2000, 21 (Suppl. A), S31–S37. [Google Scholar] [CrossRef]
- Yung, H.W.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 2008, 173, 451–462. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef]
- Katsanou, V.; Milatos, S.; Yiakouvaki, A.; Sgantzis, N.; Kotsoni, A.; Alexiou, M.; Harokopos, V.; Aidinis, V.; Hemberger, M.; Kontoyiannis, D.L. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol. Cell Biol. 2009, 29, 2762–2776. [Google Scholar] [CrossRef]
- Rappolee, D.; Xie, Y.; Slater, J.; Zhou, S.; Puscheck, E. Stress responses at the endometrial-placental interface suppress villous placental differentiation from trophoblast stem cells. Reproduction 2012. in Press. [Google Scholar] [Green Version]
- Witschi, H. Tobacco toxicology revisited. Adv. Exp. Med. Biol. 2001, 500, 471–478. [Google Scholar] [CrossRef]
- Arnould, J.P.; Verhoest, P.; Bach, V.; Libert, J.P.; Belegaud, J. Detection of benzo[a]pyrene-DNA adducts in human placenta and umbilical cord blood. Hum. Exp. Toxicol. 1997, 16, 716–721. [Google Scholar] [CrossRef]
- Zdravkovic, T.; Genbacev, O.; McMaster, M.T.; Fisher, S.J. The adverse effects of maternal smoking on the human placenta: A review. Placenta 2005, 26 (Suppl. A), S81–S86. [Google Scholar]
- Andres, R.L.; Day, M.C. Perinatal complications associated with maternal tobacco use. Semin. Neonatol. 2000, 5, 231–241. [Google Scholar] [CrossRef]
- Wang, X.; Zuckerman, B.; Pearson, C.; Kaufman, G.; Chen, C.; Wang, G.; Niu, T.; Wise, P.H.; Bauchner, H.; Xu, X. Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA 2002, 287, 195–202. [Google Scholar]
- Sanyal, M.K.; Mercan, D.; Belanger, K.; Santella, R.M. DNA adducts in human placenta exposed to ambient environment and passive cigarette smoke during pregnancy. Birth. Defects Res. A Clin. Mol. Teratol. 2007, 79, 289–294. [Google Scholar] [CrossRef]
- Filler, R.; Lew, K.J. Developmental onset of mixed-function oxidase activity in preimplantation mouse embryos. Proc. Natl. Acad. Sci. USA 1981, 78, 6991–6995. [Google Scholar] [CrossRef]
- Drukteinis, J.S.; Medrano, T.; Ablordeppey, E.A.; Kitzman, J.M.; Shiverick, K.T. Benzo[a]pyrene, but not 2,3,7,8-TCDD, induces G2/M cell cycle arrest, p21CIP1 and p53 phosphorylation in human choriocarcinoma JEG-3 cells: A distinct signaling pathway. Placenta 2005, 26 (Suppl. A), S87–S95. [Google Scholar]
- Xie, Y.; Liu, J.; Proteasa, S.; Proteasa, G.; Zhong, W.; Wang, Y.; Wang, F.; Puscheck, E.E.; Rappolee, D.A. Transient stress and stress enzyme responses have practical impacts on parameters of embryo development, from IVF to directed differentiation of stem cells. Mol. Reprod. Dev. 2008, 75, 689–697. [Google Scholar] [CrossRef]
- Du, H.J.; Tang, N.; Liu, B.C.; You, B.R.; Shen, F.H.; Ye, M.; Gao, A.; Huang, C. Benzo[a]pyrene-induced cell cycle progression is through ERKs/cyclin D1 pathway and requires the activation of JNKs and p38 mapk in human diploid lung fibroblasts. Mol. Cell Biochem. 2006, 287, 79–89. [Google Scholar] [CrossRef]
- Li, J.; Tang, M.S.; Liu, B.; Shi, X.; Huang, C. A critical role of PI-3K/Akt/JNKs pathway in benzo[a]pyrene diol-epoxide (B[a]PDE)-induced AP-1 transactivation in mouse epidermal Cl41 cells. Oncogene 2004, 23, 3932–3944. [Google Scholar] [CrossRef]
- Abdallah, M.; Xie, Y.; Puscheck, E.E.; Rappolee, D.A.; Awonuga, A.O. Benzopyrene activates SAPK and induces HAND1 that favors differentiation of trophoblast stem cells. Fertil. Steril. 2009, 92, S136–S137. [Google Scholar]
- Wang, Y.; Puscheck, E.E.; Lewis, J.J.; Trostinskaia, A.B.; Wang, F.; Rappolee, D.A. Increases in phosphorylation of SAPK/JNK and p38MAPK correlate negatively with mouse embryo development after culture in different media. Fertil. Steril. 2005, 83 (Suppl. 1), 1144–1154. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mansouri, L.; Xie, Y.; Rappolee, D.A. Adaptive and Pathogenic Responses to Stress by Stem Cells during Development. Cells 2012, 1, 1197-1224. https://doi.org/10.3390/cells1041197
Mansouri L, Xie Y, Rappolee DA. Adaptive and Pathogenic Responses to Stress by Stem Cells during Development. Cells. 2012; 1(4):1197-1224. https://doi.org/10.3390/cells1041197
Chicago/Turabian StyleMansouri, Ladan, Yufen Xie, and Daniel A. Rappolee. 2012. "Adaptive and Pathogenic Responses to Stress by Stem Cells during Development" Cells 1, no. 4: 1197-1224. https://doi.org/10.3390/cells1041197
APA StyleMansouri, L., Xie, Y., & Rappolee, D. A. (2012). Adaptive and Pathogenic Responses to Stress by Stem Cells during Development. Cells, 1(4), 1197-1224. https://doi.org/10.3390/cells1041197