Intravital Microscopy Reveals Differences in the Kinetics of Endocytic Pathways between Cell Cultures and Live Animals

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
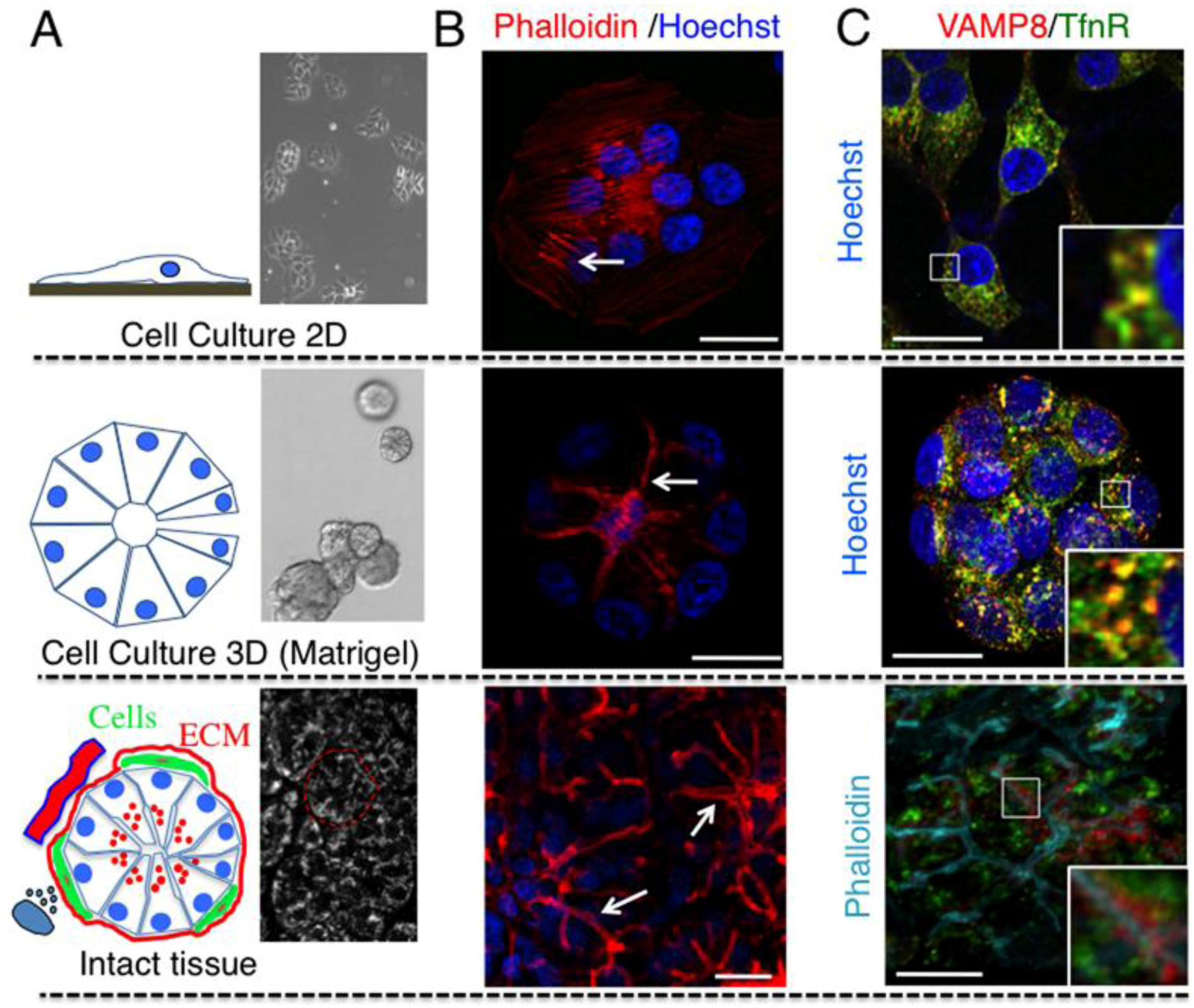
2.1. Cell Architecture and Organization of Endocytic Organelles Differ between Cell Cultures and Fully Developed Tissue
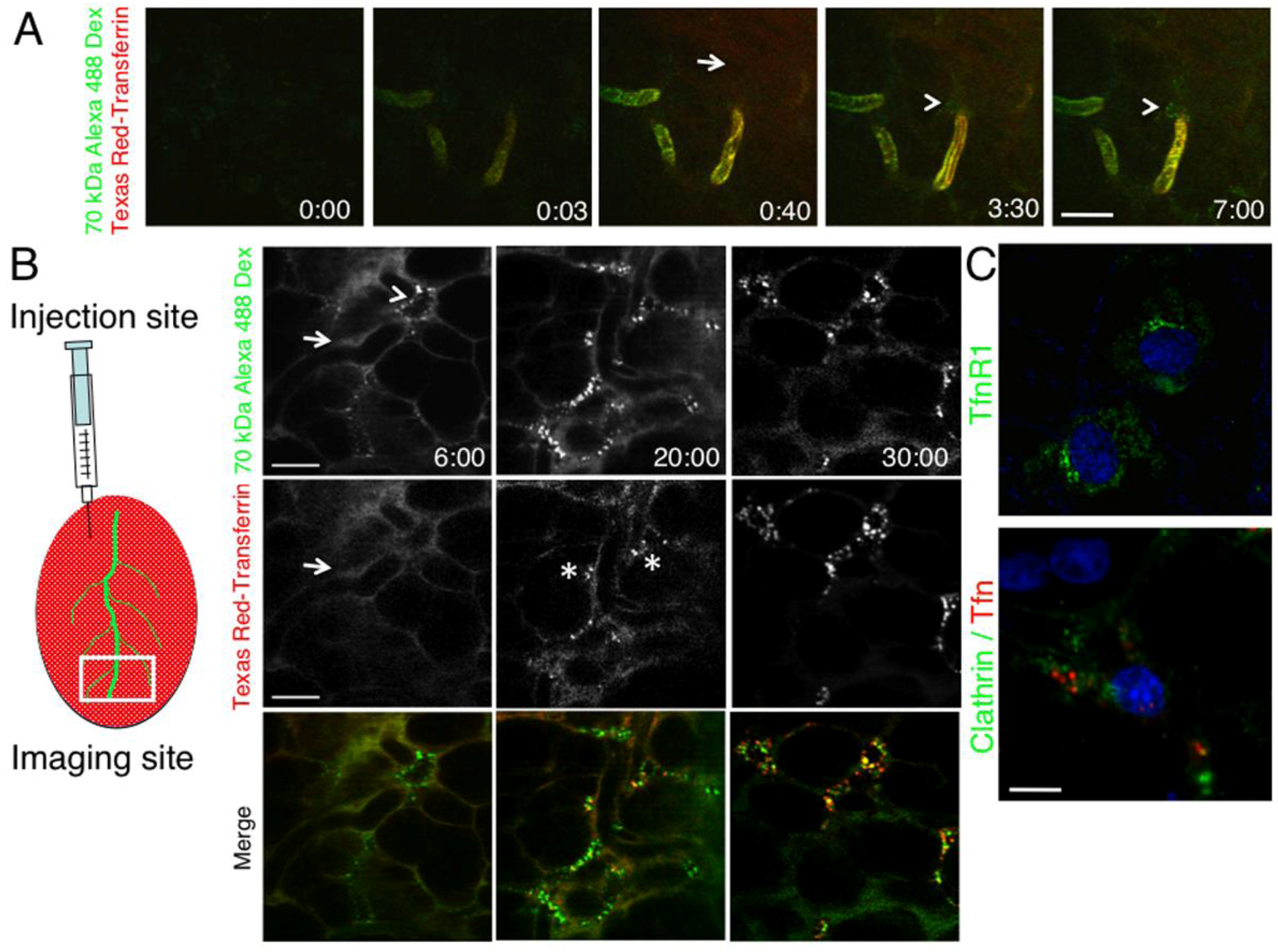
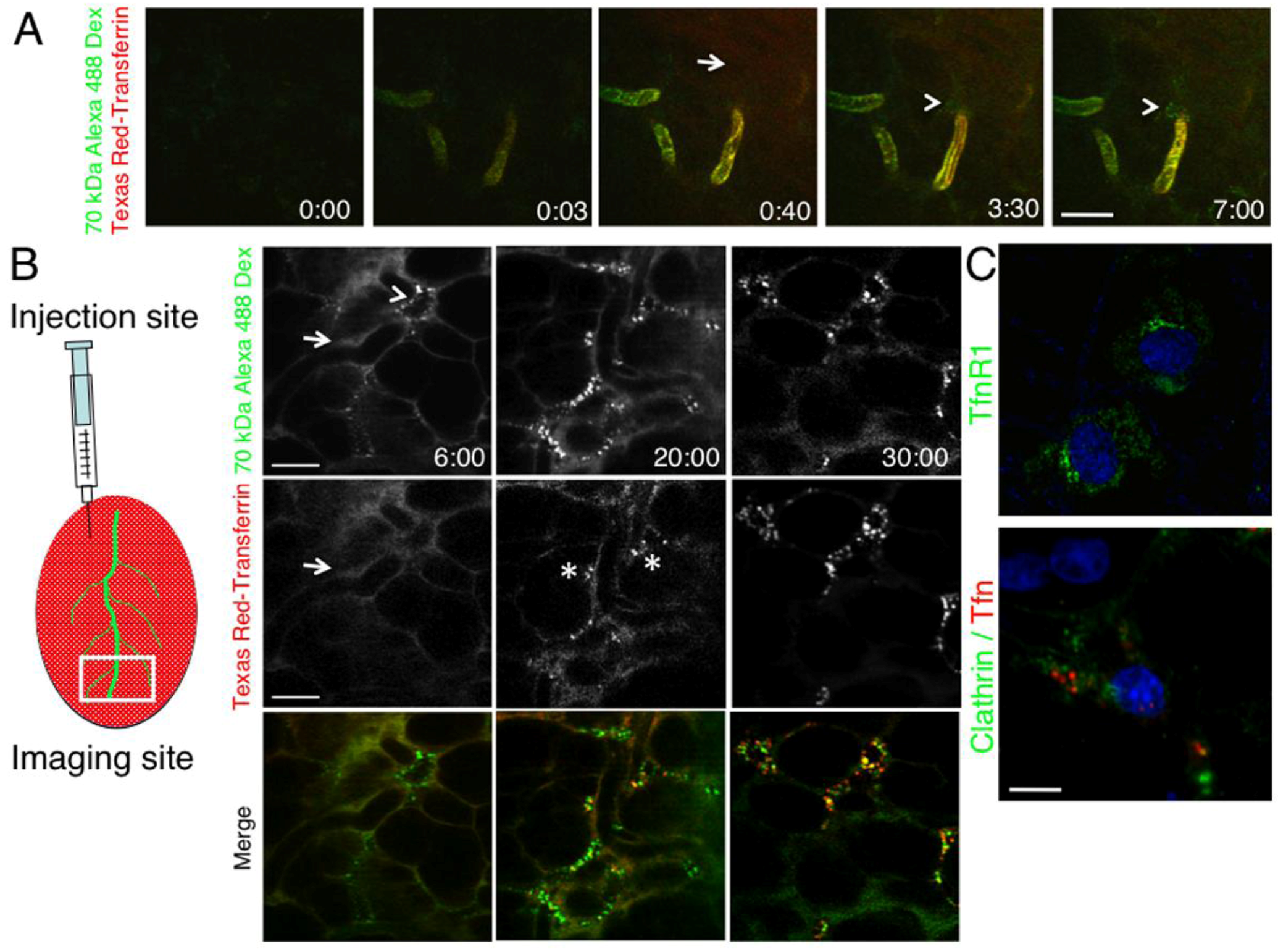
2.2. The Dynamics of Endocytosis in Live Animals Differs from that in Cell Culture


3. Experimental Section
3.1. Fluorescent Probes
3.2. Animal Procedures
3.3. Intravital and Conventional Microscopy
3.4. Image Processing
3.5. Cell Cultures in 2D and 3D
3.6. Whole Mount Immunocytochemistry
3.7. Cell Cultures from Rat Salivary Glands
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Lippincott-Schwartz, J. Bridging structure and process in developmental biology through new imaging technologies. Dev. Cell. 2011, 21, 5–10. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J. Emerging in vivo analyses of cell function using fluorescence imaging (*). Annu. Rev. Biochem. 2011, 80, 327–332. [Google Scholar] [CrossRef]
- Kedrin, D.; Gligorijevic, B.; Wyckoff, J.; Verkhusha, V.V.; Condeelis, J.; Segall, J.E.; van Rheenen, J. Intravital imaging of metastatic behavior through a mammary imaging window. Nat. Methods 2008, 5, 1019–1021. [Google Scholar]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar]
- Ghajar, C.M.; Bissell, M.J. Extracellular matrix control of mammary gland morphogenesis and tumorigenesis: insights from imaging. Histochem. Cell. Biol. 2008, 130, 1105–1118. [Google Scholar] [CrossRef]
- Xu, R.; Boudreau, A.; Bissell, M.J. Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis. Rev. 2009, 28, 167–176. [Google Scholar] [CrossRef]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar]
- Amornphimoltham, P.; Masedunskas, A.; Weigert, R. Intravital microscopy as a tool to study drug delivery in preclinical studies. Adv. Drug Deliv. Rev. 2011, 63, 119–128. [Google Scholar] [CrossRef]
- Ritsma, L.; Ponsioen, B.; van Rheenen, J. Intravital imaging of cell signaling in mice. IntraVital 2012, 1, 2–10. [Google Scholar]
- Weigert, R.; Sramkova, M.; Parente, L.; Amornphimoltham, P.; Masedunskas, A. Intravital microscopy: A novel tool to study cell biology in living animals. Histochem. Cell. Biol. 2010, 133, 481–491. [Google Scholar] [CrossRef]
- Alexander, S.; Koehl, G.E.; Hirschberg, M.; Geissler, E.K.; Friedl, P. Dynamic imaging of cancer growth and invasion: A modified skin-fold chamber model. Histochem. Cell. Biol. 2008, 130, 1147–1154. [Google Scholar] [CrossRef]
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–159. [Google Scholar]
- Pan, F.; Gan, W.B. Two-photon imaging of dendritic spine development in the mouse cortex. Dev. Neurobiol. 2008, 68, 771–778. [Google Scholar] [CrossRef]
- Pittet, M.J.; Weissleder, R. Intravital imaging. Cell 2011, 147, 983–991. [Google Scholar] [CrossRef]
- Svoboda, K.; Yasuda, R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron 2006, 50, 823–839. [Google Scholar] [CrossRef]
- Orth, J.D.; Kohler, R.H.; Foijer, F.; Sorger, P.K.; Weissleder, R.; Mitchison, T.J. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res. 2011, 71, 4608–4616. [Google Scholar] [CrossRef]
- Masedunskas, A.; Weigert, R. Intravital two-photon microscopy for studying the uptake and trafficking of fluorescently conjugated molecules in live rodents. Traffic 2008, 9, 1801–1810. [Google Scholar] [CrossRef]
- Sramkova, M.; Masedunskas, A.; Weigert, R. Plasmid DNA is internalized from the apical plasma membrane of the salivary gland epithelium in live animals. Histochem. Cell. Biol. 2012, 138, 201–213. [Google Scholar] [CrossRef]
- Sramkova, M.; Porat-Shliom, N.; Masedunskas, A.; Wigand, T.; Amornphimoltham, P.; Weigert, R. Salivary Glands: A Powerful Experimental System to Study Cell Biology in Live Animals by. In travital Microscopy. In Current Frontiers and Perspectives in Cell Biology; Najman, S., Ed.; Intech: Rijek,Croatia, 2012. [Google Scholar]
- Masedunskas, A.; Sramkova, M.; Parente, L.; Sales, K.U.; Amornphimoltham, P.; Bugge, T.H.; Weigert, R. Role for the actomyosin complex in regulated exocytosis revealed by intravital microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 13552–13557. [Google Scholar]
- Masedunskas, A.; Milber, O.; Porat-Shliom, N.; Wigand, T.; Amornphimoltham, P.; Weigert, R. Intravital Microscopy: A practical guide on imaging intracellular structures in live animals. Bioarchitecture 2012, 2, 143–157. [Google Scholar]
- Dunn, K.W.; Sandoval, R.M.; Kelly, K.J.; Dagher, P.C.; Tanner, G.A.; Atkinson, S.J.; Bacallao, R.L.; Molitoris, B.A. Functional studies of the kidney of living animals using multicolor two-photon microscopy. Am. J. Physiol. Cell. Physiol. 2002, 283, C905–C916. [Google Scholar]
- Sandoval, R.M.; Kennedy, M.D.; Low, P.S.; Molitoris, B.A. Uptake and trafficking of fluorescent conjugates of folic acid in intact kidney determined using intravital two-photon microscopy. Am. J. Physiol. Cell. Physiol. 2004, 287, C517–C526. [Google Scholar] [CrossRef]
- Masedunskas, A.; Sramkova, M.; Parente, L.; Weigert, R. Intravital Microscopy to Image Membrane Trafficking in Live Rats. Methods Mol. Biol. 2012, 931, 153–167. [Google Scholar]
- Masedunskas, A.; Porat-Shliom, N.; Weigert, R. Regulated exocytosis: Novel insights from intravital microscopy. Traffic 2012, 13, 627–634. [Google Scholar] [CrossRef]
- Masedunskas, A.; Weigert, R. Internalization of fluorescent dextrans in the submandibular salivaryglands of live animals: A study combining intravital two-photon microscopy and second harmonic generation. SPIE 2008, 6860, V68601–V68612. [Google Scholar] [CrossRef]
- Sramkova, M.; Masedunskas, A.; Parente, L.; Molinolo, A.; Weigert, R. Expression of plasmid DNA in the salivary gland epithelium: Novel approaches to study dynamic cellular processes in live animals. Am. J. Physiol. Cell. Physiol. 2009, 297, C1347–C1357. [Google Scholar] [CrossRef]
- Masedunskas, A.; Sramkova, M.; Weigert, R. Homeostasis of the apical plasma membrane during regulated exocytosis in the salivary glands of live rodents. Bioarchitecture 2011, 1, 225–229. [Google Scholar] [CrossRef]
- Wang, S.; Cukierman, E.; Swaim, W.D.; Yamada, K.M.; Baum, B.J. Extracellular matrix protein-induced changes in human salivary epithelial cell organization and proliferation on a model biological substratum. Biomaterials 1999, 11, 1043–1049. [Google Scholar]
- Royce, L.S.; Kibbey, M.C.; Mertz, P.; Kleinman, H.K.; Baum, B.J. Human neoplastic submandibular intercalated duct cells express an acinar phenotype when cultured on a basement membrane matrix. Differentiation 1993, 52, 247–255. [Google Scholar] [CrossRef]
- Hoffman, M.P.; Nomizu, M.; Roque, E.; Lee, S.; Jung, D.W.; Yamada, Y.; Kleinman, H.K. Laminin-1 and laminin-2 G-domain synthetic peptides bind syndecan-1 and are involved in acinar formation of a human submandibular gland cell line. J. Biol. Chem. 1998, 273, 28633–28641. [Google Scholar]
- Hoffman, M.P.; Kibbey, M.C.; Letterio, J.J.; Kleinman, H.K. Role of laminin-1 and TGF-beta 3 in acinar differentiation of a human submandibular gland cell line (HSG). J. Cell. Sci. 1996, 109, 2013–2021. [Google Scholar]
- Cukierman, E.; Pankov, R.; Stevens, D.R.; Yamada, K.M. Taking cell-matrix adhesions to the third dimension. Science 2001, 294, 1708–1712. [Google Scholar] [CrossRef]
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 2010, 1820, 264–281. [Google Scholar]
- Thorn, P.; Gaisano, H. Molecular control of compound Exocytosis: A key role for VAMP8. Commun. Integr. Biol. 2012, 5, 61–63. [Google Scholar] [CrossRef]
- Zhu, D.; Zhang, Y.; Lam, P.P.; Dolai, S.; Liu, Y.; Cai, E.P.; Choi, D.; Schroer, S.A.; Kang, Y.; Allister, E.M.; et al. Dual Role of VAMP8 in Regulating Insulin Exocytosis and Islet beta Cell Growth. Cell. Metab. 2012, 16, 238–249. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Porat-Shliom, N.; Cohen, L.A. Clathrin-independent endocytosis: A unique platform for cell signaling and PM remodeling. Cell. Signal. 2009, 21, 1–6. [Google Scholar] [CrossRef]
- Sandvig, K.; Pust, S.; Skotland, T.; van Deurs, B. Clathrin-independent endocytosis: Mechanisms and function. Curr. Opin. Cell. Biol. 2011, 23, 413–420. [Google Scholar] [CrossRef]
- Morgan, E.H. Transferrin and albumin distribution and turnover in the rat. Am. J. Physiol. 1966, 211, 1486–1494. [Google Scholar]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Characterization of a nonclathrin endocytic pathway: Membrane cargo and lipid requirements. Mol. Biol. Cell. 2004, 15, 3542–3552. [Google Scholar] [CrossRef]
- Rothstein, E.C.; Nauman, M.; Chesnick, S.; Balaban, R.S. Multi-photon excitation microscopy in intact animals. J. Microsc. 2006, 222, 58–64. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Masedunskas, A.; Porat-Shliom, N.; Rechache, K.; Aye, M.-P.; Weigert, R. Intravital Microscopy Reveals Differences in the Kinetics of Endocytic Pathways between Cell Cultures and Live Animals. Cells 2012, 1, 1121-1132. https://doi.org/10.3390/cells1041121
Masedunskas A, Porat-Shliom N, Rechache K, Aye M-P, Weigert R. Intravital Microscopy Reveals Differences in the Kinetics of Endocytic Pathways between Cell Cultures and Live Animals. Cells. 2012; 1(4):1121-1132. https://doi.org/10.3390/cells1041121
Chicago/Turabian StyleMasedunskas, Andrius, Natalie Porat-Shliom, Kamil Rechache, Myo-Pale' Aye, and Roberto Weigert. 2012. "Intravital Microscopy Reveals Differences in the Kinetics of Endocytic Pathways between Cell Cultures and Live Animals" Cells 1, no. 4: 1121-1132. https://doi.org/10.3390/cells1041121