Abstract
Autophagy is a housekeeping survival mechanism with a protective function against stress conditions. However, when stress severity or duration increases, it may promote cell death. Paradoxically, autophagy favors cancer development, since cancer cells could enhance their proliferation potential (thus becoming able to resist anticancer therapy) thanks to the energetic supply provided by organelle degradation typically driven by autophagy following a stepwise pathway. The main actors of the autophagic machinery as well as the features shared with apoptosis will be described. Special attention will be paid to the effects of autophagy manipulation.
1. Introduction
Cancer is one of the multifactorial and multistep complex disorders that accounts for a major cause of death all over the world, accounting 7.6 million deaths (around 13% of all deaths) in 2008 [1]; it is characterized by uncontrolled proliferation of abnormal cells that ends with the formation of a tumor. During tumor development, cancer cells can acquire many features such as sustained proliferative signaling (active oncogenes), evasion of growth suppressor functions, invasion of healthy tissues due to metastatic potential, replicative immortality, angiogenesis stimulation and resistance to cell death induced by chemotherapeutic agents [2,3]. For decades, the scientific community has been working to understand not only the molecular mechanisms at the basis of the uncontrolled proliferation of cancer cells, but also how these cells become insensitive to internal/external stimuli promoting cell death. Drug resistance of cancer cells is often correlated to an impaired activation of Programmed Cell Death (PCD), mainly occurring through the apoptotic pathway(s); accordingly, it has been assumed for a long time that the re-activation of apoptosis could be sufficient to promote the eradication of cancer cells [4,5]. Classical apoptosis implies the activation of caspases, which are in charge for extensive protein degradation [6]; this event could be also triggered by the release of proteolytic enzymes from lysosomes (lysosomal-mediated cell death) [7]. Moreover, necroptosis, “an ordered cellular explosion”, represents a cell death mechanism with morphological features resembling necrosis [8]. The scenario is even more complicated, given that a housekeeping process, i.e., autophagy, which regulates physiological functions, could also promote cancer cell survival, as illustrated below [9,10,11].
2. Main Features of Autophagy
The term Autophagy comes from the Greek words αύτος (autos) and φαγέω (fageo), which means “self-eating”, a catabolic self-degradation process for maintaining normal cell homeostasis to ensure the regular turnover of cellular components [12]. Four types of autophagy have been described in mammals: Micro-autophagy (MicroA), Chaperone-mediated autophagy (CMA), Macro-autophagy (MacroA) and Alternative Macro-Autophagy (AMA) [13]. Autophagy can selectively target organelles such as mitochondria (Mitophagy), ribosomes (Ribophagy), peroxisomes (Pexophagy), and endoplasmic reticulum (ER; Reticulophagy), thus contributing to their turnover (reviewed in [14]).
Autophagy is tightly regulated by a limited number of highly conserved genes called ATG (AuTophaGy related genes) that were first identified in Saccharomyces cerevisiae [15,16,17]. This finding facilitated the discovery of mammalian orthologues and the further definition of the autophagic machinery in other organisms [18]. Autophagy is considered as a survival mechanism, having a protective function in many cellular stress conditions [19,20,21], through the ability to counteract nutrient deprivation by recycling energy originated from macromolecule degradation. In case of prolonged starvation conditions, cells “eat” part of their own cytoplasmic components to compensate the lack of metabolites needed to synthesize essential molecules [22]. However, when stress severity or duration is extended, autophagy may participate in cell death such as type II PCD [23].
3. Execution of Autophagy
The key event in autophagy is the formation of autophagosome and autolysosome, a process that requires several sequential steps illustrated in Figure 1.
The first event is nucleation (1), where a double-membrane structure called Phagophore is formed, which derives mainly from endoplasmic reticulum, Golgi, endosomes and even mitochondria and plasma membrane [24]. In this initial step, an ubiquitin-like system regulates the formation of ATG5-ATG12 heterodimer (Figure 2), which, in the presence of an ATG16 homodimer, forms a protein complex and associates to the Phagophore Assembly Site (PAS) [24].
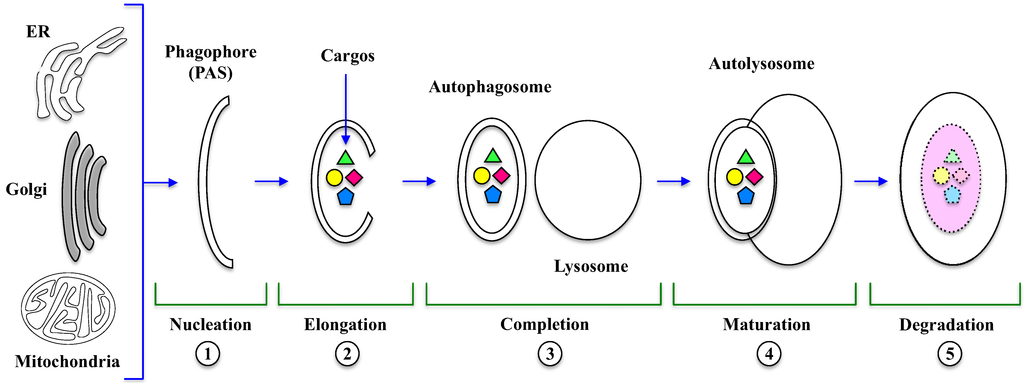
Figure 1.
Autophagosome and autolysosome formation. Several morphological changes occur during autophagy, which is stepwise regulated. In the Nucleation (1) and Elongation (2) steps, phagophore originates from membranes of organelles (ER, Golgi, mitochondria) and then encloses the cytosolic cargos, including long-lived, misfolded proteins and damaged organelles, leading to the formation of the autophagosome Completion (3). The Maturation (4) step consists in the fusion of autophagosome with lysosome to form the autolysosome. Finally, during degradation (5), lysosomal hydrolases digest autolysosomal content and release products in the cytosol. PAS: Phagophore Assembly Site; ER: endoplasmic reticulum.
In the next step, PAS expands by direct flow from a source (e.g., ER) and then seals to enclose the cytosolic cargos like long-lived, misfolded proteins and damaged organelles [12]. Ubiquitin-like (Ubl) conjugation systems are involved in vesicle elongation (2) and authophagosomal membrane completion (3). The mammalian orthologue of yeast ATG8, called LC3-I (Microtubule-Associated Protein Light Chain 3) is conjugated to the lipid PhosphatidylEthanolamine (PE), whereas ATG12 is conjugated to ATG5 [25]. At this stage, LC3-I is first cleaved, then lipidated to form LC3-II, which is incorporated into the nascent structure; for this reason, the presence of LC3-II is the most specific marker for autophagosome formation and, more in general, for autophagy occurrence. Then, the autophagosome fuses with the lysosome to form the autolysosome in a process called maturation (4) [24,25], controlled by cytoskeleton and lysosome membrane proteins [26].
The autophagosome conversion into autolysosomes can be blocked by edazol, a drug that specifically target microtubules [27]. The final step is the degradation (5) of autolysosomal content by lysosomal hydrolases that metabolize lipids, sugars, proteins and nucleotides; the degradation products are released in the cytoplasm and can be reutilized or become an energy source [24,25,28].
The entire process is tightly regulated by a cascade of kinases (Figure 2). Mitogen-Activated Protein Kinases (MAPKs), including ERK (Extracellular Signal-Related Kinase) 1/2, p38 and JNK (c-Jun N-terminal Kinase), play a fundamental role in governing the key negative regulator of autophagy mTOR (mammalian Target Of Rapamycin), a conserved Ser/Thr kinase [29,30]. DAPK (Death-Associated Protein Kinase), PI3K/AKT (Phosphatidylinositol 3-kinase/Protein Kinase B) and p53/AMPK (AMP-Activated Protein Kinase) signaling pathway also mediate the induction of autophagy through the modulation of mTOR [9,10,25,29,31,32].
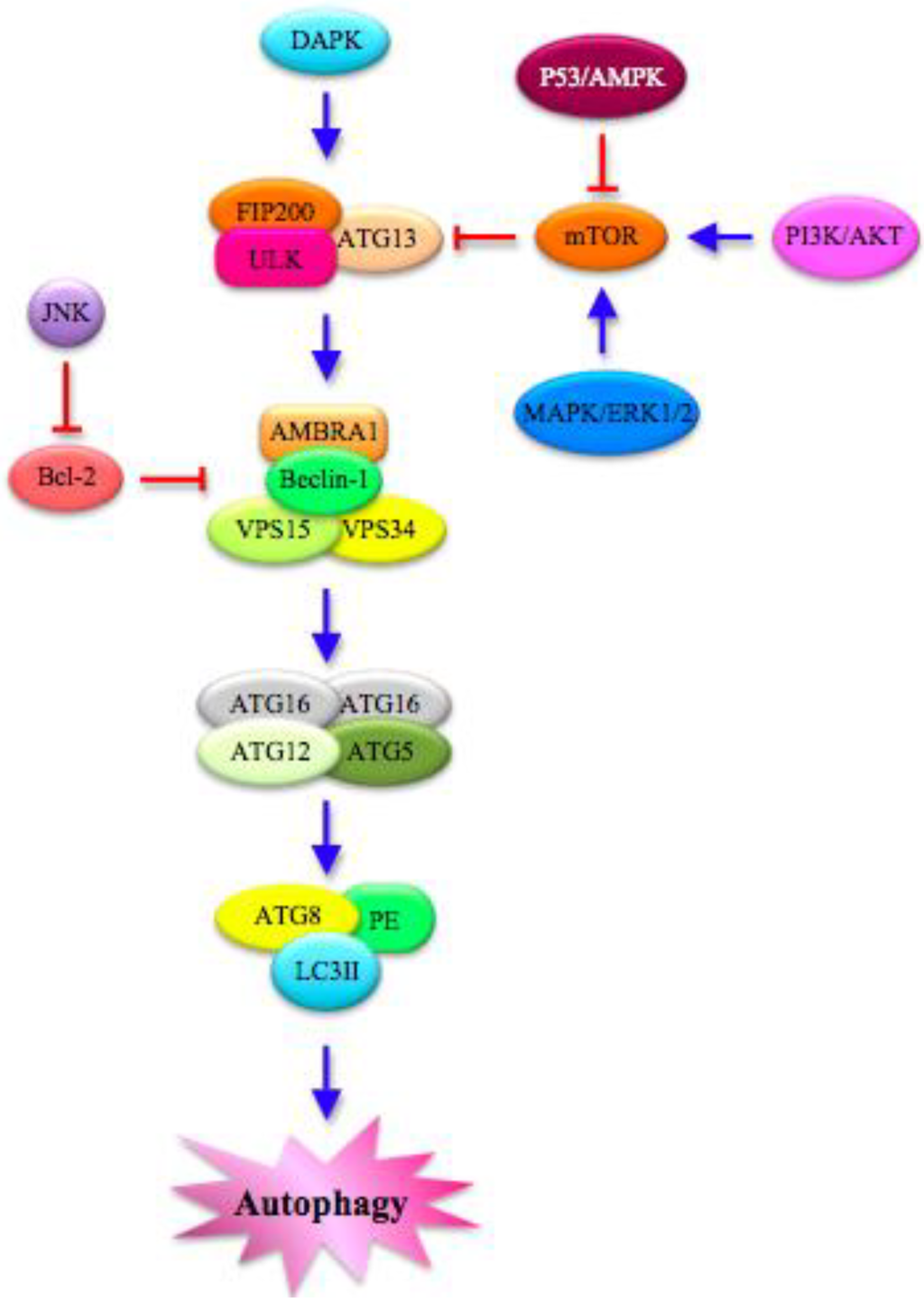
Figure 2.
Autophagy pathway: Molecular features. Several proteins in the cytoplasm interact to regulate autophagy. The different steps of the process are modulated by MAPKs (Mitogen-Activated Protein Kinases), such as ERK (Extracellular Signal-Related Kinase) 1/2 and JNK (c-Jun N-terminal Kinase), which mainly act on the key negative regulator of autophagy mTOR (mammalian Target OF Rapamycin). mTOR is also controlled directly by PI3K/AKT (PhosphatidylInositol 3-Kinase/Protein Kinase B) and p53/AMPK (AMP-Activated Protein Kinase) signaling pathways. Moreover, DAPK (Death-Associated Protein Kinase) is implicated in the autophagic cascade, which starts with the formation of ULK (Unc-51-Like Kinase) complex, which is composed of FAK (Focal Adhesion Kinase)-family Interacting Protein of 200 kDa (FIP200), ULK and ATG13. In turn, ULK complex phosphorylates AMBRA1 (Activating Molecule in Beclin-1-Regulated Autophagy), leading to the activation of a complex that includes P-AMBRA1, Beclin-1, VPS (Vacuolar Protein Sorting) 15 and 34. The final steps are characterized by the assembly of ATG complexes made by ATG factors and autophagosome proteins. PE: Phosphatidyl Ethanolamine; LC3II: lipidated form of Microtubule-Associated Protein Light Chain 3.
Under stress conditions, mTOR is inactivated, thus allowing autophagy to start through the formation of the ULK complex (Figure 2), composed of FAK (Focal Adhesion Kinase) -family Interacting Protein of 200 kDa (FIP200), Unc-51-Like Kinase (ULK) and ATG13 [33]. In the proximity of the phagophore, a multimeric PI3K (PhosphatidylInositol 3-kinase) complex is also formed, which is controlled positively by UV radiation Resistance-Associated Gene (UVRAG) and negatively by Rubicon [34]. This complex includes Beclin-1 (released at the ER level), Vacuolar Protein Sorting (VPS) 15, VPS34 and Activating Molecule in Beclin-1-Regulated Autophagy (AMBRA1) [34]. Beclin-1, a member of the Bcl-2 family and the mammalian homolog of the yeast ATG6 gene, is positively regulated by AMBRA1, which is phosphorylated and released from the dynein motor complex during autophagy initiation [35]. In addition, the dissociation of the Beclin-1/Bcl-2 complex can be promoted by p53 as an apoptotic response inhibiting the mTOR signal [36].
4. The Paradoxical Role of Autophagy in Cancer
A multitude of internal and external stimuli can persuade a healthy cell to become malignant. Once this process is activated, a series of biochemical events drive an uncontrolled proliferation status. The new progeny of transformed cells first has to evade cell death in order to sustain chronic proliferation and consolidate the tumor microenvironment. Autophagy deregulation is prevalent in many cancers and involves several autophagic genes or proteins (reviewed in [36,37]). For instance, in 40%–75% of human breast, ovarian, and prostate cancers the Beclin-1 gene is monoallelically deleted [38]. In gastric and colorectal cancers, UVRAG and others ATG genes, showed frameshift mutations [39,40,41]. Conversely, the autophagic marker LC3 is highly expressed in more than 50% of human gastric cancers [42]. Table 1 reports representative examples of the altered status of autophagic factors.

Table 1.
Deregulation of some autophagic factors in human cancers.
| Gene | Mutation | Cancer | Reference |
|---|---|---|---|
| Beclin-1 | allele deletion decreased expression | breast, ovarian, prostatic liver | [38,43,44] |
| LC3 | increased expression downregulation | gastric, esophageal melanoma | [42,45] |
| UVRAG | frameshift mutations | gastric | [39] |
| ATG8 | increased expression | colorectal | [40] |
| ATG2B, ATG5, ATG9B | frameshift mutations | colorectal, gastric | [41] |
Even in cancer cells, as it occurs during normal homeostasis control, autophagy senses stress signals and promotes the lysosomal degradation of organelles and proteins. The impact of autophagy in the yet complex network governing cancer progression could occur at different levels: (i) counteracting genome instability, thus impairing malignant transformation; (ii) protecting cancer cells from unfavorable conditions, thus promoting tumorigenesis [9,10].
(i) Autophagy can act as a tumor suppressor by removing damaged organelles and growth factors, and by facing chromosomal instability. In this respect, the autophagy factors Beclin-1 and Atg5 are considered as ‘guardians’ of the cellular genome. Mathew et al. [46] demonstrated that immortalized epithelial cells with loss of Beclin-1 or Atg5 have increased DNA damage, gene amplification and aneuploidy, in parallel with enhanced tumorigenicity. In addition, they found that defective autophagy (Beclin-1+/− and Atg5−/−) in immortalized Baby Mouse Kidney (iBMK) cells caused the accumulation of p62 protein aggregates, damaged mitochondria and misfolded proteins, driving the production of Reactive Oxygen Species (ROS) [47]. The active role of UVRAG in maintaining genomic stability has been demonstrated in UVRAG depleted cells, which were affected in centrosome stability, chromosome segregation and spindle formation [48]. This body of evidence suggests that appropriate protein quality control by autophagy contributes to contrast tumorigenesis.
(ii) Paradoxically, the cytoprotective role generally played by autophagy can be harmful in cancer cells, as it can help them resist anticancer therapy [49,50]. In fact, autophagy not only provides energy for cell division, but also has a role in eliminating damage caused by tumor microenvironment and anticancer therapies (reviewed in [51]). Indeed, several reports showed that hypoxia, a common condition in solid tumors, activates autophagy in cancer cells [52,53]; analogously, extracellular pH changes occurring during cancer development modulate autophagy [54]. Under these non-physiological conditions, autophagy responds by degrading damaged organelles, DNA and proteins and providing cancer cells with new energy, useful to sustain their proliferation. Furthermore, autophagy has been reported to increase cell survival during anoikis, which is the first step in the process of cancer cell metastatization, thus giving an advantage to migrating dangerous cells [55]. The relevance of autophagy for cell invasion is supported by the observation that the knockdown of Atg12, and the consequent inhibition of the autophagic machinery, decreases the invasiveness of glioma cells [56]. For the above reasons, autophagy could exert a dangerous function in cancer.
A further level of complexity is added by the role of p53, which, in addition to the direct control of DNA repair, also tunes the autophagic burst acting as a “rheostat” that continuously adjusts the rate of autophagy with the final aim of serve as an anticancer mechanism [57]. To do this job, p53 downregulates post-transcriptionally the autophagic protein LC3, thus keeping autophagic flux at sustainable level and avoiding excessive autophagy, potentially favorable to abnormal cancer cell proliferation [58]. However, depending on genetic and epigenetic features (e.g., p53 mutations or inactivation), the oncosuppressor effect of p53 could be abolished, rendering autophagy free from oncosuppressor control.
5. Autophagy Crosses Apoptosis
The adverse phenomenon of drug resistance often occurring in cancer cells has been correlated to an alteration of PCD machinery, mainly of apoptosis [4,5]. However, it has recently been shown that a housekeeping process, i.e., autophagy, could modulate cancer cell survival [11]. Apoptosis and autophagy are not independent processes, given that a complex crosstalk between them has been depicted, leading to the notion that they can be triggered by common upstream signals and share molecular switches [9,10,59].
Proteins that are central components of apoptosis or autophagy machinery can regulate both processes directly. Beclin-1 and Bcl-2 family members represent the best example: Bcl-2 and Bcl-XL inhibit Beclin-1 by binding to it through the Beclin-1 BH3 domain [60,61], which is atypical, lacking a hydrophobic aminoacid at position 119 (which corresponds to the polar Thr) [60,62]. This feature lowers Beclin-1 affinity for Bcl-2 compared to other BH3-containing proteins. When autophagy is essential for cell survival, the association between Bcl-2 and Beclin-1 decreases, thanks to the action of JNK-1 on Bcl-2, thus allowing Bcl-2 dissociation from Beclin-1 and autophagy promotion. Phosphorylated Bcl-2 is now free to bind the pro-apoptotic protein Bax, thus inhibiting apoptosis, which, in such context, would be promoted by loss of nutrients [63,64]. However, under extreme conditions, JNK1 hyper-phosphorylates Bcl-2, which detaches from Bax, thus facilitating apoptosis and consequently a safe cell death [63,64]. Given that the affinity of phosphorylated Bcl-2 toward pro-apoptotic proteins is higher than toward Beclin-1, a model has been hypothesized to explain Bcl-2 binding properties [63,64]. Analogously, DAPK phosphorylates Beclin-1 on Thr119, thus promoting its dissociation from Bcl-2, and autophagy activation [65]; DAPK is also involved in apoptotic bleb formation thanks to its interplay with cytoskeletal factors [66].
ERK is a kinase implicated in both apoptosis and autophagy [67,68]. ERK pathway plays a critical role in promoting apoptosis in response to several stress stimuli, both intrinsic and extrinsic. In addition, when ERK is phosphorylated, it acts as the main switch between apoptosis and autophagy, e.g., phosphorylating the α subunit of the eukaryotic Initiation Factor 2 (eIF2) to attenuate protein synthesis [69,70]. This modification determines whether the cell fate is switched to autophagy, by means of ATG5-12 complex-activated LC3, or to apoptosis through caspase activation.
Another protein strictly involved in the crosstalk is ATG5, which, other than promoting autophagy, has a role in enhancing apoptotic stimuli. In fact tumor cells overexpressing Atg5 were reported to be more sensitive to chemotherapy, while in case of gene silencing, cancer cells were partially resistant to anti-cancer drugs [71]. This occurs because ATG5, during apoptosis, is cleaved by calpains and subsequently translocated to mitochondria, where it interacts with Bcl-XL and controls cytochrome c release and caspase activation [72]. ATG3, controlled by FLICE-Inhibitory Protein (FLIP), is involved also in apoptosis, where it regulates negatively the extrinsic pathway by recognizing and binding FADD (Fas-Associated protein with Death Domain) through specific Death Effector Domains (DED). An additional aspect that supports the interplay between apoptosis and autophagy is the involvement of apoptotic caspases in the degradation, and consequent inactivation, of autophagic proteins such as Beclin-1, thus inhibiting autophagy and consequently enhancing apoptosis progression (reviewed in [9,10]). In addition, ATG12 could play an active role in both processes [73].
Several reports highlighted the role of p62 (also called sequestosome), a protein that targets other proteins for proteasome degradation or autophagic digestion, at the crossroads of autophagy, apoptosis and cancer [74]; in particular, it has been shown that LC3-II binds p62 to regulate protein packaging and delivering to the autophagosome [75,76]. In addition, p62 accumulation was described in autophagy-defective cells, which suffer from proteasome inactivation and altered NF-kB regulation, and undergo tumorigenesis [47]. This aspect is in agreement with the observation describing how p62 is involved in apoptosis: in the extrinsic pathway, the initiator caspase-8 requires p62 for its efficient polyubiquitination, aggregation and full activation [77].
Notably, it has recently been reported that autophagosome is required to activate caspase-8, which is able to interact with some autophagic factors such as p62 and ATG5; in particular caspase-8/FADD complex associates with ATG5 on ATG16- and LC3-positive structures, suggesting the role of the autophagosomal membrane as a platform for the formation of a dual-armed DISC (Death-Inducing Signaling Complex) that facilitates the activation of caspase-8 and initiation of apoptosis [78]. Conversely, it has been found that caspases cleave and inactivate Beclin-1, thus inhibiting autophagy and enhancing apoptosis, given that the proteolytic Beclin-1 C-terminal fragment induces the release of apoptotic factors from mitochondria [79,80,81]. These observations strongly support the dependence of apoptosis from autophagy and vice versa [78] and further stimulates the discussion about the real distinct identity of these processes [82].
6. Autophagy Manipulation
The intricate relation between apoptosis and autophagy leads to opposite situations: suppression of apoptosis could induce autophagy, while autophagy inhibition causes apoptosis [9,10,60,83].
The complex impact of autophagy on cancer cell metabolism is schematized in Figure 3. A possible way to take advantage of autophagy manipulation is the idea of battery-operated tumor growth, according to which inhibiting or forcing autophagic machinery would be useful in drug cancer treatment [84].
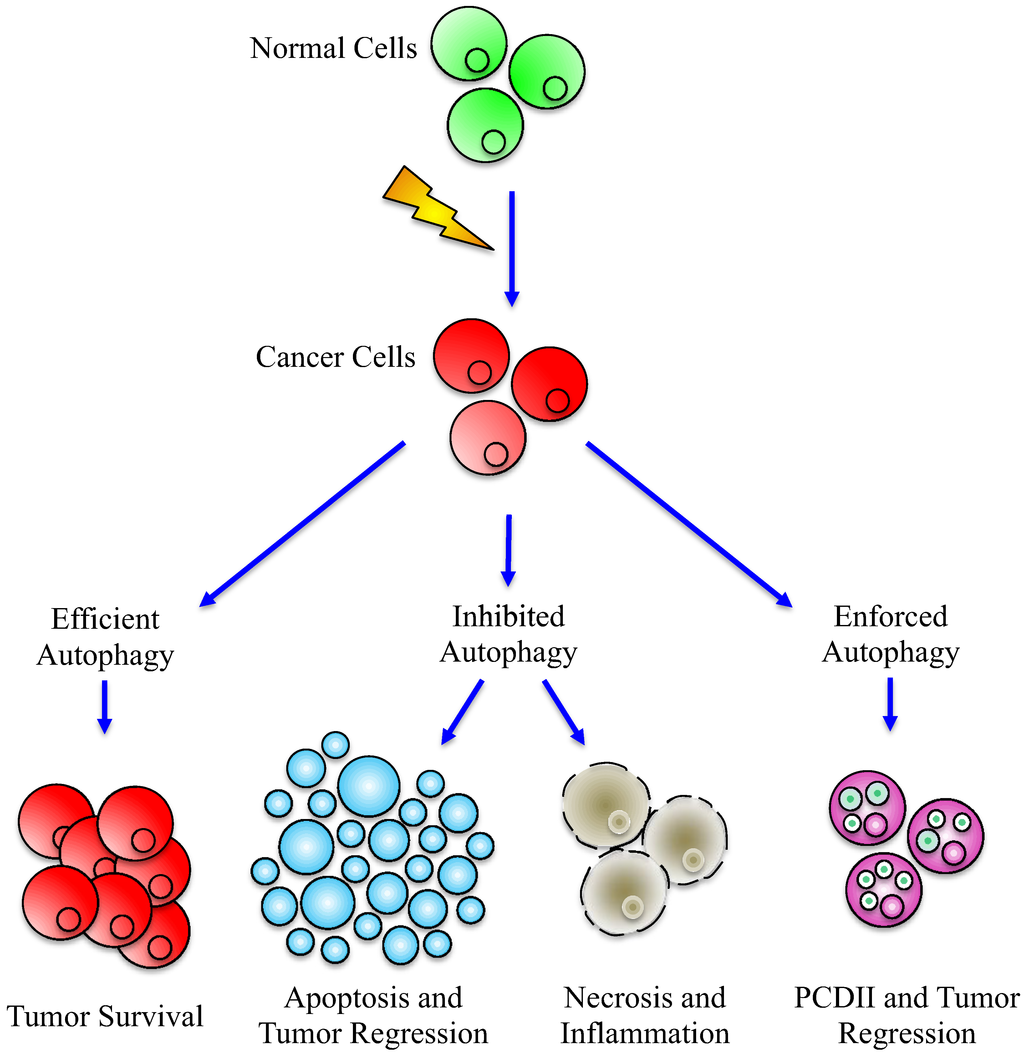
Figure 3.
Effects of efficient or deregulated Autophagy in cancer development. Alteration of autophagy may have different effects on cancer cells: when efficient, the autophagic machinery could help cancer cells to survive and proliferate (left part), while, when inhibited, autophagy cannot anymore sustain cancer progression, leading to the activation of apoptosis or to necrosis (central part) and, by consequence, tumor regression. Paradoxically, the same end point can be reached after enforced activation of autophagy (right part), which can act as type II PCD (Programmed Cell Death).
The possibility to manipulate autophagy for fighting cancer is extremely intriguing: many groups attempted to sensitize cancer cells to treatments through the use of inhibitors/activators of the autophagic machinery [85]. In this respect, on the one hand, inhibitors of autophagy applied in combination with anticancer agents could improve the efficacy of classical drugs; on the other hand, activators of autophagy could enforce the cell death potential of autophagy itself.
For example, it has been shown that after anti-angiogenesis therapies, cancer cells respond to hypoxia by activating autophagy and, in this way, they can survive rendering the therapy no more effective [86]. This observation suggests that the inhibition of autophagy may cooperate with anti-angiogenic factors to avoid drug resistance. Conversely, the activation of autophagy could represent a powerful strategy to kill cancer cells, as reported for some Triple-Negative Breast Cancer (TNBC) -derived cell lines, where the treatment with an mTOR inhibitor kills cancer cell, providing the evidence that the release of autophagy inhibition could be useful to counteract cancer growth [87].
These reports are representative (and contradictory) examples of the rationale basis for manipulating autophagy in order to interfere with cancer cell metabolism. An exhaustive list of the methods for developing autophagy-based therapies, together with “Pros and Cons” of these strategies, has been recently drawn [36,49,85 and references therein]. On the whole, the knowledge of the molecular bases of autophagy has encouraged many attempts to modulate autophagy in order to identify new tools for elaborating an efficient action plan against cancer. In fact, the existence of more than 20 ongoing clinical trials based on inhibition/stimulation of autophagy [88] supports the growing interest toward the impact of this process and the possible applications in clinics.
As a further matter of debate, although the development of autophagy-based anticancer strategies is promising, this approach has to be carefully examined with respect to undesirable effects on non-cancer cells [36]. On the one hand, strategies able to manipulating autophagy could be a novel weapon to treat cancer; on the other hand, they could have a noxious impact on normal cells, such as neurons. For example, it has been shown that genetic inhibition of autophagy allows the occurrence of neurodegenerative hallmarks possibly culminating in aging [89,90]. In fact, the role of autophagy in neurodegenerative disorders has not been fully elucidated, being either beneficial or detrimental, depending on the disease features [91,92]. Notably, attempts to pharmacologically modulate autophagy through the use of the mTOR inhibitor rapamycin (and analogs), allowed neuroprotection in several experimental models of neurodegenerative diseases, due to the contribute to the clearance of intracellular protein aggregates [93,94,95]. In this respect, autophagic dysfunction has been described in neurodegenerative disorders, thus opening new perspectives for clinical treatments [96].
7. Conclusions
The aim of this review is to delineate the impact of the so complex and amazing autophagic process on cancer cell survival. In this respect, the investigation of the role played by autophagy in the complex network of cell death(s) as well as the in-depth examination of its intricate connection with apoptosis, could help in understanding when and how autophagy switches from a survival to a death function [92]. Taking into account the two faces of Janus of autophagy [91], the attempt to define the autophagic function in a univocal manner is a hard duty. This housekeeping process, due to its enrollment in several basic functions, assumes more than one connotation, and for this reason it can be defined only within the frame of the specific biological context [51,97,98]. The data collected in this field could be crucial in defining the cascade of events transforming a normal cell in a tumoral one and eventually confer metastatic potential to it. In conclusion, the “take home message” from the above considerations is that the frontier between the opposite functions of autophagy is not sharp but multi-faceted and depends on the subtle regulation of survival/death signals, including those governing cancer development, drug resistance and neuronal deficits.
Acknowledgments
AIS laboratory is granted by Italian Ministry of Health (Project SEpiAs) and Regione Lombardia, (Project Plant Cell) supporting also FA. LMGO is a PhD student (Dottorato in Genetica, Biologia Cellulare e Molecolare (University of Pavia, Italy) supported by SENESCYT (Quito, Ecuador) and UTPL (Loja, Ecuador). GV was supported by an Investigator Fellowship from Collegio Ghislieri, Pavia, Italy.
Conflict of Interest
The authors declare no conflict of interest.
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10.
- Hanahan, D.; Weinberg, R. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Philchenkov, A. Apoptosis, cancer, and beyond. Cell Death Differ. 2006, 13, 2004–2005. [Google Scholar] [CrossRef]
- Call, J.A.; Eckhardt, S.; Camidge, D.R. Targeted manipulation of apoptosis in cancer treatment. Lancet Oncol. 2008, 9, 1002–1011. [Google Scholar] [CrossRef]
- Mondello, C.; Scovassi, A.I. Apoptosis: A way to maintain healthy individuals. Subcell. Biochem. 2010, 50, 307–323. [Google Scholar] [CrossRef]
- Guamán Ortiz, L.M. Chronicles of a silent death: Apoptosis. Res. Cell Biol. 2012, 1, 1–7. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Giansanti, V.; Tillhon, M.; Mazzini, G.; Prosperi, E.; Lombardi, P.; Scovassi, A.I. Killing of tumor cells: A drama in two acts. Biochem. Pharmacol. 2011, 82, 1304–1310. [Google Scholar]
- Giansanti, V.; Torriglia, A.; Scovassi, A.I. Conversation between apoptosis and autophagy: “Is it your turn or mine?”. Apoptosis 2011, 16, 321–333. [Google Scholar] [CrossRef]
- Scovassi, A.I. Defective Apoptosis and Efficient Autophagy: Two ways to protect cancer cells from death. Biochem. Pharmacol. 2012, 1, e114. [Google Scholar]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Todde, V.; Veenhuis, M.; van der Klei, I.J. Autophagy: Principles and significance in health and disease. Biochim. Biophys. Acta 2009, 1792, 3–13. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Huang, W.P.; Klionsky, D.J. Autophagy in yeast: A review of the molecular machinery. Cell Struct. Funct. 2002, 27, 409–420. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular mechanism of autophagy in yeast, Saccharomyces cerevisiae. Philos. Trans. R Soc. Lond. B. Biol. Sci. 1999, 354, 1577–1580. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Levine, B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar]
- Rubinsztein, D.C. Autophagy--alias self-eating--appetite and ageing. EMBO Rep. 2012, 13, 173–174. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy-unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Rubinsztein, D.C. Microtubule disruption inhibits autophagosome-lysosome fusion: Implications for studying the roles of aggresomes in polyglutamine diseases. Int. J. Biochem. Cell Biol. 2004, 36, 2541–2550. [Google Scholar] [CrossRef]
- Burman, C.; Ktistakis, N.T. Autophagosome formation in mammalian cells. Semin. Immunopathol. 2010, 32, 397–413. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, X.; Wang, J.; Fan, J.; Kong, Q.; Yu, X. Aristolochic acid I induced autophagy extenuates cell apoptosis via ERK 1/2 pathway in renal tubular epithelial cells. PLoS One 2012, 7, e30312. [Google Scholar]
- Weichhart, T. Mammalian target of rapamycin: A signaling kinase for every aspect of cellular life. Methods Mol. Biol. 2012, 821, 1–14. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Jing, K.; Song, K.S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.R.; Park, J.H.; Seo, K.S.; Heo, J.Y.; Han, J.; et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef]
- Neufeld, T.P. TOR-dependent control of autophagy: Biting the hand that feeds. Curr. Opin. Cell Biol. 2010, 22, 157–168. [Google Scholar] [CrossRef]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin-1-VPS34 complex at the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef]
- Fimia, G.M.; Di Bartolomeo, S.; Piacentini, M.; Cecconi, F. Unleashing the Ambra1-Beclin-1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy 2011, 7, 115–117. [Google Scholar] [CrossRef]
- Wu, W.K.; Coffelt, S.B.; Cho, C.H.; Wang, X.J.; Lee, C.W.; Chan, F.K.; Yu, J.; Sung, J.J. The autophagic paradox in cancer therapy. Oncogene 2012, 31, 939–953. [Google Scholar] [CrossRef]
- Wong, A.S.; Cheung, Z.H.; Ip, N.Y. Molecular machinery of macroautophagy and its deregulation in diseases. Biochim. Biophys. Acta 2011, 1812, 1490–1497. [Google Scholar] [CrossRef]
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and genomic organization of Beclin-1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar]
- Kim, M.S.; Jeong, E.G.; Ahn, C.H.; Kim, S.S.; Lee, S.H.; Yoo, N.J. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum. Pathol. 2008, 39, 1059–1063. [Google Scholar] [CrossRef]
- Miao, Y.; Zhang, Y.; Chen, Y.; Chen, L.; Wang, F. GABARAP is overexpressed in colorectal carcinoma and correlates with shortened patient survival. Hepatogastroenterology 2010, 57, 257–261. [Google Scholar]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef]
- Yoshioka, A.; Miyata, H.; Doki, Y.; Yamasaki, M.; Sohma, I.; Gotoh, K.; Takiguchi, S.; Fujiwara, Y.; Uchiyama, Y.; Monden, M. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int. J. Oncol. 2008, 33, 461–468. [Google Scholar]
- Li, Z.; Chen, B.; Wu, Y.; Jin, F.; Xia, Y.; Liu, X. Genetic and epigenetic silencing of the Beclin-1 gene in sporadic breast tumors. BMC Cancer 2010, 10. [Google Scholar]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar]
- Miracco, C.; Cevenini, G.; Franchi, A.; Luzi, P.; Cosci, E.; Mourmouras, V.; Monciatti, I.; Mannucci, S.; Biagioli, M.; Toscano, M.; et al. Beclin-1 and LC3 autophagic gene expression in cutaneous melanocytic lesions. Hum. Pathol. 2010, 41, 503–512. [Google Scholar] [CrossRef]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar]
- Zhao, Z.; Oh, S.; Li, D.; Ni, D.; Dolatshahi Pirooz, S.; Lee, J.; Yang, S.; Lee, J.; Ghozalli, I.; Costanzo, V.; et al. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev. Cell 2012, 22, 1001–1016. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef]
- Roy, S.; Debnath, J. Autophagy and tumorigenesis. Semin. Immunopathol. 2010, 32, 383–396. [Google Scholar] [CrossRef]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3 and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar]
- Xu, T.; Su, H.; Ganapathy, S.; Yuan, Z.M. Modulation of autophagic activity by extracellular pH. Autophagy 2011, 7, 1316–1322. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar]
- Macintosh, R.L.; Timpson, P.; Thorburn, J.; Anderson, K.I.; Thorburn, A.; Ryan, K.M. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012, 11, 2022–2029. [Google Scholar]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Fimia, G.M.; Piacentini, M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell. Mol. Life Sci. 2010, 67, 1581–1588. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar]
- Feng, W.; Huang, S.; Wu, H.; Zhang, M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 2007, 372, 223–235. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of Beclin-1 promotes dissociation of Beclin-1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Bovellan, M.; Fritzsche, M.; Stevens, C.; Charras, G. Death-associated protein kinase (DAPK) and signal transduction: blebbing in programmed cell death. FEBS J. 2010, 277, 58–65. [Google Scholar]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Corcelle, E.; Djerbi, N.; Mari, M.; Nebout, M.; Fiorini, C.; Fénichel, P.; Hofman, P.; Poujeol, P.; Mograbi, B. Control of the autophagy maturation step by the MAPK ERK and p38: Lessons from environmental carcinogens. Autophagy 2007, 3, 57–59. [Google Scholar]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar]
- Liu, J.; Mao, W.; Ding, B.; Liang, C.S. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1956–H1965. [Google Scholar]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Bhutia, S.K.; Dash, R.; Das, S.K.; Azab, B.; Su, Z.Z.; Lee, S.G.; Grant, S.; Yacoub, A.; Dent, P.; Curiel, D.T.; et al. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Res. 2010, 70, 3667–3676. [Google Scholar]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. Feedback on fat: p62-mTORC1-autophagy connections. Cell 2011, 147, 724–727. [Google Scholar]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signalling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated Caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar]
- Cho, D.H.; Jo, Y.K.; Hwang, J.J.; Lee, Y.M.; Roh, S.A.; Kim, J.C. Caspase-mediated cleavage of ATG6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett. 2009, 274, 95–100. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Kersse, K; Cornelis, S.; Claerhout, S.;Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Maiuri, M.C.; Kroemer, G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin-1. Oncogene 2010, 29, 1717–1719. [Google Scholar]
- Chonghaile, T.N.; Letai, A. Who put the “A” in Atg12: Autophagy or apoptosis? Mol. Cell 2011, 44, 844–845. [Google Scholar] [CrossRef]
- Platini, F.; Perez-Tomas, R.; Ambrosio, S.; Tessitore, L. Understanding autophagy in cell death control. Curr. Pharm. Des. 2010, 16, 101–113. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Pavlides, S.; Chiavarina, B.; Bonuccelli, G.; Casey, T.; Tsirigos, A.; Migneco, G.; Witkiewicz, A.; Balliet, R.; et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle 2010, 9, 4297–4306. [Google Scholar]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef]
- Hu, Y.L.; Jahangiri, A.; De Lay, M.; Aghi, M.K. Hypoxia-induced tumor cell autophagy mediates resistance to anti-angiogenic therapy. Autophagy. 2012, 8. Available online: http://dx.doi.org/10.4161/auto.20232.
- Yunokawa, M.; Koizumi, F.; Kitamura, Y.; Katanasaka, Y.; Okamoto, N.; Kodaira, M.; Yonemori, K.; Shimizu, C.; Ando, M.; Masutomi, K.; et al. Efficacy of everolimus, a novel mTOR inhibitor, against basal-like triple-negative breast cancer cells. Cancer Sci. 2012. [Google Scholar]
- Homepage of ClinicalTrials.gov. Available online: http://clinicaltrials.gov/ (accessed on 27 July 2012).
- Madeo, F.; Tavernarakis, N.; Kroemer, G. Can autophagy promote longevity? Nat. Cell Biol. 2010, 12, 842–846. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Vellai, T.; Tóth, M.L.; Kovács, A.L. Janus-faced autophagy: A dual role of cellular self-eating in neurodegeneration? Autophagy 2007, 3, 461–463. [Google Scholar]
- Amelio, I.; Melino, G.; Knight, R.A. Cell death pathology: Cross-talk with autophagy and its clinical implications. Biochem. Biophys. Res. Commun. 2011, 414, 277–281. [Google Scholar] [CrossRef]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar]
- Garelick, M.G.; Kennedy, B.K. TOR on the brain. Exp. Gerontol. 2011, 46, 155–163. [Google Scholar] [CrossRef]
- Mariño, G.; Madeo, F.; Kroemer, G. Autophagy for tissue homeostasis and neuroprotection. Curr. Opin. Cell Biol. 2011, 23, 198–206. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar]
- Denton, D.; Nicholson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef]
- Lozy, F.; Karantza, V. Autophagy and cancer cell metabolism. Semin. Cell Dev. Biol. 2012, 23, 395–401. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).