Regulation of the Autophagic Bcl-2/Beclin 1 Interaction

Abstract
:1. Introduction
2. The Phosphatidylinositol 3-Kinase Complex III

{kind=link}
{kind=link}
| Mice | Phenotypes | References |
|---|---|---|
| Beclin 1-/- | Prenatal embryonic lethality | [19,20] |
| Beclin 1+/- | Increased tumor incidence | [19,20] |
| Abnormal ovary morphology | [38] | |
| Increased cell proliferation | [19,39] | |
| Premature death | [19] | |
| Increased angiogenesis | [39] | |
| Decreased cardiac injury | [40,41] | |
| Neurodegeneration | [42] | |
| Renal fibrosis | [43] | |
| Bcl-2-/- | Partial postnatal lethality | [44,45,46,47,48,49,50] |
| Abnormal kidneys/Polycystic kidney disease | [44,45,46,47,48,49,50,51] | |
| Abnormal lymphoid system | [44,45,46,48,50] | |
| Abnormal spleen morphology | [46,48,50,52] | |
| Abnormal thymus morphology | [48,50] | |
| Small ears | [44,45,48,50] | |
| Abnormal nose morphology | [44,48] | |
| Decreased body size/weight | [44,45,46,47,48,50] | |
| Abnormal hair pigmentation | [44,45,48,50] | |
| Abnormal neuron morphology/Neuron degeneration | [47,53] | |
| Abnormal retinal vasculature morphology/Pericyte morphology | [54] | |
| Abnormal skeletal muscle fiber type ratio | [55] | |
| Abnormal small intestine morphology | [50] | |
| Abnormal osteoblast morphology | [56] | |
| Bcl-XL-/- | Prenatal lethality | [57,58,59] |
| Bcl-XL-/- (CKO) | Abnormal platelet morphology/physiology | [57] |
| Neuron degeneration | [58,59] | |
| Liver fibrosis | [60] | |
| Anemia/Splenomegaly | [61] | |
| Increased bone resorption | [62] | |
| Mcl-1-/- | Peri-implantation embryonic lethality | [63] |
| Mcl-1-/- (CKO) | Decreased T cell number/Abnormal T cell morphology | [64,65] |
| Decreased neutrophil number | [66] | |
| Decreased mast cell and basophil number | [67] | |
| Decreased thymocyte number | [68] | |
| Neuron degeneration | [69] | |
| Abnormal liver morphology | [70,71,72] | |
| Bcl-w-/- | Male infertility | [73,74,75] |
| Abnormal neurological behavior | [76] | |
| Decreased cellular stress protection | [77,78] |
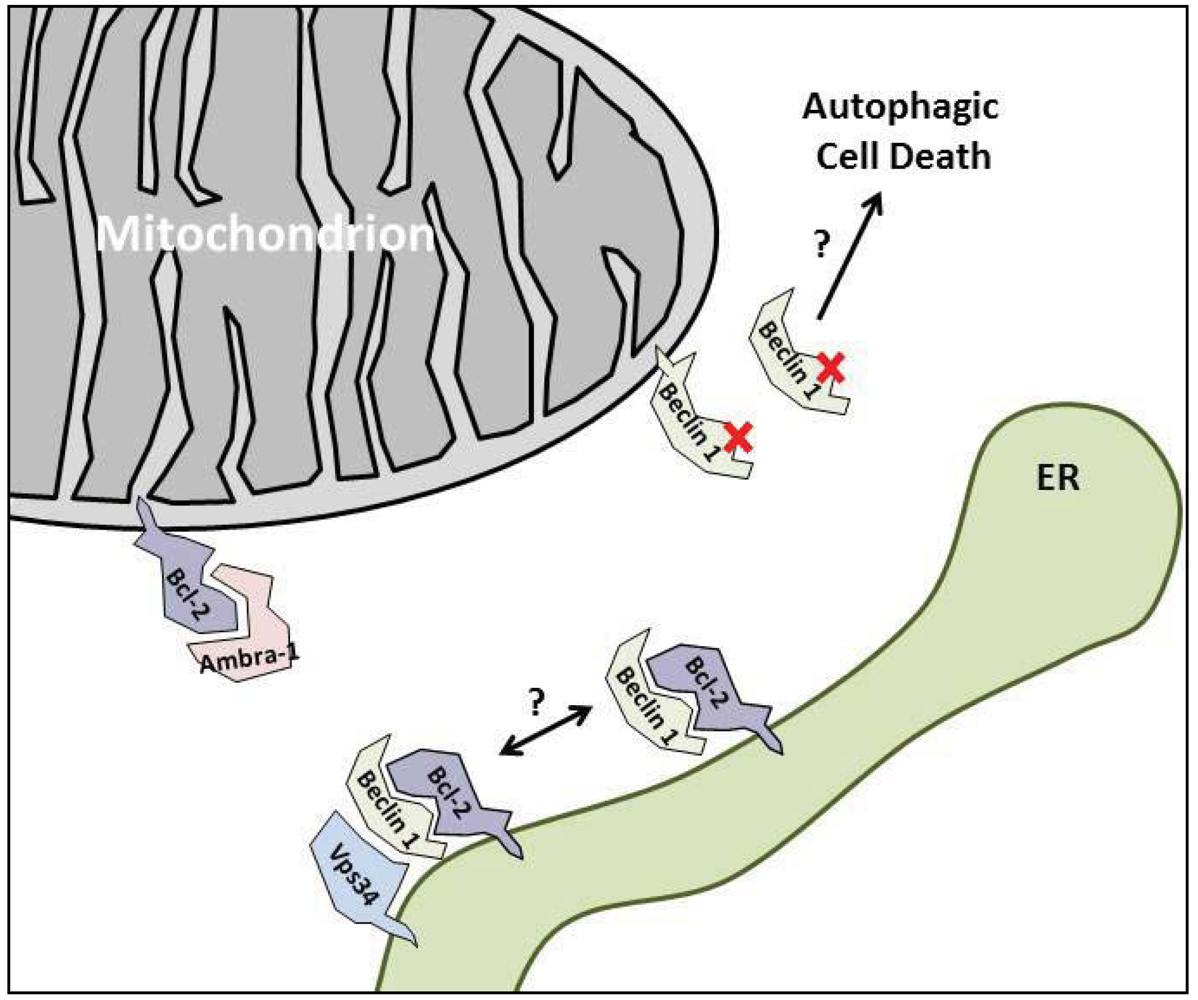
3. The Anti-Autophagic Bcl-2-Family Proteins
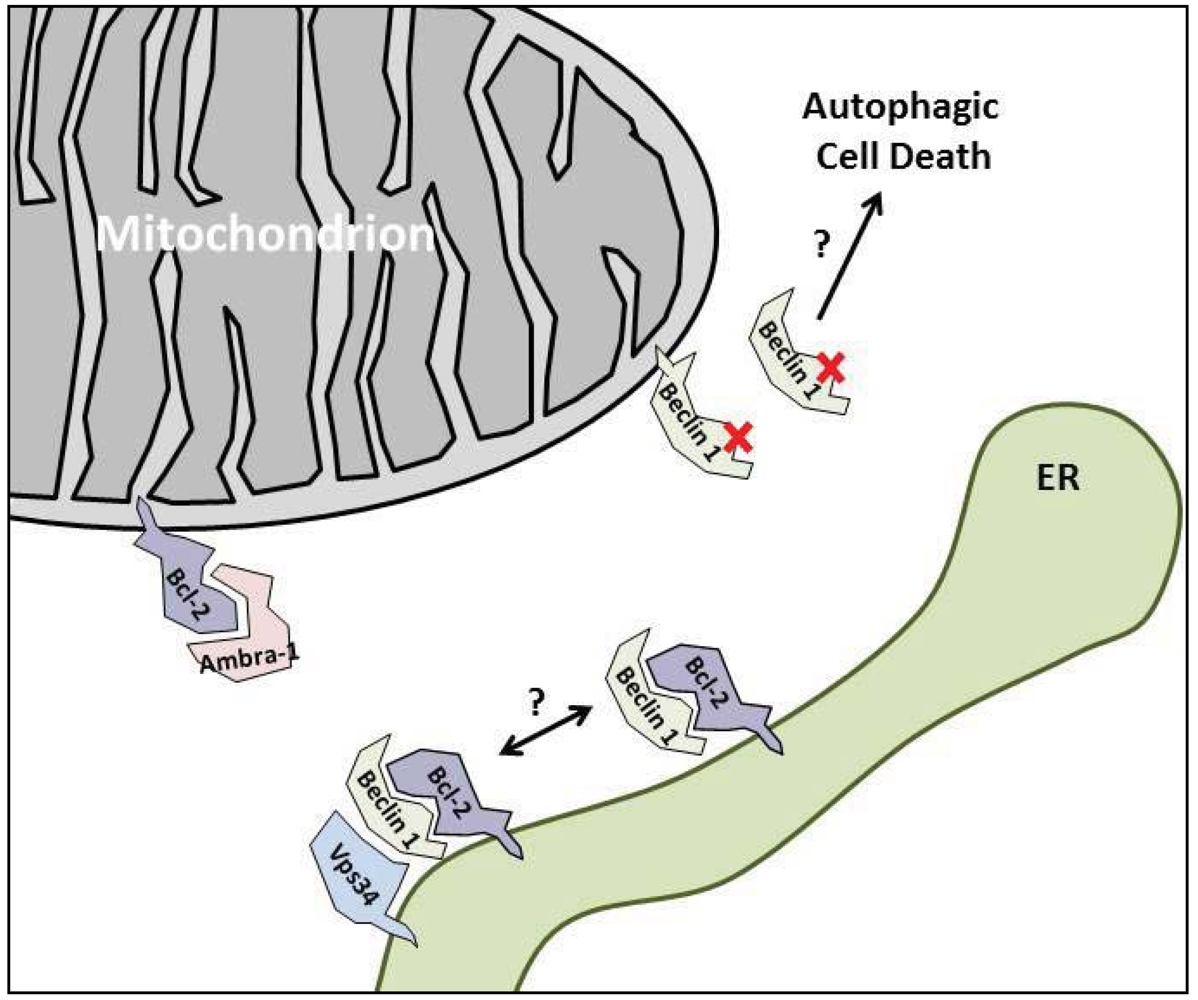
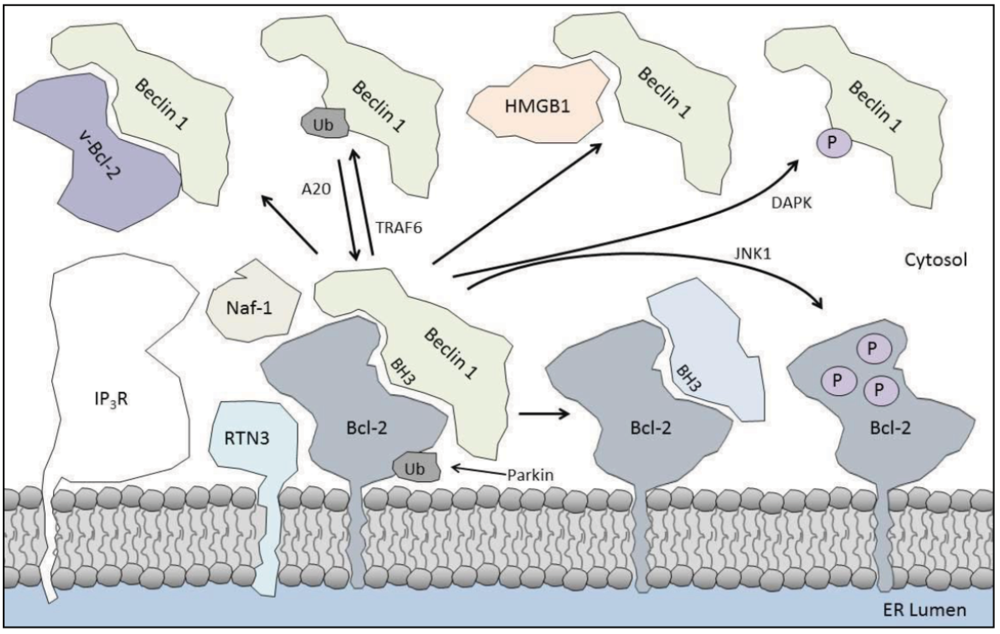
4. Enhancing the Bcl-2/Beclin 1 Interaction
4.1. Parkin
4.2. IP3R
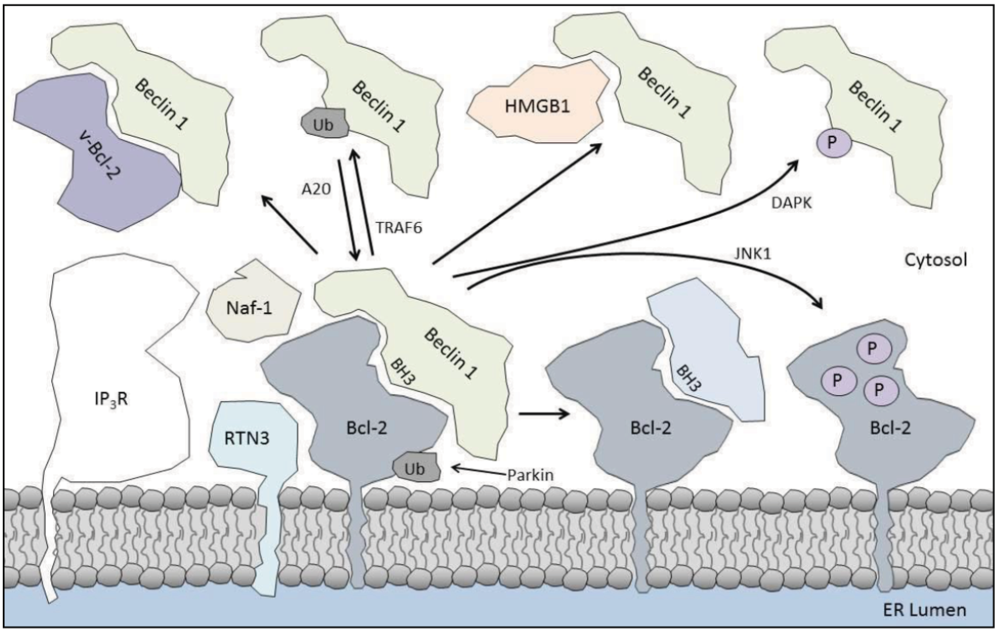
4.3. Naf-1
4.4. Reticulon 3
4.5. Beclin 1 Self-Association
5. Inhibiting the Bcl-2/Beclin 1 Interaction
5.1. Kinases
5.2. Ambra-1
5.3. HMGB1
5.4. ARF
5.5. TRAF6
5.6. Caspases
5.7. BH3-Only Proteins
5.8. Viral Proteins
6. Affecting the Bcl-2/Beclin 1 Interaction as Therapy
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Marino, G.; Madeo, F.; Kroemer, G. Autophagy for tissue homeostasis and neuroprotection. Curr. Opin. Cell Biol. 2011, 23, 198–206. [Google Scholar] [CrossRef]
- Debnath, J.; Baehrecke, E.H.; Kroemer, G. Does autophagy contribute to cell death? Autophagy 2005, 1, 66–74. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Yen, W.L.; Shintani, T.; Nair, U.; Cao, Y.; Richardson, B.C.; Li, Z.; Hughson, F.M.; Baba, M.; Klionsky, D.J. The conserved oligomeric golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010, 188, 101–114. [Google Scholar] [CrossRef]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-Vps34 complex—At the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef]
- Meiling-Wesse, K.; Barth, H.; Voss, C.; Eskelinen, E.L.; Epple, U.D.; Thumm, M. Atg21 is required for effective recruitment of Atg8 to the preautophagosomal structure during the Cvt pathway. J. Biol. Chem. 2004, 279, 37741–37750. [Google Scholar]
- Proikas-Cezanne, T.; Ruckerbauer, S.; Stierhof, Y.D.; Berg, C.; Nordheim, A. Human WIPI-1 puncta-formation: A novel assay to assess mammalian autophagy. FEBS Lett. 2007, 581, 3396–3404. [Google Scholar]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef]
- Backer, J.M. The regulation and function of class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef]
- Nobukuni, T.; Kozma, S.C.; Thomas, G. hVps34, an ancient player, enters a growing game: mTOR complex1/S6K1 signaling. Curr. Opin. Cell Biol. 2007, 19, 135–141. [Google Scholar] [CrossRef]
- Yoon, M.S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III PI-3-kinase activates phospholipase d in an amino acid-sensing mtorc1 pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef]
- Ktistakis, N.T.; Manifava, M.; Schoenfelder, P.; Rotondo, S. How phosphoinositide 3-phosphate controls growth downstream of amino acids and autophagy downstream of amino acid withdrawal. Biochem. Soc. Trans. 2012, 40, 37–43. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-x(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Goemans, G.C.; Skepper, J.N.; Tolkovsky, A.M. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009, 28, 2128–2141. [Google Scholar] [CrossRef]
- Scarlatti, F.; Maffei, R.; Beau, I.; Codogno, P.; Ghidoni, R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008, 15, 1318–1329. [Google Scholar] [CrossRef]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef]
- Tian, S.; Lin, J.; Jun Zhou, J.; Wang, X.; Li, Y.; Ren, X.; Yu, W.; Zhong, W.; Xiao, J.; Sheng, F.; et al. Beclin 1-independent autophagy induced by a Bcl-xL/Bcl-2 targeting compound, z18. Autophagy 2010, 6, 1032–1041. [Google Scholar] [CrossRef]
- Gui, Y.X.; Fan, X.N.; Wang, H.M.; Wang, G.; Chen, S.D. Glyphosate induced cell death through apoptotic and autophagic mechanisms. Neurotoxicol. Teratol. 2012, 34, 344–349. [Google Scholar] [CrossRef]
- Seillier, M.; Peuget, S.; Gayet, O.; Gauthier, C.; N’Guessan, P.; Monte, M.; Carrier, A.; Iovanna, J.L.; Dusetti, N.J. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012. [Google Scholar] [CrossRef]
- Li, P.; Du, Q.; Cao, Z.; Guo, Z.; Evankovich, J.; Yan, W.; Chang, Y.; Shao, L.; Stolz, D.B.; Tsung, A.; et al. Interferon-gamma induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012, 314, 213–222. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal sindbis virus encephalitis by Beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-xL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar]
- Feng, W.; Huang, S.; Wu, H.; Zhang, M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 2007, 372, 223–235. [Google Scholar] [CrossRef]
- Gawriluk, T.R.; Hale, A.N.; Flaws, J.A.; Dillon, C.P.; Green, D.R.; Rucker, E.B., 3rd. Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction 2011, 141, 759–765. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 2007, 117, 1782–1793. [Google Scholar]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein Beclin 1 shows reduced expression in early alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 2008, 118, 2190–2199. [Google Scholar]
- Kim, S.I.; Na, H.J.; Ding, Y.; Wang, Z.; Lee, S.J.; Choi, M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J. Biol. Chem. 2012, 287, 11677–11688. [Google Scholar]
- Nakayama, K.; Negishi, I.; Kuida, K.; Sawa, H.; Loh, D.Y. Targeted disruption of Bcl-2 alpha beta in mice: Occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc. Natl. Acad. Sci. USA 1994, 91, 3700–3704. [Google Scholar]
- Bouillet, P.; Cory, S.; Zhang, L.C.; Strasser, A.; Adams, J.M. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist bim. Dev. Cell 2001, 1, 645–653. [Google Scholar] [CrossRef]
- Merino, D.; Giam, M.; Hughes, P.D.; Siggs, O.M.; Heger, K.; O’Reilly, L.A.; Adams, J.M.; Strasser, A.; Lee, E.F.; Fairlie, W.D.; et al. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J. Cell Biol. 2009, 186, 355–362. [Google Scholar] [CrossRef]
- Michaelidis, T.M.; Sendtner, M.; Cooper, J.D.; Airaksinen, M.S.; Holtmann, B.; Meyer, M.; Thoenen, H. Inactivation of Bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron 1996, 17, 75–89. [Google Scholar] [CrossRef]
- Veis, D.J.; Sorenson, C.M.; Shutter, J.R.; Korsmeyer, S.J. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993, 75, 229–240. [Google Scholar] [CrossRef]
- Fedorov, L.M.; Schmittwolf, C.; Amann, K.; Thomas, W.H.; Muller, A.M.; Schubert, H.; Domen, J.; Kneitz, B. Renal failure causes early death of Bcl-2 deficient mice. Mech. Ageing Dev. 2006, 127, 600–609. [Google Scholar] [CrossRef]
- Kamada, S.; Shimono, A.; Shinto, Y.; Tsujimura, T.; Takahashi, T.; Noda, T.; Kitamura, Y.; Kondoh, H.; Tsujimoto, Y. Bcl-2 deficiency in mice leads to pleiotropic abnormalities: Accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer Res. 1995, 55, 354–359. [Google Scholar]
- Nagata, M.; Nakauchi, H.; Nakayama, K.; Loh, D.; Watanabe, T. Apoptosis during an early stage of nephrogenesis induces renal hypoplasia in Bcl-2-deficient mice. Am. J. Pathol. 1996, 148, 1601–1611. [Google Scholar]
- Wellmer, A.; von Mering, M.; Spreer, A.; Diem, R.; Eiffert, H.; Noeske, C.; Bunkowski, S.; Gold, R.; Nau, R. Experimental pneumococcal meningitis: Impaired clearance of bacteria from the blood due to increased apoptosis in the spleen in Bcl-2-deficient mice. Infect. Immun. 2004, 72, 3113–3119. [Google Scholar]
- Hilton, M.; Middleton, G.; Davies, A.M. Bcl-2 influences axonal growth rate in embryonic sensory neurons. Curr. Biol. 1997, 7, 798–800. [Google Scholar] [CrossRef]
- Wang, S.; Sorenson, C.M.; Sheibani, N. Attenuation of retinal vascular development and neovascularization during oxygen-induced ischemic retinopathy in bcl-2-/- mice. Dev. Biol. 2005, 279, 205–219. [Google Scholar] [CrossRef]
- Dominov, J.A.; Houlihan-Kawamoto, C.A.; Swap, C.J.; Miller, J.B. Pro- and anti-apoptotic members of the Bcl-2 family in skeletal muscle: A distinct role for Bcl-2 in later stages of myogenesis. Dev. Dyn. 2001, 220, 18–26. [Google Scholar] [CrossRef]
- Boot-Handford, R.P.; Michaelidis, T.M.; Hillarby, M.C.; Zambelli, A.; Denton, J.; Hoyland, J.A.; Freemont, A.J.; Grant, M.E.; Wallis, G.A. The Bcl-2 knockout mouse exhibits marked changes in osteoblast phenotype and collagen deposition in bone as well as a mild growth plate phenotype. Int. J. Exp. Pathol. 1998, 79, 329–335. [Google Scholar]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Shindler, K.S.; Latham, C.B.; Roth, K.A. Bax deficiency prevents the increased cell death of immature neurons in Bcl-x-deficient mice. J. Neurosci. 1997, 17, 3112–3119. [Google Scholar]
- Motoyama, N.; Wang, F.; Roth, K.A.; Sawa, H.; Nakayama, K.; Negishi, I.; Senju, S.; Zhang, Q.; Fujii, S. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995, 267, 1506–1510. [Google Scholar]
- Takehara, T.; Tatsumi, T.; Suzuki, T.; Rucker, E.B., 3rd; Hennighausen, L.; Jinushi, M.; Miyagi, T.; Kanazawa, Y.; Hayashi, N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology 2004, 127, 1189–1197. [Google Scholar] [CrossRef]
- Rhodes, M.M.; Kopsombut, P.; Bondurant, M.C.; Price, J.O.; Koury, M.J. Bcl-x(L) prevents apoptosis of late-stage erythroblasts but does not mediate the antiapoptotic effect of erythropoietin. Blood 2005, 106, 1857–1863. [Google Scholar] [CrossRef]
- Iwasawa, M.; Miyazaki, T.; Nagase, Y.; Akiyama, T.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Yasui, T.; Matsumoto, T.; Nakamura, T.; et al. The antiapoptotic protein Bcl-xL negatively regulates the bone-resorbing activity of osteoclasts in mice. J. Clin. Invest. 2009, 119, 3149–3159. [Google Scholar]
- Rinkenberger, J.L.; Horning, S.; Klocke, B.; Roth, K.; Korsmeyer, S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000, 14, 23–27. [Google Scholar]
- Opferman, J.T.; Letai, A.; Beard, C.; Sorcinelli, M.D.; Ong, C.C.; Korsmeyer, S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic Mcl-1. Nature 2003, 426, 671–676. [Google Scholar] [CrossRef]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic Mcl-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar]
- Dzhagalov, I.; St John, A.; He, Y.W. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 2007, 109, 1620–1626. [Google Scholar] [CrossRef]
- Lilla, J.N.; Chen, C.C.; Mukai, K.; BenBarak, M.J.; Franco, C.B.; Kalesnikoff, J.; Yu, M.; Tsai, M.; Piliponsky, A.M.; Galli, S.J. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 2011, 118, 6930–6938. [Google Scholar]
- Dunkle, A.; Dzhagalov, I.; He, Y.W. Mcl-1 promotes survival of thymocytes by inhibition of bak in a pathway separate from Bcl-2. Cell Death Differ. 2010, 17, 994–1002. [Google Scholar] [CrossRef]
- Arbour, N.; Vanderluit, J.L.; Le Grand, J.N.; Jahani-Asl, A.; Ruzhynsky, V.A.; Cheung, E.C.; Kelly, M.A.; MacKenzie, A.E.; Park, D.S.; Opferman, J.T.; et al. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J. Neurosci. 2008, 28, 6068–6078. [Google Scholar]
- Weng, S.Y.; Yang, C.Y.; Li, C.C.; Sun, T.P.; Tung, S.Y.; Yen, J.J.; Tsai, T.F.; Chen, C.M.; Chen, S.H.; Hsiao, M.; et al. Synergism between p53 and Mcl-1 in protecting from hepatic injury, fibrosis and cancer. J. Hepatol. 2011, 54, 685–694. [Google Scholar]
- Weber, A.; Boger, R.; Vick, B.; Urbanik, T.; Haybaeck, J.; Zoller, S.; Teufel, A.; Krammer, P.H.; Opferman, J.T.; Galle, P.R.; et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 2010, 51, 1226–1236. [Google Scholar] [CrossRef]
- Vick, B.; Weber, A.; Urbanik, T.; Maass, T.; Teufel, A.; Krammer, P.H.; Opferman, J.T.; Schuchmann, M.; Galle, P.R.; Schulze-Bergkamen, H. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology 2009, 49, 627–636. [Google Scholar] [CrossRef]
- Ross, A.J.; Waymire, K.G.; Moss, J.E.; Parlow, A.F.; Skinner, M.K.; Russell, L.D.; MacGregor, G.R. Testicular degeneration in Bclw-deficient mice. Nat. Genet. 1998, 18, 251–256. [Google Scholar]
- Russell, L.D.; Warren, J.; Debeljuk, L.; Richardson, L.L.; Mahar, P.L.; Waymire, K.G.; Amy, S.P.; Ross, A.J.; MacGregor, G.R. Spermatogenesis in Bclw-deficient mice. Biol. Reprod. 2001, 65, 318–332. [Google Scholar] [CrossRef]
- Print, C.G.; Loveland, K.L.; Gibson, L.; Meehan, T.; Stylianou, A.; Wreford, N.; de Kretser, D.; Metcalf, D.; Kontgen, F.; Adams, J.M.; et al. Apoptosis regulator Bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. USA 1998, 95, 12424–12431. [Google Scholar]
- Liu, Q.A.; Shio, H. Mitochondrial morphogenesis, dendrite development, and synapse formation in cerebellum require both Bcl-w and the glutamate receptor delta2. PLoS Genet. 2008, 4, e1000097. [Google Scholar] [CrossRef]
- Murphy, B.; Dunleavy, M.; Shinoda, S.; Schindler, C.; Meller, R.; Bellver-Estelles, C.; Hatazaki, S.; Dicker, P.; Yamamoto, A.; Koegel, I.; et al. Bcl-w protects hippocampus during experimental status epilepticus. Am. J. Pathol. 2007, 171, 1258–1268. [Google Scholar] [CrossRef]
- Pritchard, D.M.; Print, C.; O’Reilly, L.; Adams, J.M.; Potten, C.S.; Hickman, J.A. Bcl-w is an important determinant of damage-induced apoptosis in epithelia of small and large intestine. Oncogene 2000, 19, 3955–3959. [Google Scholar] [CrossRef]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; Fiebig, A.A.; Andrews, D.; Molgo, J.; Diaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Brady, N.R.; Hamacher-Brady, A.; Yuan, H.; Gottlieb, R.A. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007, 274, 3184–3197. [Google Scholar] [CrossRef]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.L.; et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef]
- Yue, Z.; Horton, A.; Bravin, M.; DeJager, P.L.; Selimi, F.; Heintz, N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: Implications for neurodegeneration in lurcher mice. Neuron 2002, 35, 921–933. [Google Scholar] [CrossRef]
- Takacs-Vellai, K.; Vellai, T.; Puoti, A.; Passannante, M.; Wicky, C.; Streit, A.; Kovacs, A.L.; Muller, F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. Elegans. Curr. Biol. 2005, 15, 1513–1517. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickman, J.A.; Geneste, O.; Kroemer, G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-x(L). Autophagy 2007, 3, 374–376. [Google Scholar]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced Bcl2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell Biol. 2009, 29, 2594–2608. [Google Scholar] [CrossRef]
- Wang, B.; Ling, S.; Lin, W.C. 14-3-3tau regulates Beclin 1 and is required for autophagy. PLoS One 2010, 5, e10409. [Google Scholar]
- West, A.B.; Maidment, N.T. Genetics of parkin-linked disease. Hum. Genet. 2004, 114, 327–336. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. Pink1- and parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Chen, D.; Gao, F.; Li, B.; Wang, H.; Xu, Y.; Zhu, C.; Wang, G. Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J. Biol. Chem. 2010, 285, 38214–38223. [Google Scholar]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca2+ in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef]
- Khan, M.T.; Joseph, S.K. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J. Biol. Chem. 2010, 285, 16912–16920. [Google Scholar]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Chen, R.; Valencia, I.; Zhong, F.; McColl, K.S.; Roderick, H.L.; Bootman, M.D.; Berridge, M.J.; Conway, S.J.; Holmes, A.B.; Mignery, G.A.; et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell Biol. 2004, 166, 193–203. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The endoplasmic reticulum gateway to apoptosis by Bcl-x(L) modulation of the InsP3R. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol. Chem. 2010, 285, 13678–13684. [Google Scholar]
- Rong, Y.P.; Aromolaran, A.S.; Bultynck, G.; Zhong, F.; Li, X.; McColl, K.; Matsuyama, S.; Herlitze, S.; Roderick, H.L.; Bootman, M.D.; et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol. Cell 2008, 31, 255–265. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgo, J.; Agostinis, P.; Missiaen, L.; de Smedt, H.; Parys, J.B.; et al. IP3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Parys, J.B.; de Smedt, H. Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar] [CrossRef]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by Bcl-2 at the endoplasmic reticulum requires Naf-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Bourdon, J.; Risse, P.A.; Martin, J.; Danialou, G.; Rizzuto, R.; Petrof, B.J.; Shore, G.C. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012, 21, 2277–2287. [Google Scholar] [CrossRef]
- Wakana, Y.; Koyama, S.; Nakajima, K.; Hatsuzawa, K.; Nagahama, M.; Tani, K.; Hauri, H.P.; Melancon, P.; Tagaya, M. Reticulon 3 is involved in membrane trafficking between the endoplasmic reticulum and golgi. Biochem. Biophys. Res. Commun. 2005, 334, 1198–1205. [Google Scholar] [CrossRef]
- Hu, J.; Shibata, Y.; Voss, C.; Shemesh, T.; Li, Z.; Coughlin, M.; Kozlov, M.M.; Rapoport, T.A.; Prinz, W.A. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 2008, 319, 1247–1250. [Google Scholar]
- Voeltz, G.K.; Prinz, W.A.; Shibata, Y.; Rist, J.M.; Rapoport, T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006, 124, 573–586. [Google Scholar] [CrossRef]
- He, W.; Lu, Y.; Qahwash, I.; Hu, X.Y.; Chang, A.; Yan, R. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat. Med. 2004, 10, 959–965. [Google Scholar] [CrossRef]
- Chen, R.; Jin, R.; Wu, L.; Ye, X.; Yang, Y.; Luo, K.; Wang, W.; Wu, D.; Huang, L.; Huang, T.; et al. Reticulon 3 attenuates the clearance of cytosolic prion aggregates via inhibiting autophagy. Autophagy 2011, 7, 205–216. [Google Scholar] [CrossRef]
- Zhu, L.; Xiang, R.; Dong, W.; Liu, Y.; Qi, Y. Anti-apoptotic activity of Bcl-2 is enhanced by its interaction with RTN3. Cell Biol. Int. 2007, 31, 825–830. [Google Scholar] [CrossRef]
- Kuang, E.; Wan, Q.; Li, X.; Xu, H.; Liu, Q.; Qi, Y. ER Ca2+ depletion triggers apoptotic signals for endoplasmic reticulum (ER) overload response induced by overexpressed reticulon 3 (RTN3/HAP). J. Cell Physiol. 2005, 204, 549–559. [Google Scholar] [CrossRef]
- Noble, C.G.; Dong, J.M.; Manser, E.; Song, H. Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. J. Biol. Chem. 2008, 283, 26274–26282. [Google Scholar]
- Ku, B.; Woo, J.S.; Liang, C.; Lee, K.H.; Jung, J.U.; Oh, B.H. An insight into the mechanistic role of Beclin 1 and its inhibition by prosurvival Bcl-2 family proteins. Autophagy 2008, 4, 519–520. [Google Scholar]
- Adi-Harel, S.; Erlich, S.; Schmukler, E.; Cohen-Kedar, S.; Segev, O.; Mizrachy, L.; Hirsch, J.A.; Pinkas-Kramarski, R. Beclin 1 self-association is independent of autophagy induction by amino acid deprivation and rapamycin treatment. J. Cell Biochem. 2010, 110, 1262–1271. [Google Scholar] [CrossRef]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of Bcl-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef]
- Ventura, J.J.; Hubner, A.; Zhang, C.; Flavell, R.A.; Shokat, K.M.; Davis, R.J. Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell 2006, 21, 701–710. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of Beclin 1 promotes dissociation of Beclin 1 from Bcl-xL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886. [Google Scholar] [CrossRef] [Green Version]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-xL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of Ambra1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef]
- Strappazzon, F.; Vietri-Rudan, M.; Campello, S.; Nazio, F.; Florenzano, F.; Fimia, G.M.; Piacentini, M.; Levine, B.; Cecconi, F. Mitochondrial Bcl-2 inhibits Ambra1-induced autophagy. EMBO J. 2011, 30, 1195–1208. [Google Scholar] [CrossRef]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Hoppe, G.; Talcott, K.E.; Bhattacharya, S.K.; Crabb, J.W.; Sears, J.E. Molecular basis for the redox control of nuclear transport of the structural chromatin protein HMGB1. Exp. Cell Res. 2006, 312, 3526–3538. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Zeh, H.J., 3rd; Lotze, M.T. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid. Redox Signal. 2011, 15, 2185–2195. [Google Scholar] [CrossRef]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J., 3rd; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef]
- Zhao, M.; Yang, M.; Yang, L.; Yu, Y.; Xie, M.; Zhu, S.; Kang, R.; Tang, D.; Jiang, Z.; Yuan, W.; et al. HMGB1 regulates autophagy through increasing transcriptional activities of JNK and ERK in human myeloid leukemia cells. BMB Rep. 2011, 44, 601–606. [Google Scholar] [CrossRef]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef]
- Abida, W.M.; Gu, W. p53-dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. ARF induces autophagy by virtue of interaction with Bcl-xL. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Monaco, G.; Bultynck, G.; Missiaen, L.; de Smedt, H.; Parys, J.B. The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 1813, 1003–1013. [Google Scholar]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, 42. [Google Scholar]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar]
- Huang, G.; Yao, J.; Zeng, W.; Mizuno, Y.; Kamm, K.E.; Stull, J.T.; Harding, H.P.; Ron, D.; Muallem, S. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from perk-knockout mice. J. Cell Sci. 2006, 119, 153–161. [Google Scholar] [CrossRef]
- Lebiedzinska, M.; Szabadkai, G.; Jones, A.W.; Duszynski, J.; Wieckowski, M.R. Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int. J. Biochem. Cell Biol. 2009, 41, 1805–1816. [Google Scholar] [CrossRef]
- Gordy, C.; He, Y.W. The crosstalk between autophagy and apoptosis: Where does this lead? Protein Cell 2012, 3, 17–27. [Google Scholar] [CrossRef]
- Cho, D.H.; Jo, Y.K.; Hwang, J.J.; Lee, Y.M.; Roh, S.A.; Kim, J.C. Caspase-mediated cleavage of Atg6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett. 2009, 274, 95–100. [Google Scholar] [CrossRef]
- Rohn, T.T.; Wirawan, E.; Brown, R.J.; Harris, J.R.; Masliah, E.; Vandenabeele, P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the alzheimer's disease brain. Neurobiol. Dis. 2011, 43, 68–78. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef]
- Li, H.; Wang, P.; Sun, Q.; Ding, W.X.; Yin, X.M.; Sobol, R.W.; Stolz, D.B.; Yu, J.; Zhang, L. Following cytochrome c release, autophagy is inhibited during chemotherapy-induced apoptosis by caspase 8-mediated cleavage of Beclin 1. Cancer Res. 2011, 71, 3625–3634. [Google Scholar]
- Chen, G.; Ray, R.; Dubik, D.; Shi, L.; Cizeau, J.; Bleackley, R.C.; Saxena, S.; Gietz, R.D.; Greenberg, A.H. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. 1997, 186, 1975–1983. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of Bnip3 and Bnip3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lan, S.H.; Cheng, D.E.; Chen, W.K.; Shen, C.H.; Lee, Y.R.; Zuchini, R.; Liu, H.S. Ras-related tumorigenesis is suppressed by Bnip3-mediated autophagy through inhibition of cell proliferation. Neoplasia 2011, 13, 1171–1182. [Google Scholar]
- Byun, J.Y.; Yoon, C.H.; An, S.; Park, I.C.; Kang, C.M.; Kim, M.J.; Lee, S.J. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic ras. Carcinogenesis 2009, 30, 1880–1888. [Google Scholar] [CrossRef]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 2011, 42, 23–35. [Google Scholar] [CrossRef]
- Tang, Y.; Hamed, H.A.; Cruickshanks, N.; Fisher, P.B.; Grant, S.; Dent, P. Obatoclax and lapatinib interact to induce toxic autophagy through Noxa. Mol. Pharmacol. 2012, 81, 527–540. [Google Scholar] [CrossRef]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Belec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes Mcl-1 and overcomes Mcl-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar]
- Munz, C. Beclin-1 targeting for viral immune escape. Viruses 2011, 3, 1166–1178. [Google Scholar] [CrossRef]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of intracellular Shigella from autophagy. Science 2005, 307, 727–731. [Google Scholar]
- Sinha, S.; Colbert, C.L.; Becker, N.; Wei, Y.; Levine, B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 2008, 4, 989–997. [Google Scholar]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef]
- Jiang, H.; White, E.J.; Rios-Vicil, C.I.; Xu, J.; Gomez-Manzano, C.; Fueyo, J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J. Virol. 2011, 85, 4720–4729. [Google Scholar]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 2012, 86, 2571–2584. [Google Scholar]
- Ku, B.; Woo, J.S.; Liang, C.; Lee, K.H.; Hong, H.S.; Kim, K.S.; Jung, J.U.; Oh, B.H. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral Bcl-2 of murine gamma-herpesvirus 68. PLoS Pathog. 2008, 4, e25. [Google Scholar] [CrossRef]
- Piya, S.; White, E.J.; Klein, S.R.; Jiang, H.; McDonnell, T.J.; Gomez-Manzano, C.; Fueyo, J. The E1B19K oncoprotein complexes with Beclin 1 to regulate autophagy in adenovirus-infected cells. PLoS One 2011, 6, e29467. [Google Scholar]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Gannage, M.; Ramer, P.C.; Munz, C. Targeting Beclin 1 for viral subversion of macroautophagy. Autophagy 2010, 6, 166–167. [Google Scholar] [CrossRef]
- Zhou, S.; Kuang, M.; Zhang, B.; Zhao, L.; Liang, Z.; Yi, T.; Huang, C.; Wei, Y.; Zhao, X. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 2012. [Google Scholar]
- Schrijvers, D.M.; de Meyer, G.R.; Martinet, W. Autophagy in atherosclerosis: A potential drug target for plaque stabilization. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2787–2791. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kume, S.; Kitada, M.; Kanasaki, K.; Uzu, T.; Maegawa, H.; Koya, D. Autophagy as a therapeutic target in diabetic nephropathy. Exp. Diabetes Res. 2012. [Google Scholar] [CrossRef]
- Nemchenko, A.; Chiong, M.; Turer, A.; Lavandero, S.; Hill, J.A. Autophagy as a therapeutic target in cardiovascular disease. J. Mol. Cell. Cardiol. 2011, 51, 584–593. [Google Scholar] [CrossRef]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2012, 8, 108–117. [Google Scholar]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Hogarty, M.D. Small-molecule BH3 mimetics to antagonize Bcl-2-homolog survival functions in cancer. Curr. Opin. Investig. Drugs 2009, 10, 559–571. [Google Scholar]
- Chonghaile, T.N.; Letai, A. Mimicking the BH3 domain to kill cancer cells. Oncogene 2008, 27, S149–S157. [Google Scholar] [CrossRef]
- Lian, J.; Karnak, D.; Xu, L. The Bcl-2-Beclin 1 interaction in (-)-gossypol-induced autophagy versus apoptosis in prostate cancer cells. Autophagy 2010, 6, 1201–1203. [Google Scholar] [CrossRef]
- Lin, J.; Zheng, Z.; Li, Y.; Yu, W.; Zhong, W.; Tian, S.; Zhao, F.; Ren, X.; Xiao, J.; Wang, N.; et al. A novel Bcl-xL inhibitor z36 that induces autophagic cell death in hela cells. Autophagy 2009, 5, 314–320. [Google Scholar] [CrossRef]
- Malik, S.A.; Orhon, I.; Morselli, E.; Criollo, A.; Shen, S.; Marino, G.; BenYounes, A.; Benit, P.; Rustin, P.; Maiuri, M.C.; et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 2011, 30, 3918–3929. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Decuypere, J.-P.; Parys, J.B.; Bultynck, G. Regulation of the Autophagic Bcl-2/Beclin 1 Interaction. Cells 2012, 1, 284-312. https://doi.org/10.3390/cells1030284
Decuypere J-P, Parys JB, Bultynck G. Regulation of the Autophagic Bcl-2/Beclin 1 Interaction. Cells. 2012; 1(3):284-312. https://doi.org/10.3390/cells1030284
Chicago/Turabian StyleDecuypere, Jean-Paul, Jan B. Parys, and Geert Bultynck. 2012. "Regulation of the Autophagic Bcl-2/Beclin 1 Interaction" Cells 1, no. 3: 284-312. https://doi.org/10.3390/cells1030284