Modulation of Autophagy-Like Processes by Tumor Viruses

Abstract
:1. Introduction
1.1. Human Oncogenic Viruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Associated cancer types | Mechanism of interference with autophagy and/or autophagy-regulating pathways |
|---|---|
| Epstein–Barr virus (EBV) Herpesviridae | |
| Burkitt’s and Hodgkin- and non-Hodgkin lymphomas, nasopharyngeal carcinoma, lymphoproliferative diseases | BILF1 ➔ PKR inhibition ➔ may inhibit autophagy [12] |
| LMP1 ➔ JNK activation ➔ may promote autophagy [13,14,15] | |
| LMP1 ➔ NF-κB activation ➔ inhibits autophagy in B cells [16] | |
| LMP1 ➔ p38 activation ➔ may inhibit autophagy [17] | |
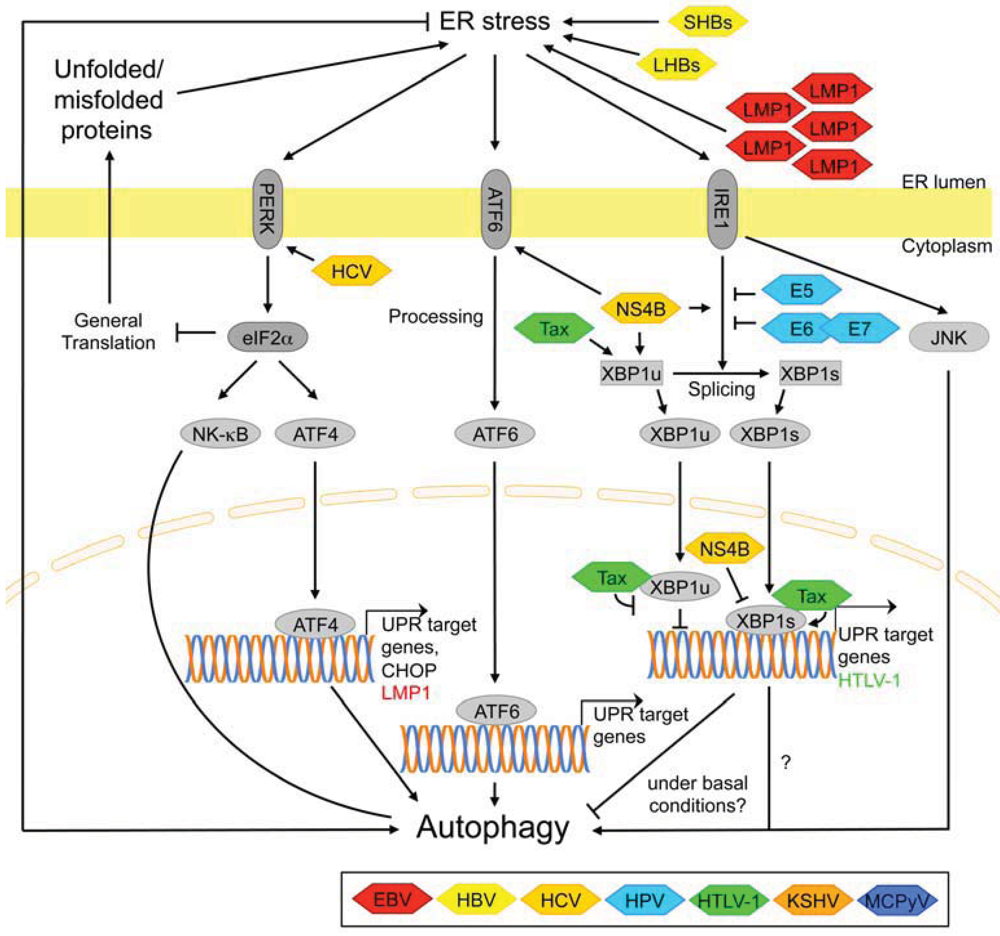
| LMP1 ➔ activation of UPR signaling ➔ autophagic markers [18,19] | |
| LMP1/LMP2A ➔ PI3K/mTOR activation ➔ may inhibit autophagy [20,21,22,23,24] | |
| ZTA ➔ NF-κB inhibition ➔ ? [25] | |
| Hepatitis B virus (HBV) Hepadnaviridae | |
| Hepatocellular carcinoma | HBx ➔ increased Beclin-1 transcription ➔ autophagic markers [26] |
| HBx ➔ increased LC3-lipidation and VPS34 activity ➔ incomplete autophagic response [27] | |
| HBx ➔ interacts with p53 ➔ may inhibit autophagy [28] | |
| HBx ➔ p38 activation ➔ may inhibit autophagy [29] | |
| HBx/LHBs/MHBs ➔ ERK activation ➔ may inhibit autophagy [30,31,32] | |
| LHBs ➔ PI3K/AKT/mTOR activation ➔ may inhibit autophagy [32,33,34] | |
| LHBs/MHBs ➔ NF-κB activation ➔ ? [31,35] | |
| SHBs ➔ activation of UPR signaling ➔ incomplete autophagic response [33,36] | |
| SHBs ➔ interacts with LC3 ➔ incomplete autophagic response [36] | |
| Hepatitis C virus (HCV) Flaviviridae | |
| Hepatocellular carcinoma | Core/NS3 ➔ ERK activation ➔ may inhibit autophagy [37,38] |
| Core/NS3 ➔ JNK activation ➔ may promote autophagy [37,38] | |
| Core/NS3/NS4B/NS5A ➔ NF-κB activation ➔ ? [37,39,40] | |
| Core/NS3 ➔ p38 activation ➔ may inhibit autophagy [37,38] | |
| Core/NS3/NS5A ➔ interact with p53 ➔ may inhibit autophagy [41,42,43,44,45] | |
| Core/NS4B ➔ AKT activation ➔ may inhibit autophagy [46] | |
| NS3 ➔ interacts with IRGM ➔ increases autophagic markers [47] | |
| NS4B ➔ activation of UPR signaling ➔ autophagic markers [48] | |
| NS4B ➔ interacts with Rab5 and VPS34 ➔ incomplete autophagic response [48] | |
| NS5A ➔ PI3K/mTOR activation ➔ may inhibit autophagy [49,50] | |
| NS5A ➔ ERK inhibition ➔ may activate autophagy [50,51] | |
| NS5A ➔ PKR inhibition ➔ may inhibit autophagy [52] | |
| NS5A ➔ p38 inhibition ➔ may promote autophagy [53] | |
| NS5B ➔ interacts with ATG5 ➔ ? [54] | |
| ? ➔ increased Beclin-1 expression ➔ autophagic markers [55] | |
| Human papillomavirus, high-risk types (HPV) Papillomaviridae | |
| Cervical, anal and penile cancers, head and neck cancers | E5 ➔ p38 activation ➔ may inhibit autophagy [56] |
| E5/E6/E7 ➔ inhibit XBP1-splicing under basal conditions ➔ ? [57] | |
| E6 ➔ sustained AKT/mTORC1 activity ➔ may inhibit autophagy [58,59] | |
| E6 ➔ inhibits p53 ➔ may inhibit autophagy [60] | |
| E6/E7 ➔ ERK activation ➔ may inhibit autophagy [61] | |
| E7 ➔ AKT activation ➔ may inhibit autophagy [62,63] | |
| E7 ➔ NF-κB inhibition ➔ ? [64,65,66] | |
| E7 ➔ JNK inhibition ➔ may inhibit autophagy [67] | |
| E7 ➔ ? ➔ autophagic markers [68] | |
| Human T-lymphotropic virus (HTLV-1) Retroviridae | |
| Adult T-cell leukemia | Tax ➔ JNK activation ➔ may promote autophagy [69] |
| Tax ➔ sustained AKT/mTORC1 activity ➔ may inhibit autophagy [70,71] | |
| Tax ➔ p38 activation ➔ may inhibit autophagy [69] | |
| Tax ➔ activation of UPR signaling ➔ may promote autophagy [72] | |
| Tax ➔ inhibits p53 ➔ may inhibit autophagy [73] | |
| Tax+HBZ ➔ NF-κB activation ➔ ? [74,75,76,77] | |
| Kaposi’s sarcoma associated herpesvirus (KSHV) Herpesviridae | |
| Kaposi’s sarcoma, pleural effusion lymphoma, multicentric Castleman’s disease | K1 ➔ PI3K/AKT/mTOR activation ➔ may inhibit autophagy [78,79,80] |
| K15 ➔ ERK activation ➔ may inhibit autophagy [81] | |
| K15 ➔ p38 activation ➔ may inhibit autophagy [81] | |
| LANA ➔ inhibits p53 ➔ may inhibit autophagy [82] | |
| ORF45 ➔ sustained ERK/RSK activity ➔ autophagy [83,84] | |
| ORF49 ➔ JNK activation➔ may promote autophagy [85] | |
| ORF49/vGPCR ➔ p38 activation➔ may inhibit autophagy [85,86] | |
| RTA ➔? ➔ increased autophagy [87] | |
| vBCL-2 ➔ interacts with Beclin-1 ➔ autophagy inhibition [88,89] | |
| vFLIP ➔ interacts with ATG3 ➔ autophagy inhibition [90] | |
| vFLIP/K15/ORF75/miR-K1/vGPCR7/vIRF3 ➔ NF-κB activation ➔ ? [81,91,92,93,94,95,96] | |
| vGPCR ➔ PI3Kγ/mTORC1 activation ➔ may inhibit autophagy [97] | |
| vIRF2/vIRF3 ➔ PKR inhibition ➔ may inhibit autophagy [98,99] | |
| vIRF3 ➔ NF-κB inhibition ➔ ? [93,100] | |
| Merkel cell polyomavirus (MCPyV) Polyomaviridae | |
| Merkel cell carcinoma | Small T ➔ mTORC1 activation ➔ may inhibit autophagy [75] |
1.2. Autophagy—Basic Function and Role in Human Disease
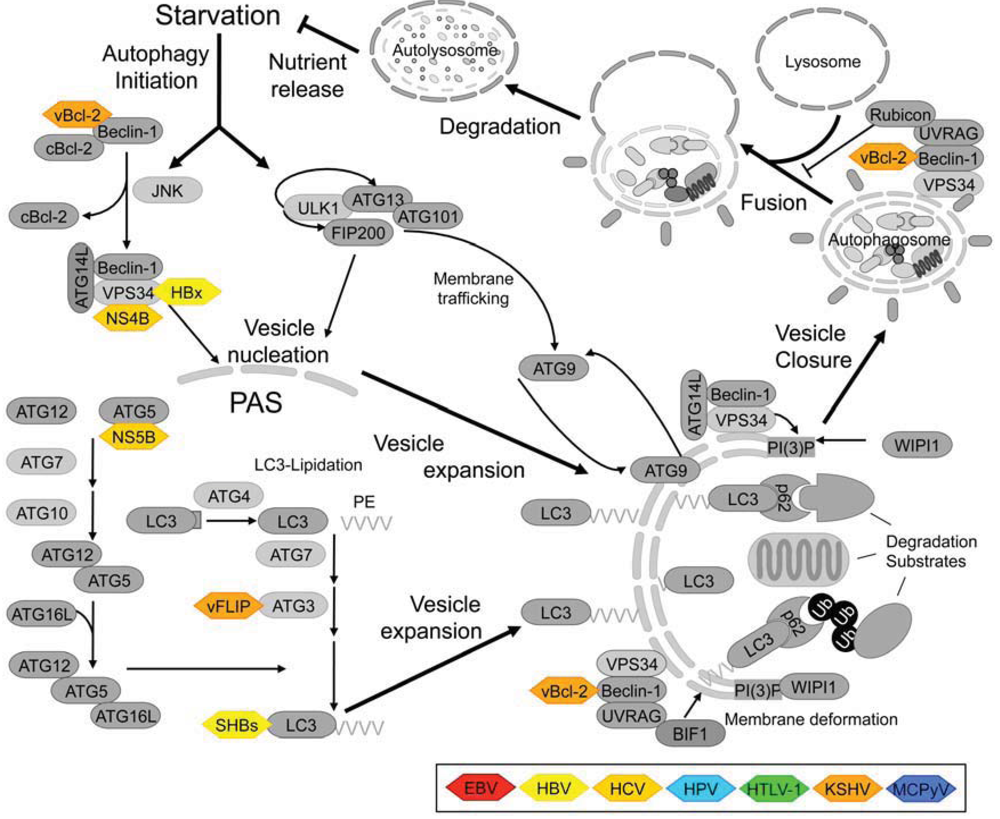
1.3. Components of the Mammalian Autophagy Machinery
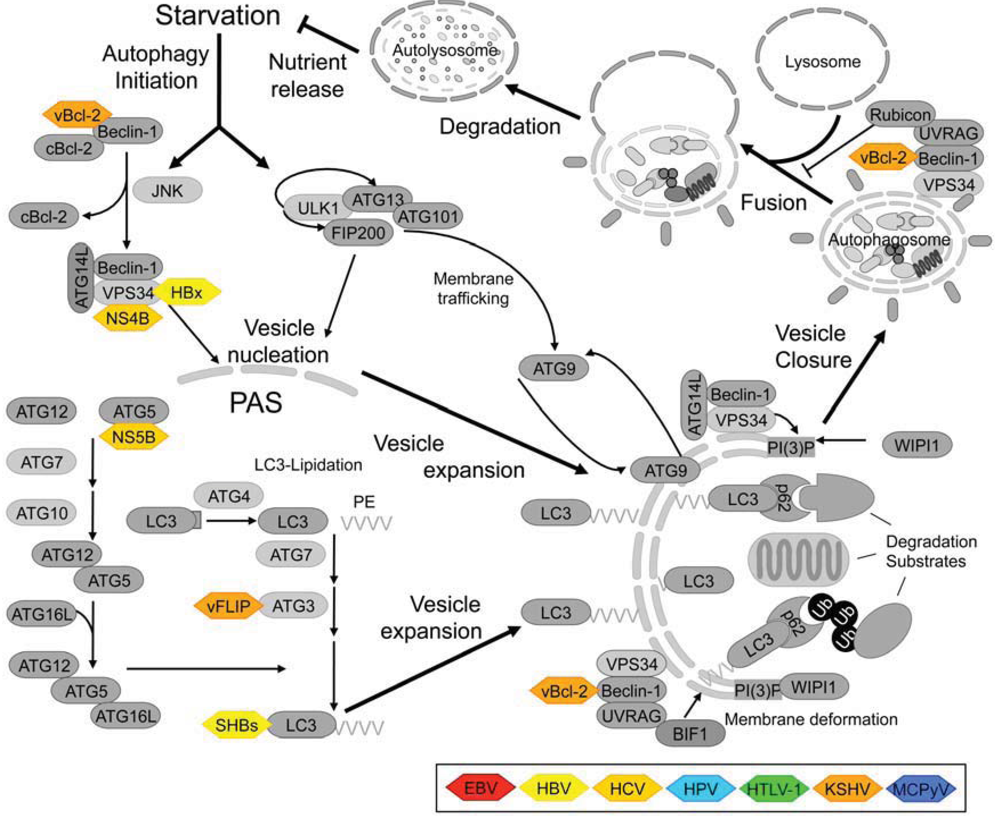
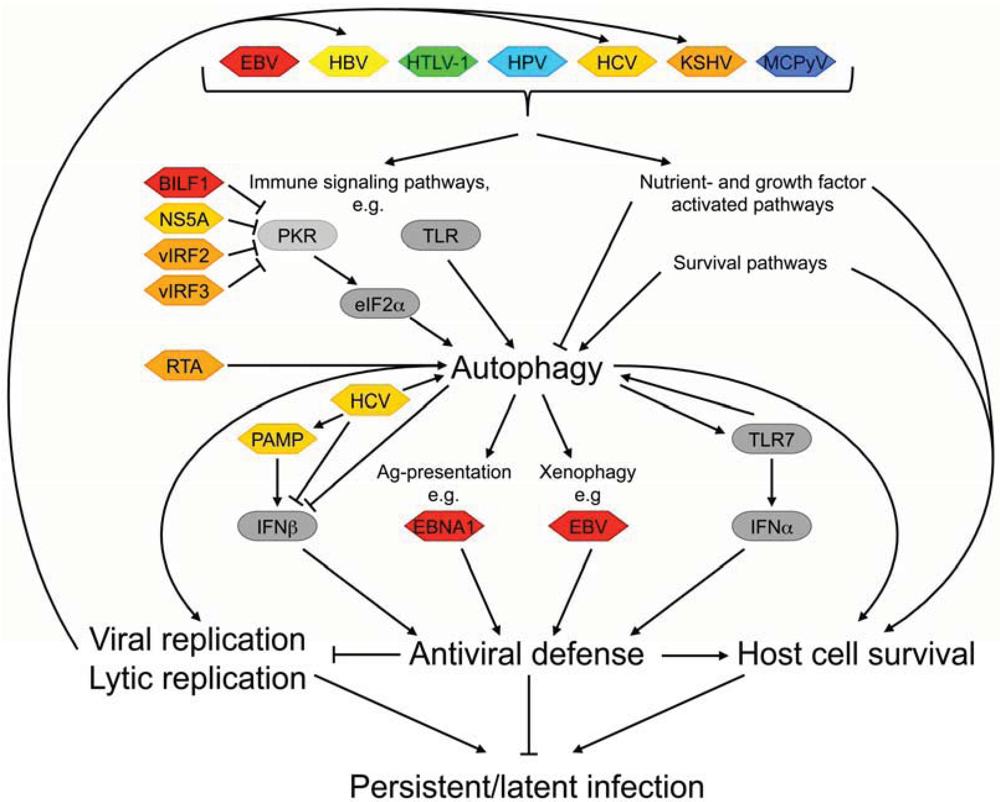
2. Benefits of Autophagy Modulation on the Viral Life Cycle
2.1. Viral Modulation of Autophagy-Mediated Immune Defense Mechanisms
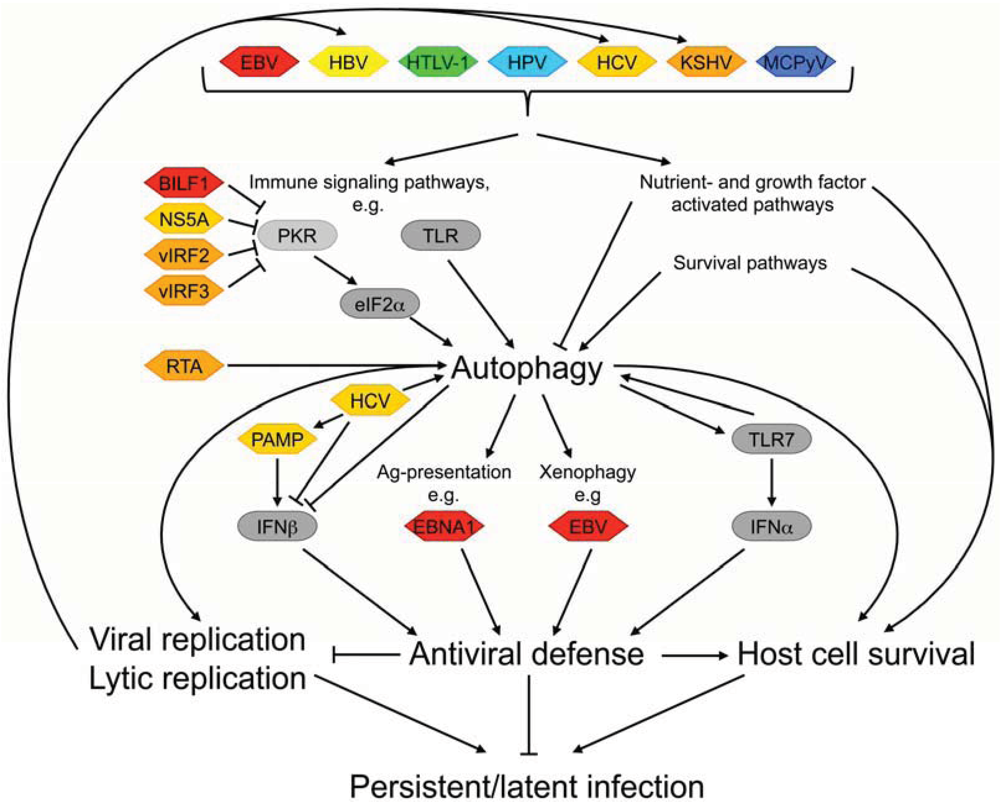
2.2. Virally Induced Autophagy and Replication
3. Mechanisms of Viral Interference with Autophagy
3.1. Viral Oncoproteins Directly Targeting the Autophagy Machinery
3.1.1. Beclin-1—A Popular Target with Viral Proteins
3.1.2. ATG3 Binding to FLIP-Proteins
3.1.3. Interactions of Viral Proteins with Other Autophagy-Regulating Proteins
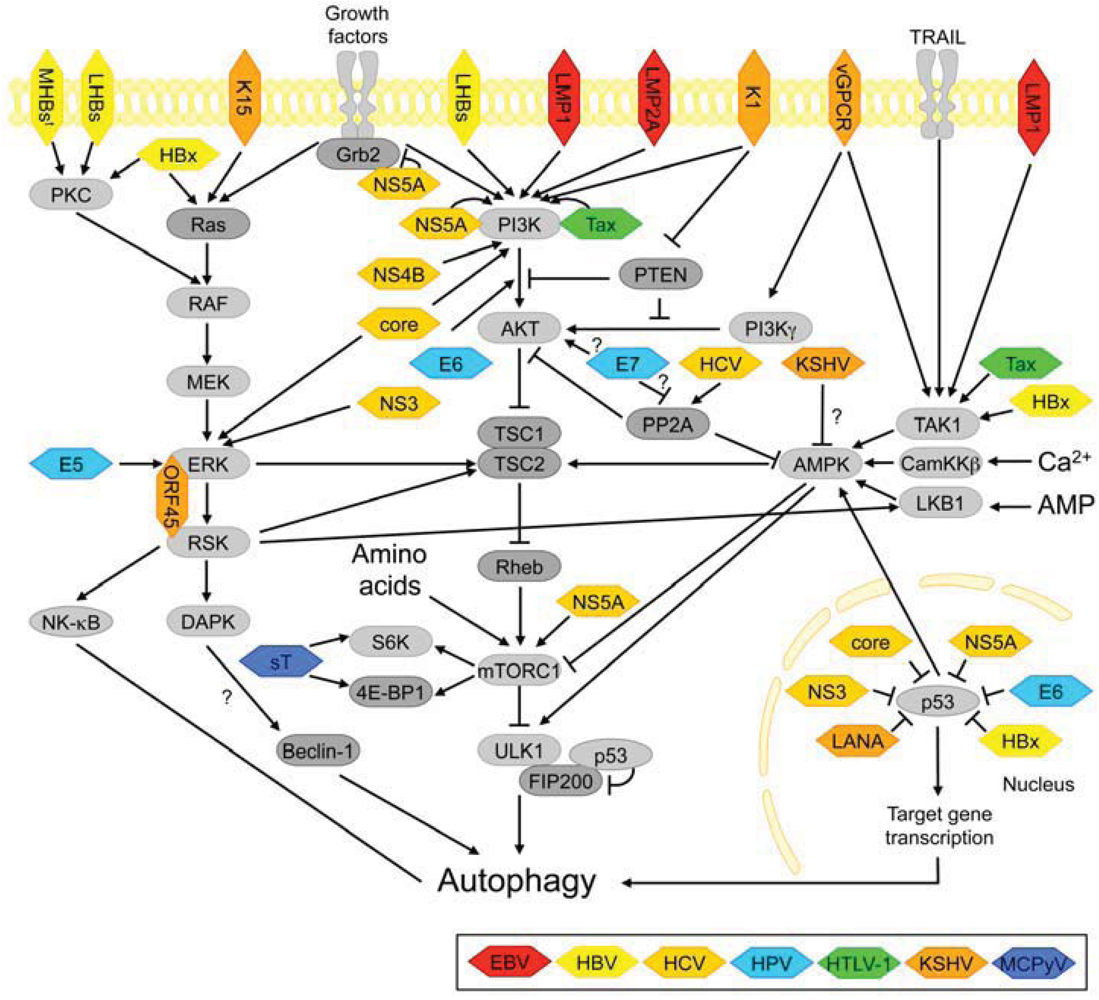
3.2. Autophagy-Regulating Signaling Pathways Targeted by Viral Oncoproteins
3.2.1. PI3K-AKT and mTORC1 Signaling
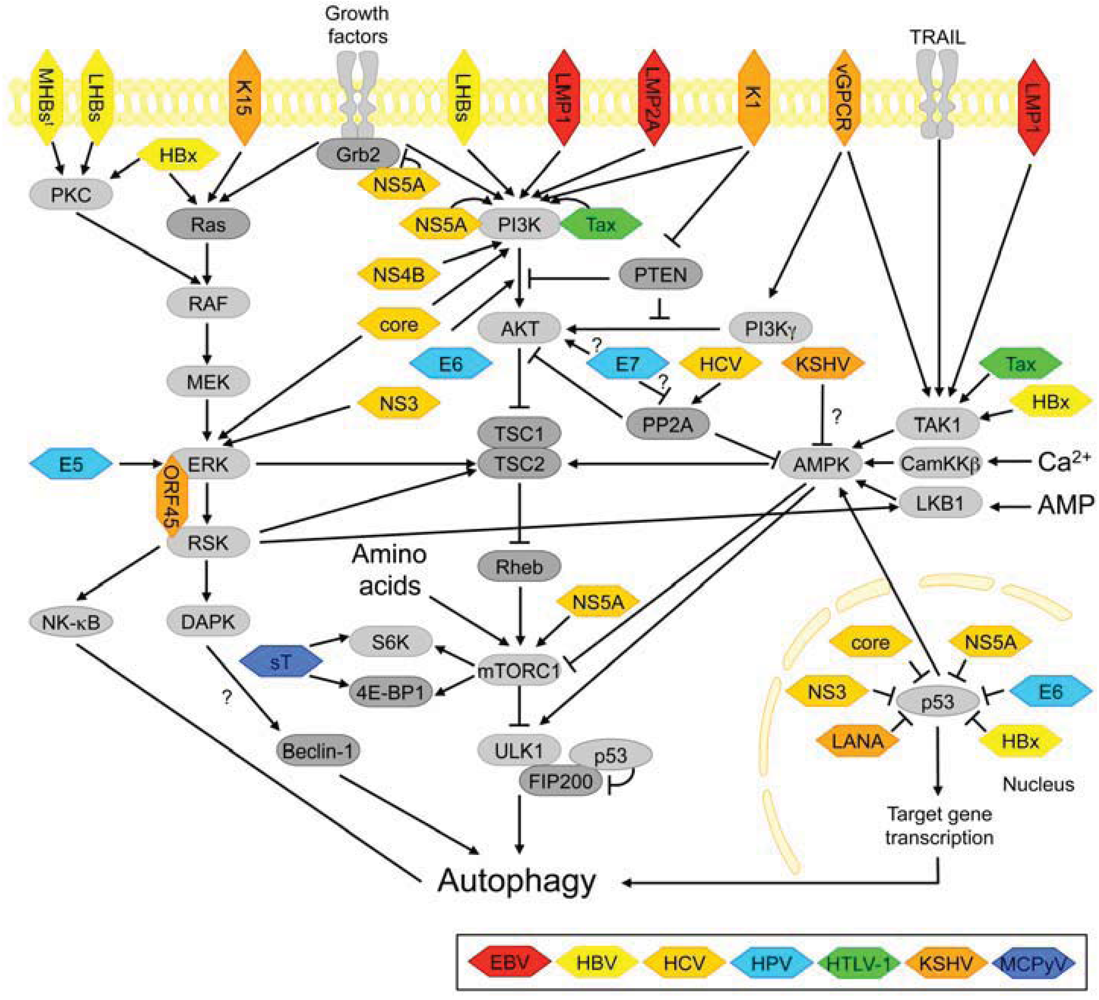
3.2.2. ERK Signaling
3.2.3. AMPK-Signaling
3.2.4. Other Kinases Involved in Starvation-Induced Autophagy
3.2.5. The p53 Tumor Suppressor
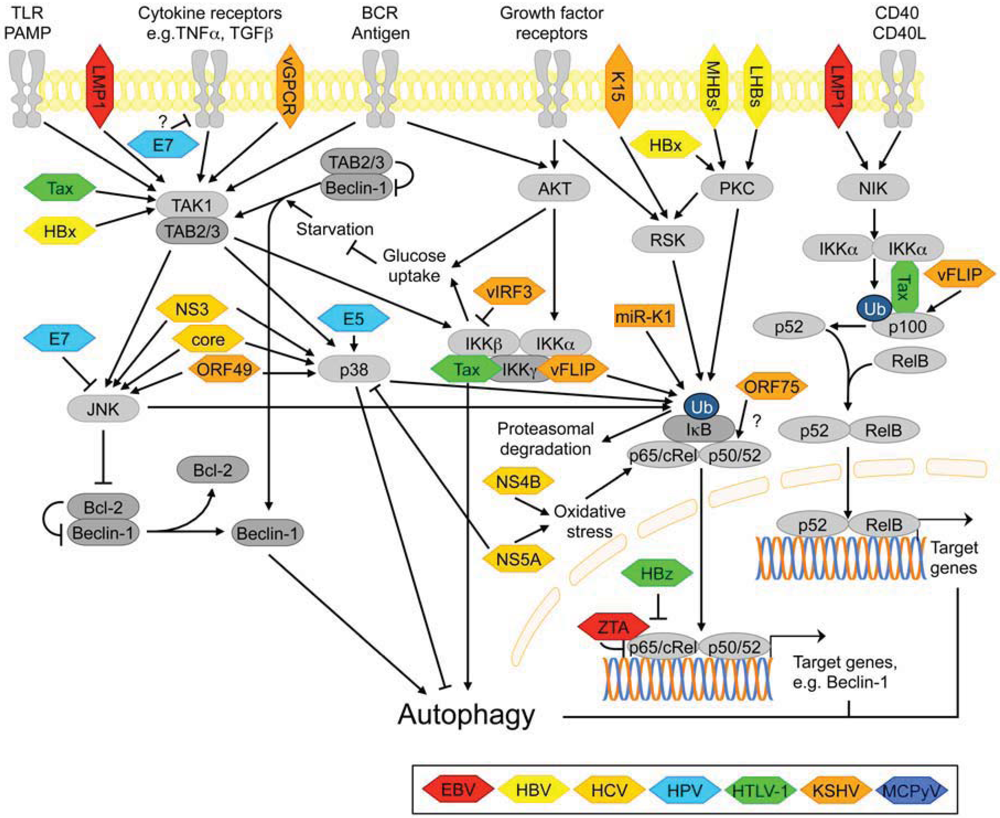
3.2.6. NF-κB Signaling
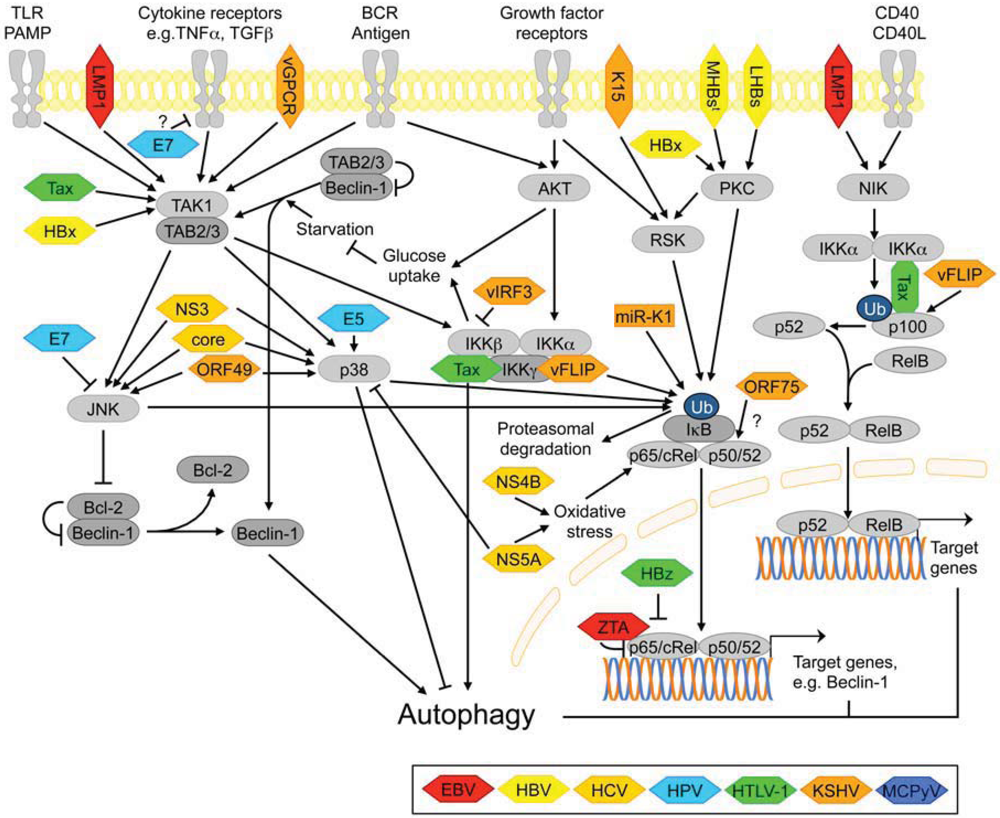
3.2.7. Signaling Pathways Activated by ER Stress
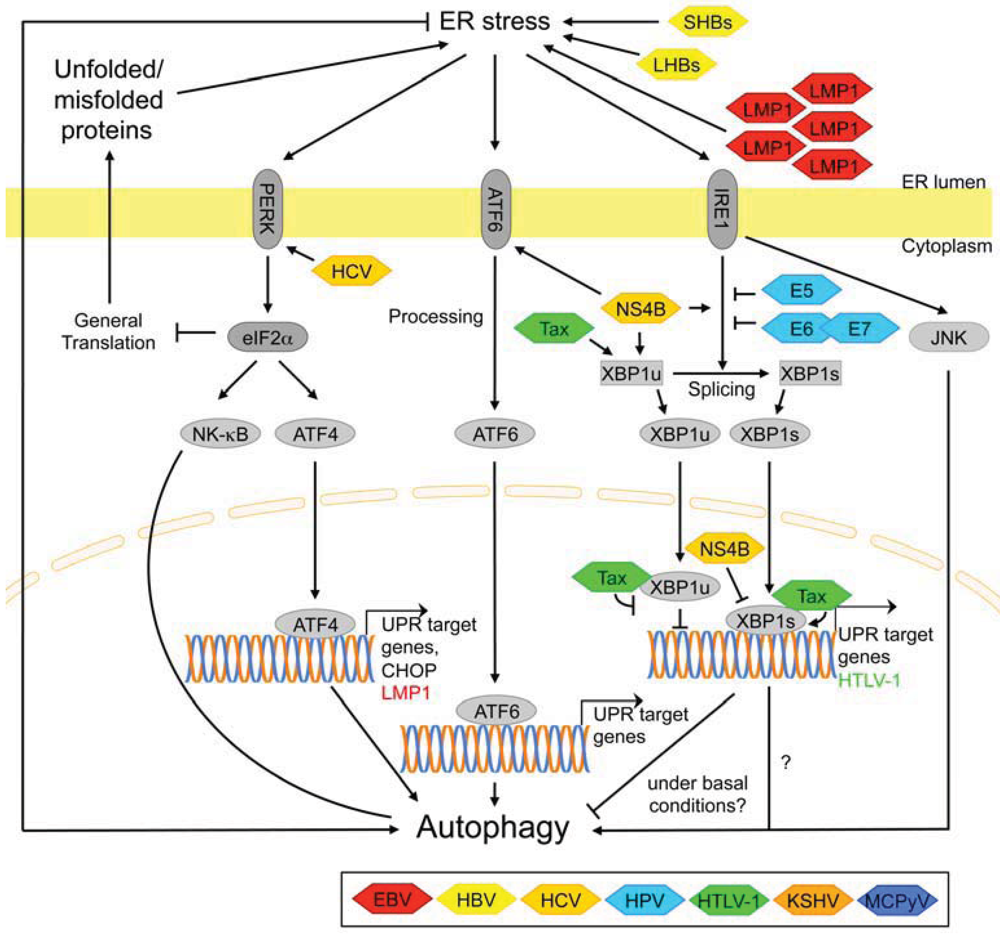
4. Concluding Remarks
Acknowledgements
Conflict of Interest
References
- Kochel, T.; Aguilar, P.; Felices, V.; Comach, G.; Cruz, C.; Alava, A.; Vargas, J.; Olson, J.; Blair, P. Molecular epidemiology of dengue virus type 3 in Northern South America: 2000–2005. Infect. Genet. Evol. 2008, 8, 682–688. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Shah, K.M.; Young, L.S. Epstein-Barr virus and carcinogenesis: Beyond Burkitt's lymphoma. Clin. Microbiol. Infect. 2009, 15, 982–988. [Google Scholar] [CrossRef]
- Tsai, W.L.; Chung, R.T. Viral hepatocarcinogenesis. Oncogene 2010, 29, 2309–2324. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: Viral infectivity, Tax, HBZ and therapy. Oncogene 2011, 30, 1379–1389. [Google Scholar] [CrossRef]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. Viruses associated with human cancer. Biochim. Biophys. Acta 2008, 1782, 127–150. [Google Scholar]
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; McGregor Dallas, S.R.; Smit, M.; Soroceanu, L.; Cobbs, C.S. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro. Oncol. 2012, 14, 246–255. [Google Scholar] [CrossRef]
- Martin, D.; Gutkind, J.S. Human tumor-associated viruses and new insights into the molecular mechanisms of cancer. Oncogene 2008, 2, S31–S42. [Google Scholar] [CrossRef]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar]
- Beisser, P.S.; Verzijl, D.; Gruijthuijsen, Y.K.; Beuken, E.; Smit, M.J.; Leurs, R.; Bruggeman, C.A.; Vink, C. The Epstein–Barr virus BILF1 gene encodes a G protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J. Virol. 2005, 79, 441–449. [Google Scholar]
- Eliopoulos, A.G.; Blake, S.M.; Floettmann, J.E.; Rowe, M.; Young, L.S. Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J. Virol. 1999, 73, 1023–1035. [Google Scholar]
- Eliopoulos, A.G.; Young, L.S. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene 1998, 16, 1731–1742. [Google Scholar]
- Uemura, N.; Kajino, T.; Sanjo, H.; Sato, S.; Akira, S.; Matsumoto, K.; Ninomiya-Tsuji, J. TAK1 is a component of the Epstein-Barr virus LMP1 complex and is essential for activation of JNK but not of NF-kappaB. J. Biol. Chem. 2006, 281, 7863–7872. [Google Scholar]
- Sommermann, T.G.; O'Neill, K.; Plas, D.R.; Cahir-McFarland, E. IKKbeta and NF-kappaB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Res. 2011, 71, 7291–7300. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Gallagher, N.J.; Blake, S.M.; Dawson, C.W.; Young, L.S. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem. 1999, 274, 16085–16096. [Google Scholar]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef]
- Lee, D.Y.; Sugden, B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 2008, 111, 2280–2289. [Google Scholar] [CrossRef]
- Chen, J.; Hu, C.F.; Hou, J.H.; Shao, Q.; Yan, L.X.; Zhu, X.F.; Zeng, Y.X.; Shao, J.Y. Epstein-Barr virus encoded latent membrane protein 1 regulates mTOR signaling pathway genes which predict poor prognosis of nasopharyngeal carcinoma. J. Transl. Med. 2010, 8, 30. [Google Scholar] [CrossRef]
- Lambert, S.L.; Martinez, O.M. Latent membrane protein 1 of EBV activates phosphatidylinositol 3-kinase to induce production of IL-10. J. Immunol. 2007, 179, 8225–8234. [Google Scholar]
- Moody, C.A.; Scott, R.S.; Amirghahari, N.; Nathan, C.O.; Young, L.S.; Dawson, C.W.; Sixbey, J.W. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J. Virol. 2005, 79, 5499–5506. [Google Scholar]
- Morrison, J.A.; Klingelhutz, A.J.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J. Virol. 2003, 77, 12276–12284. [Google Scholar]
- Scholle, F.; Bendt, K.M.; Raab-Traub, N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol. 2000, 74, 10681–10689. [Google Scholar] [CrossRef]
- Morrison, T.E.; Kenney, S.C. BZLF1, an Epstein-Barr virus immediate-early protein, induces p65 nuclear translocation while inhibiting p65 transcriptional function. Virology 2004, 328, 219–232. [Google Scholar] [CrossRef]
- Tang, H.; Da, L.; Mao, Y.; Li, Y.; Li, D.; Xu, Z.; Li, F.; Wang, Y.; Tiollais, P.; Li, T.; et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 2009, 49, 60–71. [Google Scholar] [CrossRef]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4383–4388. [Google Scholar]
- Feitelson, M.A.; Zhu, M.; Duan, L.X.; London, W.T. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene 1993, 8, 1109–1117. [Google Scholar]
- Tarn, C.; Zou, L.; Hullinger, R.L.; Andrisani, O.M. Hepatitis B virus X protein activates the p38 mitogen-activated protein kinase pathway in dedifferentiated hepatocytes. J. Virol. 2002, 76, 9763–9772. [Google Scholar]
- Benn, J.; Schneider, R.J. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 10350–10354. [Google Scholar] [CrossRef]
- Hildt, E.; Munz, B.; Saher, G.; Reifenberg, K.; Hofschneider, P.H. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002, 21, 525–535. [Google Scholar]
- Liu, H.; Xu, J.; Zhou, L.; Yun, X.; Chen, L.; Wang, S.; Sun, L.; Wen, Y.; Gu, J. Hepatitis B virus large surface antigen promotes liver carcinogenesis by activating the Src/PI3K/Akt pathway. Cancer Res. 2011, 71, 7547–7557. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Chang, W.W.; Lei, H.Y.; Lai, M.D.; Chang, W.T.; Huang, W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef]
- Teng, C.F.; Wu, H.C.; Tsai, H.W.; Shiah, H.S.; Huang, W.; Su, I.J. Novel feedback inhibition of surface antigen synthesis by mammalian target of rapamycin (mTOR) signal and its implication for hepatitis B virus tumorigenesis and therapy. Hepatology 2011, 54, 1199–1207. [Google Scholar] [CrossRef]
- Meyer, M.; Caselmann, W.H.; Schluter, V.; Schreck, R.; Hofschneider, P.H.; Baeuerle, P.A. Hepatitis B virus transactivator MHBst: Activation of NF-kappa B, selective inhibition by antioxidants and integral membrane localization. EMBO J. 1992, 11, 2991–3001. [Google Scholar]
- Li, J.; Liu, Y.; Wang, Z.; Liu, K.; Wang, Y.; Liu, J.; Ding, H.; Yuan, Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 2011, 85, 6319–6333. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Erhardt, A.; Hassan, M.; Heintges, T.; Haussinger, D. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology 2002, 292, 272–284. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 9599–9604. [Google Scholar]
- Li, S.; Ye, L.; Yu, X.; Xu, B.; Li, K.; Zhu, X.; Liu, H.; Wu, X.; Kong, L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology 2009, 391, 257–264. [Google Scholar] [CrossRef]
- Ishido, S.; Hotta, H. Complex formation of the nonstructural protein 3 of hepatitis C virus with the p53 tumor suppressor. FEBS Lett. 1998, 438, 258–262. [Google Scholar] [CrossRef]
- Kao, C.F.; Chen, S.Y.; Chen, J.Y.; Wu Lee, Y.H. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 2004, 23, 2472–2483. [Google Scholar] [CrossRef]
- Lu, W.; Lo, S.Y.; Chen, M.; Wu, K.; Fung, Y.K.; Ou, J.H. Activation of p53 tumor suppressor by hepatitis C virus core protein. Virology 1999, 264, 134–141. [Google Scholar] [CrossRef]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef]
- Otsuka, M.; Kato, N.; Lan, K.; Yoshida, H.; Kato, J.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 2000, 275, 34122–34130. [Google Scholar]
- Park, C.Y.; Jun, H.J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef]
- Tan, S.L.; Nakao, H.; He, Y.; Vijaysri, S.; Neddermann, P.; Jacobs, B.L.; Mayer, B.J.; Katze, M.G. NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5533–5538. [Google Scholar]
- Gale, M., Jr.; Blakely, C.M.; Kwieciszewski, B.; Tan, S.L.; Dossett, M.; Tang, N.M.; Korth, M.J.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: Molecular mechanisms of kinase regulation. Mol. Cell Biol. 1998, 18, 5208–5218. [Google Scholar]
- Mankouri, J.; Dallas, M.L.; Hughes, M.E.; Griffin, S.D.; Macdonald, A.; Peers, C.; Harris, M. Suppression of a pro-apoptotic K+ channel as a mechanism for hepatitis C virus persistence. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 15903–15908. [Google Scholar]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef]
- Crusius, K.; Rodriguez, I.; Alonso, A. The human papillomavirus type 16 E5 protein modulates ERK1/2 and p38 MAP kinase activation by an EGFR-independent process in stressed human keratinocytes. Virus Genes 2000, 20, 65–69. [Google Scholar] [CrossRef]
- Sudarshan, S.R.; Schlegel, R.; Liu, X. The HPV-16 E5 protein represses expression of stress pathway genes XBP-1 and COX-2 in genital keratinocytes. Biochem. Biophys. Res. Commun. 2010, 399, 617–622. [Google Scholar] [CrossRef]
- Lu, Z.; Hu, X.; Li, Y.; Zheng, L.; Zhou, Y.; Jiang, H.; Ning, T.; Basang, Z.; Zhang, C.; Ke, Y. Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J. Biol. Chem. 2004, 279, 35664–35670. [Google Scholar]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Dannenberg, A.J. Cyclooxygenase-2 transcription is regulated by human papillomavirus 16 E6 and E7 oncoproteins: Evidence of a corepressor/coactivator exchange. Cancer Res. 2007, 67, 3976–3985. [Google Scholar] [CrossRef]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef]
- Basile, J.R.; Zacny, V.; Munger, K. The cytokines tumor necrosis factor-alpha (TNF-alpha ) and TNF-related apoptosis-inducing ligand differentially modulate proliferation and apoptotic pathways in human keratinocytes expressing the human papillomavirus-16 E7 oncoprotein. J. Biol. Chem. 2001, 276, 22522–22528. [Google Scholar]
- Pietenpol, J.A.; Stein, R.W.; Moran, E.; Yaciuk, P.; Schlegel, R.; Lyons, R.M.; Pittelkow, M.R.; Munger, K.; Howley, P.M.; Moses, H.L. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 1990, 61, 777–785. [Google Scholar] [CrossRef]
- Vandermark, E.R.; Deluca, K.A.; Gardner, C.R.; Marker, D.F.; Schreiner, C.N.; Strickland, D.A.; Wilton, K.M.; Mondal, S.; Woodworth, C.D. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 2012, 425, 53–60. [Google Scholar] [CrossRef]
- Mileo, A.M.; Abbruzzese, C.; Mattarocci, S.; Bellacchio, E.; Pisano, P.; Federico, A.; Maresca, V.; Picardo, M.; Giorgi, A.; Maras, B.; et al. Human papillomavirus-16 E7 interacts with glutathione S-transferase P1 and enhances its role in cell survival. PLoS One 2009, 4, e7254. [Google Scholar]
- Zhou, X.; Munger, K. Expression of the human papillomavirus type 16 E7 oncoprotein induces an autophagy-related process and sensitizes normal human keratinocytes to cell death in response to growth factor deprivation. Virology 2009, 385, 192–197. [Google Scholar] [CrossRef]
- Suzuki, S.; Singhirunnusorn, P.; Mori, A.; Yamaoka, S.; Kitajima, I.; Saiki, I.; Sakurai, H. Constitutive activation of TAK1 by HTLV-1 tax-dependent overexpression of TAB2 induces activation of JNK-ATF2 but not IKK-NF-kappaB. J. Biol. Chem. 2007, 282, 25177–25181. [Google Scholar]
- Peloponese, J.M., Jr.; Jeang, K.T. Role for Akt/protein kinase B and activator protein-1 in cellular proliferation induced by the human T-cell leukemia virus type 1 tax oncoprotein. J. Biol. Chem. 2006, 281, 8927–8938. [Google Scholar] [CrossRef]
- Yoshita, M.; Higuchi, M.; Takahashi, M.; Oie, M.; Tanaka, Y.; Fujii, M. Activation of mTOR by human T-cell leukemia virus type 1 Tax is important for the transformation of mouse T cells to interleukin-2-independent growth. Cancer Sci. 2011, 103, 369–374. [Google Scholar]
- Ku, S.C.; Lee, J.; Lau, J.; Gurumurthy, M.; Ng, R.; Lwa, S.H.; Lee, J.; Klase, Z.; Kashanchi, F.; Chao, S.H. XBP-1, a novel human T-lymphotropic virus type 1 (HTLV-1) tax binding protein, activates HTLV-1 basal and tax-activated transcription. J. Virol. 2008, 82, 4343–4353. [Google Scholar]
- Jeong, S.J.; Radonovich, M.; Brady, J.N.; Pise-Masison, C.A. HTLV-I Tax induces a novel interaction between p65/RelA and p53 that results in inhibition of p53 transcriptional activity. Blood 2004, 104, 1490–1497. [Google Scholar]
- Geleziunas, R.; Ferrell, S.; Lin, X.; Mu, Y.; Cunningham, E.T., Jr.; Grant, M.; Connelly, M.A.; Hambor, J.E.; Marcu, K.B.; Greene, W.C. Human T-cell leukemia virus type 1 Tax induction of NF-kappaB involves activation of the IkappaB kinase alpha (IKKalpha) and IKKbeta cellular kinases. Mol. Cell Biol. 1998, 18, 5157–5165. [Google Scholar]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Invest. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Uhlik, M.; Good, L.; Xiao, G.; Harhaj, E.W.; Zandi, E.; Karin, M.; Sun, S.C. NF-kappaB-inducing kinase and IkappaB kinase participate in human T-cell leukemia virus I Tax-mediated NF-kappaB activation. J. Biol. Chem. 1998, 273, 21132–21136. [Google Scholar]
- Zhao, T.; Yasunaga, J.; Satou, Y.; Nakao, M.; Takahashi, M.; Fujii, M.; Matsuoka, M. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood 2009, 113, 2755–2764. [Google Scholar] [CrossRef]
- Tomlinson, C.C.; Damania, B. The K1 protein of Kaposi's sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol. 2004, 78, 1918–1927. [Google Scholar] [CrossRef]
- Wang, L.; Damania, B. Kaposi's sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 2008, 68, 4640–4648. [Google Scholar] [CrossRef]
- Wang, L.; Dittmer, D.P.; Tomlinson, C.C.; Fakhari, F.D.; Damania, B. Immortalization of primary endothelial cells by the K1 protein of Kaposi's sarcoma-associated herpesvirus. Cancer Res. 2006, 66, 3658–3666. [Google Scholar]
- Brinkmann, M.M.; Glenn, M.; Rainbow, L.; Kieser, A.; Henke-Gendo, C.; Schulz, T.F. Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 2003, 77, 9346–9358. [Google Scholar] [CrossRef]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar]
- Kuang, E.; Fu, B.; Liang, Q.; Myoung, J.; Zhu, F. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J. Biol. Chem. 2011, 286, 41171–41182. [Google Scholar]
- Kuang, E.; Tang, Q.; Maul, G.G.; Zhu, F. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi's sarcoma-associated herpesvirus and its role in viral lytic replication. J. Virol. 2008, 82, 1838–1850. [Google Scholar] [CrossRef]
- Gonzalez, C.M.; Wong, E.L.; Bowser, B.S.; Hong, G.K.; Kenney, S.; Damania, B. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 2006, 80, 3062–3070. [Google Scholar] [CrossRef]
- Bais, C.; Santomasso, B.; Coso, O.; Arvanitakis, L.; Raaka, E.G.; Gutkind, J.S.; Asch, A.S.; Cesarman, E.; Gershengorn, M.C.; Mesri, E.A. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 1998, 391, 86–89. [Google Scholar]
- Wen, H.J.; Yang, Z.; Zhou, Y.; Wood, C. Enhancement of autophagy during lytic replication by the Kaposi's sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 2010, 84, 7448–7458. [Google Scholar]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef]
- Cannon, M.L.; Cesarman, E. The KSHV G protein-coupled receptor signals via multiple pathways to induce transcription factor activation in primary effusion lymphoma cells. Oncogene 2004, 23, 514–523. [Google Scholar]
- Guasparri, I.; Keller, S.A.; Cesarman, E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 2004, 199, 993–1003. [Google Scholar] [CrossRef]
- Konrad, A.; Wies, E.; Thurau, M.; Marquardt, G.; Naschberger, E.; Hentschel, S.; Jochmann, R.; Schulz, T.F.; Erfle, H.; Brors, B.; et al. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-kappaB activation. J. Virol. 2009, 83, 2563–2574. [Google Scholar]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef]
- Martin, D.; Galisteo, R.; Ji, Y.; Montaner, S.; Gutkind, J.S. An NF-kappaB gene expression signature contributes to Kaposi's sarcoma virus vGPCR-induced direct and paracrine neoplasia. Oncogene 2008, 27, 1844–1852. [Google Scholar] [CrossRef]
- Seo, T.; Park, J.; Lim, C.; Choe, J. Inhibition of nuclear factor kappaB activity by viral interferon regulatory factor 3 of Kaposi's sarcoma-associated herpesvirus. Oncogene 2004, 23, 6146–6155. [Google Scholar] [CrossRef]
- Martin, D.; Galisteo, R.; Molinolo, A.A.; Wetzker, R.; Hirsch, E.; Gutkind, J.S. PI3Kgamma mediates kaposi's sarcoma-associated herpesvirus vGPCR-induced sarcomagenesis. Cancer Cell 2011, 19, 805–813. [Google Scholar] [CrossRef]
- Burysek, L.; Pitha, P.M. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J. Virol. 2001, 75, 2345–2352. [Google Scholar] [CrossRef]
- Esteban, M.; Garcia, M.A.; Domingo-Gil, E.; Arroyo, J.; Nombela, C.; Rivas, C. The latency protein LANA2 from Kaposi's sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol. 2003, 84, 1463–1470. [Google Scholar] [CrossRef]
- Lee, B.S.; Paulose-Murphy, M.; Chung, Y.H.; Connlole, M.; Zeichner, S.; Jung, J.U. Suppression of tetradecanoyl phorbol acetate-induced lytic reactivation of Kaposi's sarcoma-associated herpesvirus by K1 signal transduction. J. Virol. 2002, 76, 12185–12199. [Google Scholar]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2012, 8. in press.. [Google Scholar]
- Webber, J.L.; Tooze, S.A. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2010, 29, 27–40. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Shen, S.; Kepp, O.; Kroemer, G. The end of autophagic cell death? Autophagy 2012, 8, 1–3. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar] [CrossRef]
- Kanki, T.; Wang, K.; Baba, M.; Bartholomew, C.R.; Lynch-Day, M.A.; Du, Z.; Geng, J.; Mao, K.; Yang, Z.; Yen, W.L.; et al. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol. Biol. Cell 2009, 20, 4730–4738. [Google Scholar] [CrossRef]
- Suzuki, K.; Kondo, C.; Morimoto, M.; Ohsumi, Y. Selective transport of alpha-mannosidase by autophagic pathways: Identification of a novel receptor, Atg34p. J. Biol. Chem. 2010, 285, 30019–30025. [Google Scholar]
- Nazarko, V.Y.; Nazarko, T.Y.; Farre, J.C.; Stasyk, O.V.; Warnecke, D.; Ulaszewski, S.; Cregg, J.M.; Sibirny, A.A.; Subramani, S. Atg35, a micropexophagy-specific protein that regulates micropexophagic apparatus formation in Pichia pastoris. Autophagy 2011, 7, 375–385. [Google Scholar] [CrossRef]
- Meijer, W.H.; van der Klei, I.J.; Veenhuis, M.; Kiel, J.A. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007, 3, 106–116. [Google Scholar]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar]
- Zhao, Z.; Fux, B.; Goodwin, M.; Dunay, I.R.; Strong, D.; Miller, B.C.; Cadwell, K.; Delgado, M.A.; Ponpuak, M.; Green, K.G.; et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 2008, 4, 458–469. [Google Scholar] [CrossRef]
- Levine, B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar]
- Valencia, S.M.; Hutt-Fletcher, L.M. Important but differential roles for actin in trafficking of Epstein-Barr virus in B cells and epithelial cells. J. Virol. 2012, 86, 2–10. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar]
- Schmid, D.; Pypaert, M.; Munz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Muller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 7922–7927. [Google Scholar]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2165–2170. [Google Scholar]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef]
- Martin, H.J.; Lee, J.M.; Walls, D.; Hayward, S.D. Manipulation of the toll-like receptor 7 signaling pathway by Epstein-Barr virus. J. Virol. 2007, 81, 9748–9758. [Google Scholar] [CrossRef]
- Mizobe, T.; Tsukada, J.; Higashi, T.; Mouri, F.; Matsuura, A.; Tanikawa, R.; Minami, Y.; Yoshida, Y.; Tanaka, Y. Constitutive association of MyD88 to IRAK in HTLV-I-transformed T cells. Exp. Hematol. 2007, 35, 1812–1822. [Google Scholar] [CrossRef]
- West, J.; Damania, B. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J. Virol. 2008, 82, 5440–5449. [Google Scholar] [CrossRef]
- Balachandran, S.; Barber, G.N. PKR in innate immunity, cancer, and viral oncolysis. Methods Mol. Biol. 2007, 383, 277–301. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.t.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 190–195. [Google Scholar]
- Orvedahl, A.; Levine, B. Viral evasion of autophagy. Autophagy 2008, 4, 280–285. [Google Scholar]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011, 121, 37–56. [Google Scholar] [CrossRef]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef]
- Estrabaud, E.; De Muynck, S.; Asselah, T. Activation of unfolded protein response and autophagy during HCV infection modulates innate immune response. J. Hepatol. 2011, 55, 1150–1153. [Google Scholar] [CrossRef]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14046–14051. [Google Scholar]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar]
- Khakpoor, A.; Panyasrivanit, M.; Wikan, N.; Smith, D.R. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 2009, 90, 1093–1103. [Google Scholar] [CrossRef]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 2009, 90, 448–456. [Google Scholar]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar]
- Tian, Y.; Sir, D.; Kuo, C.F.; Ann, D.K.; Ou, J.H. Autophagy required for hepatitis B virus replication in transgenic mice. J. Virol. 2011, 85, 13453–13456. [Google Scholar] [CrossRef]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia-Monzon, C.; et al. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 2012, 142, 644–653 e643. [Google Scholar]
- Sun, R.; Lin, S.F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar]
- Cheng, E.H.; Nicholas, J.; Bellows, D.S.; Hayward, G.S.; Guo, H.G.; Reitz, M.S.; Hardwick, J.M. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 690–694. [Google Scholar]
- Sarid, R.; Sato, T.; Bohenzky, R.A.; Russo, J.J.; Chang, Y. Kaposi's sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat. Med. 1997, 3, 293–298. [Google Scholar] [CrossRef]
- Virgin, H.W.t.; Latreille, P.; Wamsley, P.; Hallsworth, K.; Weck, K.E.; Dal Canto, A.J.; Speck, S.H. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 1997, 71, 5894–5904. [Google Scholar]
- Liang, C.; E, X.; Jung, J.U. Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy 2008, 4, 268–272. [Google Scholar]
- Pearson, G.R.; Luka, J.; Petti, L.; Sample, J.; Birkenbach, M.; Braun, D.; Kieff, E. Identification of an Epstein-Barr virus early gene encoding a second component of the restricted early antigen complex. Virology 1987, 160, 151–161. [Google Scholar] [CrossRef]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schroter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef]
- Dazert, E.; Hall, M.N. mTOR signaling in disease. Curr. Opin. Cell Biol. 2011, 23, 744–755. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- Cen, O.; Longnecker, R. Rapamycin reverses splenomegaly and inhibits tumor development in a transgenic model of Epstein-Barr virus-related Burkitt's lymphoma. Mol. Cancer Ther. 2011, 10, 679–686. [Google Scholar] [CrossRef]
- Nichols, L.A.; Adang, L.A.; Kedes, D.H. Rapamycin blocks production of KSHV/HHV8: Insights into the anti-tumor activity of an immunosuppressant drug. PLoS One 2011, 6, e14535. [Google Scholar]
- Stallone, G.; Schena, A.; Infante, B.; Di Paolo, S.; Loverre, A.; Maggio, G.; Ranieri, E.; Gesualdo, L.; Schena, F.P.; Grandaliano, G. Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N. Engl. J. Med. 2005, 352, 1317–1323. [Google Scholar]
- Severi, T.; van Malenstein, H.; Verslype, C.; van Pelt, J.F. Tumor initiation and progression in hepatocellular carcinoma: Risk factors, classification, and therapeutic targets. Acta Pharmacol. Sin. 2010, 31, 1409–1420. [Google Scholar] [CrossRef]
- Wagner, D.; Kniepeiss, D.; Schaffellner, S.; Jakoby, E.; Mueller, H.; Fahrleitner-Pammer, A.; Stiegler, P.; Tscheliessnigg, K.H.; Iberer, F. Sirolimus has a potential to influent viral recurrence in HCV positive liver transplant candidates. Int. Immunopharmacol. 2010, 10, 990–993. [Google Scholar] [CrossRef]
- Stelzer, M.K.; Pitot, H.C.; Liem, A.; Lee, D.; Kennedy, G.D.; Lambert, P.F. Rapamycin inhibits anal carcinogenesis in two preclinical animal models. Cancer Prev. Res. (Phila) 2010, 3, 1542–1551. [Google Scholar] [CrossRef]
- Darwiche, N.; Sinjab, A.; Abou-Lteif, G.; Chedid, M.B.; Hermine, O.; Dbaibo, G.; Bazarbachi, A. Inhibition of mammalian target of rapamycin signaling by everolimus induces senescence in adult T-cell leukemia/lymphoma and apoptosis in peripheral T-cell lymphomas. Int. J. Cancer 2011, 129, 993–1004. [Google Scholar] [CrossRef]
- Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Yang, Y.; Kuwayama, Y.; Adachi, Y.; Takeuchi, T.; Koeffler, H.P.; Taguchi, H. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk. Res. 2007, 31, 673–682. [Google Scholar] [CrossRef]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef]
- Kaye, K.M.; Izumi, K.M.; Kieff, E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 9150–9154. [Google Scholar]
- Wang, D.; Liebowitz, D.; Kieff, E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 1985, 43, 831–840. [Google Scholar]
- Kulwichit, W.; Edwards, R.H.; Davenport, E.M.; Baskar, J.F.; Godfrey, V.; Raab-Traub, N. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 11963–11968. [Google Scholar]
- Longnecker, R.; Kieff, E. A second Epstein-Barr virus membrane protein (LMP2) is expressed in latent infection and colocalizes with LMP1. J. Virol. 1990, 64, 2319–2326. [Google Scholar]
- Speck, P.; Kline, K.A.; Cheresh, P.; Longnecker, R. Epstein-Barr virus lacking latent membrane protein 2 immortalizes B cells with efficiency indistinguishable from that of wild-type virus. J. Gen. Virol. 1999, 80, 2193–2203. [Google Scholar]
- Shair, K.H.; Bendt, K.M.; Edwards, R.H.; Nielsen, J.N.; Moore, D.T.; Raab-Traub, N. Epstein-Barr virus encoded LMP1 and LMP2A function co-operatively to promote carcinoma development in a mouse carcinogenesis model. J. Virol. 2012, 86, 5352–5365. [Google Scholar] [CrossRef]
- Portis, T.; Longnecker, R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 2004, 23, 8619–8628. [Google Scholar] [CrossRef]
- Burkhardt, A.L.; Bolen, J.B.; Kieff, E.; Longnecker, R. An Epstein-Barr virus transformation-associated membrane protein interacts with src family tyrosine kinases. J. Virol. 1992, 66, 5161–5167. [Google Scholar]
- Miller, C.L.; Lee, J.H.; Kieff, E.; Longnecker, R. An integral membrane protein (LMP2) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 772–776. [Google Scholar]
- Panousis, C.G.; Rowe, D.T. Epstein-Barr virus latent membrane protein 2 associates with and is a substrate for mitogen-activated protein kinase. J. Virol. 1997, 71, 4752–4760. [Google Scholar]
- Rommel, C.; Camps, M.; Ji, H. PI3K delta and PI3K gamma: Partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol. 2007, 7, 191–201. [Google Scholar] [CrossRef]
- Sodhi, A.; Chaisuparat, R.; Hu, J.; Ramsdell, A.K.; Manning, B.D.; Sausville, E.A.; Sawai, E.T.; Molinolo, A.; Gutkind, J.S.; Montaner, S. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 2006, 10, 133–143. [Google Scholar] [CrossRef]
- Lee, H.; Guo, J.; Li, M.; Choi, J.K.; DeMaria, M.; Rosenzweig, M.; Jung, J.U. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi's sarcoma-associated herpesvirus. Mol. Cell Biol. 1998, 18, 5219–5228. [Google Scholar]
- Lagunoff, M.; Majeti, R.; Weiss, A.; Ganem, D. Deregulated signal transduction by the K1 gene product of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5704–5709. [Google Scholar] [CrossRef]
- Lee, B.S.; Connole, M.; Tang, Z.; Harris, N.L.; Jung, J.U. Structural analysis of the Kaposi's sarcoma-associated herpesvirus K1 protein. J. Virol. 2003, 77, 8072–8086. [Google Scholar] [CrossRef]
- Cross, J.C.; Wen, P.; Rutter, W.J. Transactivation by hepatitis B virus X protein is promiscuous and dependent on mitogen-activated cellular serine/threonine kinases. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 8078–8082. [Google Scholar]
- Peng, L.; Liang, D.; Tong, W.; Li, J.; Yuan, Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J. Biol. Chem. 2010, 285, 20870–20881. [Google Scholar]
- Guo, H.; Zhou, T.; Jiang, D.; Cuconati, A.; Xiao, G.H.; Block, T.M.; Guo, J.T. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway. J. Virol. 2007, 81, 10072–10080. [Google Scholar]
- Mannova, P.; Beretta, L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell survival and viral replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef]
- Ishida, H.; Li, K.; Yi, M.; Lemon, S.M. p21-activated kinase 1 is activated through the mammalian target of rapamycin/p70 S6 kinase pathway and regulates the replication of hepatitis C virus in human hepatoma cells. J. Biol. Chem. 2007, 282, 11836–11848. [Google Scholar]
- Shao, R.X.; Zhang, L.; Peng, L.F.; Sun, E.; Chung, W.J.; Jang, J.Y.; Tsai, W.L.; Hyppolite, G.; Chung, R.T. Suppressor of cytokine signaling 3 suppresses hepatitis C virus replication in an mTOR-dependent manner. J. Virol. 2010, 84, 6060–6069. [Google Scholar]
- George, A.; Panda, S.; Kudmulwar, D.; Chhatbar, S.P.; Nayak, S.C.; Krishnan, H.H. Hepatitis C Virus NS5A Binds to the mRNA Cap-binding Eukaryotic Translation Initiation 4F (eIF4F) Complex and Up-regulates Host Translation Initiation Machinery through eIF4E-binding Protein 1 Inactivation. J. Biol. Chem. 2012, 287, 5042–5058. [Google Scholar]
- Coito, C.; Diamond, D.L.; Neddermann, P.; Korth, M.J.; Katze, M.G. High-throughput screening of the yeast kinome: Identification of human serine/threonine protein kinases that phosphorylate the hepatitis C virus NS5A protein. J. Virol. 2004, 78, 3502–3513. [Google Scholar]
- Zheng, L.; Ding, H.; Lu, Z.; Li, Y.; Pan, Y.; Ning, T.; Ke, Y. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 2008, 13, 285–294. [Google Scholar] [CrossRef]
- Zhou, X.; Spangle, J.M.; Munger, K. Expression of a viral oncoprotein in normal human epithelial cells triggers an autophagy-related process: Is autophagy an "Achilles' heel" of human cancers? Autophagy 2009, 5, 578–579. [Google Scholar] [CrossRef]
- Zwerschke, W.; Mazurek, S.; Massimi, P.; Banks, L.; Eigenbrodt, E.; Jansen-Durr, P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 1291–1296. [Google Scholar]
- Nardi, V.; Song, Y.C.; Santamaria-Barria, J.A.; Cosper, A.K.; Lam, Q.; Faber, A.C.; Boland, G.M.; Yeap, B.Y.; Bergethon, K.; Scialabba, V.L.; et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin. Cancer Res. 2012, 18, 1227–1236. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 1773, 1263–1284. [Google Scholar]
- Anjum, R.; Roux, P.P.; Ballif, B.A.; Gygi, S.P.; Blenis, J. The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr. Biol. 2005, 15, 1762–1767. [Google Scholar]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Zheng, B.; Jeong, J.H.; Asara, J.M.; Yuan, Y.Y.; Granter, S.R.; Chin, L.; Cantley, L.C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell 2009, 33, 237–247. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Schouten, G.J.; Vertegaal, A.C.; Whiteside, S.T.; Israel, A.; Toebes, M.; Dorsman, J.C.; van der Eb, A.J.; Zantema, A. IkappaB alpha is a target for the mitogen-activated 90 kDa ribosomal S6 kinase. EMBO J. 1997, 16, 3133–3144. [Google Scholar]
- Zhu, F.X.; Cusano, T.; Yuan, Y. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5556–5567. [Google Scholar]
- Kuang, E.; Wu, F.; Zhu, F. Mechanism of sustained activation of ribosomal S6 kinase (RSK) and ERK by kaposi sarcoma-associated herpesvirus ORF45: Multiprotein complexes retain active phosphorylated ERK AND RSK and protect them from dephosphorylation. J. Biol. Chem. 2009, 284, 13958–13968. [Google Scholar] [CrossRef]
- Kang-Park, S.; Lee, J.H.; Shin, J.H.; Lee, Y.I. Activation of the IGF-II gene by HBV-X protein requires PKC and p44/p42 map kinase signalings. Biochem. Biophys. Res. Commun. 2001, 283, 303–307. [Google Scholar] [CrossRef]
- Yun, C.; Cho, H.; Kim, S.J.; Lee, J.H.; Park, S.Y.; Chan, G.K.; Cho, H. Mitotic aberration coupled with centrosome amplification is induced by hepatitis B virus X oncoprotein via the Ras-mitogen-activated protein/extracellular signal-regulated kinase-mitogen-activated protein pathway. Mol. Cancer Res. 2004, 2, 159–169. [Google Scholar]
- Chung, T.W.; Lee, Y.C.; Kim, C.H. Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERK and PI-3K/AKT pathways: Involvement of invasive potential. FASEB J. 2004, 18, 1123–1125. [Google Scholar]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2010, 331, 456–461. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 4788–4793. [Google Scholar]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010, 5, e15394. [Google Scholar]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Kochl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef]
- Mack, H.I.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8. in press.. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Herrero-Martin, G.; Hoyer-Hansen, M.; Garcia-Garcia, C.; Fumarola, C.; Farkas, T.; Lopez-Rivas, A.; Jaattela, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar]
- Kumar, S.H.; Rangarajan, A. Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J. Virol. 2009, 83, 8565–8574. [Google Scholar]
- Sommermann, T.G.; Mack, H.I.; Cahir-McFarland, E. Autophagy prolongs survival after NFkappaB inhibition in B-cell lymphomas. Autophagy 2012, 8, 265–267. [Google Scholar] [CrossRef]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar]
- Mankouri, J.; Harris, M. Viruses and the fuel sensor: The emerging link between AMPK and virus replication. Rev. Med. Virol. 2011, 21, 205–212. [Google Scholar] [CrossRef]
- Mankouri, J.; Tedbury, P.R.; Gretton, S.; Hughes, M.E.; Griffin, S.D.; Dallas, M.L.; Green, K.A.; Hardie, D.G.; Peers, C.; Harris, M. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 2011, 107, 11549–11554. [Google Scholar]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 2561–2566. [Google Scholar]
- Bernsmeier, C.; Duong, F.H.; Christen, V.; Pugnale, P.; Negro, F.; Terracciano, L.; Heim, M.H. Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J. Hepatol. 2008, 49, 429–440. [Google Scholar] [CrossRef]
- Bottero, V.; Kerur, N.; Sadagopan, S.; Patel, K.; Sharma-Walia, N.; Chandran, B. Phosphorylation and polyubiquitination of transforming growth factor beta-activated kinase 1 are necessary for activation of NF-kappaB by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J. Virol. 2011, 85, 1980–1993. [Google Scholar]
- Zhou, Y.; Wang, S.; Ma, J.W.; Lei, Z.; Zhu, H.F.; Lei, P.; Yang, Z.S.; Zhang, B.; Yao, X.X.; Shi, C.; et al. Hepatitis B virus protein X-induced expression of the CXC chemokine IP-10 is mediated through activation of NF-kappaB and increases migration of leukocytes. J. Biol. Chem. 2010, 285, 12159–12168. [Google Scholar]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar] [CrossRef]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O'Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Malik, S.A.; Morselli, E.; Kepp, O.; Criollo, A.; Mouchel, P.L.; Carnuccio, R.; Kroemer, G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 2009, 8, 1571–1576. [Google Scholar]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res 2007, 67, 3043–3053. [Google Scholar]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D'Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.A.; Marino, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.C.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- O'Shea, C.C.; Fried, M. Modulation of the ARF-p53 pathway by the small DNA tumor viruses. Cell Cycle 2005, 4, 449–452. [Google Scholar] [CrossRef]
- Wang, E.H.; Friedman, P.N.; Prives, C. The murine p53 protein blocks replication of SV40 DNA in vitro by inhibiting the initiation functions of SV40 large T antigen. Cell 1989, 57, 379–392. [Google Scholar] [CrossRef]
- Qing, G.; Yan, P.; Qu, Z.; Liu, H.; Xiao, G. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 2007, 17, 520–530. [Google Scholar] [CrossRef]
- Qing, G.; Yan, P.; Xiao, G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res. 2006, 16, 895–901. [Google Scholar] [CrossRef]
- Criollo, A.; Chereau, F.; Malik, S.A.; Niso-Santano, M.; Marino, G.; Galluzzi, L.; Maiuri, M.C.; Baud, V.; Kroemer, G. Autophagy is required for the activation of NFkappaB. Cell Cycle 2012, 11, 194–199. [Google Scholar] [CrossRef]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell Biol. 2009, 29, 2594–2608. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besancon, F.; Bauvy, C.; Souquere, S.; Pierron, G.; Codogno, P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar]
- Niso-Santano, M.; Criollo, A.; Malik, S.A.; Michaud, M.; Morselli, E.; Marino, G.; Lachkar, S.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Direct molecular interactions between Beclin 1 and the canonical NFkappaB activation pathway. Autophagy 2012, 8, 268–270. [Google Scholar] [CrossRef]
- Chu, Z.L.; Shin, Y.A.; Yang, J.M.; DiDonato, J.A.; Ballard, D.W. IKKgamma mediates the interaction of cellular IkappaB kinases with the tax transforming protein of human T cell leukemia virus type 1. J. Biol. Chem. 1999, 274, 15297–15300. [Google Scholar]
- Field, N.; Low, W.; Daniels, M.; Howell, S.; Daviet, L.; Boshoff, C.; Collins, M. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 2003, 116, 3721–3728. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Sun, S.C. IKKgamma serves as a docking subunit of the IkappaB kinase (IKK) and mediates interaction of IKK with the human T-cell leukemia virus Tax protein. J. Biol. Chem. 1999, 274, 22911–22914. [Google Scholar] [CrossRef]
- Jin, D.Y.; Giordano, V.; Kibler, K.V.; Nakano, H.; Jeang, K.T. Role of adapter function in oncoprotein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I Tax interacts directly with IkappaB kinase gamma. J. Biol. Chem. 1999, 274, 17402–17405. [Google Scholar]
- Liu, L.; Eby, M.T.; Rathore, N.; Sinha, S.K.; Kumar, A.; Chaudhary, P.M. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 2002, 277, 13745–13751. [Google Scholar]
- Matta, H.; Chaudhary, P.M. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9399–9404. [Google Scholar] [CrossRef]
- Matta, H.; Mazzacurati, L.; Schamus, S.; Yang, T.; Sun, Q.; Chaudhary, P.M. Kaposi's sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IkappaB kinase complex to selectively activate NF-kappaB without JNK activation. J. Biol. Chem. 2007, 282, 24858–24865. [Google Scholar]
- Xiao, G.; Cvijic, M.E.; Fong, A.; Harhaj, E.W.; Uhlik, M.T.; Waterfield, M.; Sun, S.C. Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: Evidence for the involvement of IKKalpha. EMBO J. 2001, 20, 6805–6815. [Google Scholar] [CrossRef]
- de Oliveira, D.E.; Ballon, G.; Cesarman, E. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 2010, 18, 248–257. [Google Scholar] [CrossRef]
- Cahir McFarland, E.D.; Izumi, K.M.; Mosialos, G. Epstein-barr virus transformation: Involvement of latent membrane protein 1-mediated activation of NF-kappaB. Oncogene 1999, 18, 6959–6964. [Google Scholar]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 2010, 138, 2509–2518, 2518 e2501. [Google Scholar]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar]
- Yan, P.; Qing, G.; Qu, Z.; Wu, C.C.; Rabson, A.; Xiao, G. Targeting autophagic regulation of NFkappaB in HTLV-I transformed cells by geldanamycin: Implications for therapeutic interventions. Autophagy 2007, 3, 600–603. [Google Scholar]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Izumi, K.M.; Kieff, E.D. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. USA 1997, 94, 12592–12597. [Google Scholar] [CrossRef]
- Lam, N.; Sandberg, M.L.; Sugden, B. High physiological levels of LMP1 result in phosphorylation of eIF2 alpha in Epstein-Barr virus-infected cells. J. Virol. 2004, 78, 1657–1664. [Google Scholar] [CrossRef]
- Lee, D.Y.; Lee, J.; Sugden, B. The unfolded protein response and autophagy: Herpesviruses rule! J. Virol. 2009, 83, 1168–1172. [Google Scholar] [CrossRef]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell Biol. 2004, 24, 10161–10168. [Google Scholar]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell Biol. 2003, 23, 5651–5663. [Google Scholar]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef]
- Matus, S.; Nassif, M.; Glimcher, L.H.; Hetz, C. XBP-1 deficiency in the nervous system reveals a homeostatic switch to activate autophagy. Autophagy 2009, 5, 1226–1228. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, B.; Ye, L.; Kong, L.; Jing, W.; Yang, X.; Wu, Z.; Ye, L. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005, 43, 529–536. [Google Scholar]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar]
- McPherson, S.; Powell, E.E.; Barrie, H.D.; Clouston, A.D.; McGuckin, M.; Jonsson, J.R. No evidence of the unfolded protein response in patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2011, 26, 319–327. [Google Scholar]
- Asselah, T.; Bieche, I.; Mansouri, A.; Laurendeau, I.; Cazals-Hatem, D.; Feldmann, G.; Bedossa, P.; Paradis, V.; Martinot-Peignoux, M.; Lebrec, D.; et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mack, H.I.D.; Munger, K. Modulation of Autophagy-Like Processes by Tumor Viruses. Cells 2012, 1, 204-247. https://doi.org/10.3390/cells1030204
Mack HID, Munger K. Modulation of Autophagy-Like Processes by Tumor Viruses. Cells. 2012; 1(3):204-247. https://doi.org/10.3390/cells1030204
Chicago/Turabian StyleMack, Hildegard I. D., and Karl Munger. 2012. "Modulation of Autophagy-Like Processes by Tumor Viruses" Cells 1, no. 3: 204-247. https://doi.org/10.3390/cells1030204
APA StyleMack, H. I. D., & Munger, K. (2012). Modulation of Autophagy-Like Processes by Tumor Viruses. Cells, 1(3), 204-247. https://doi.org/10.3390/cells1030204