
Biodegradable Polyphosphazene Based Peptide-Polymer Hybrids

 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Tetra Peptide Synthesis (Gly-Phe-Leu-Gly-OtBu)
2.2. Synthesis of Cl3PNSiMe3
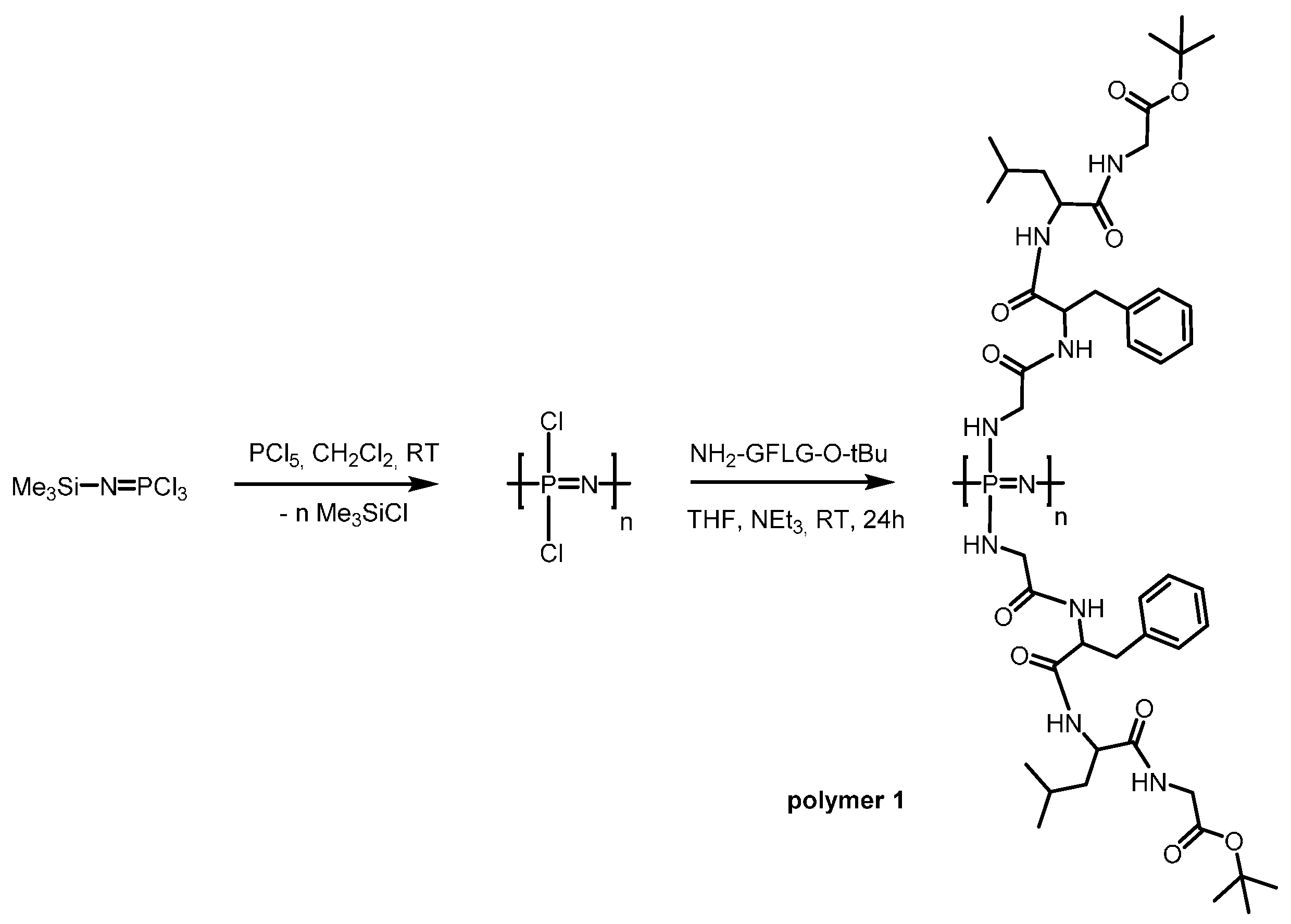
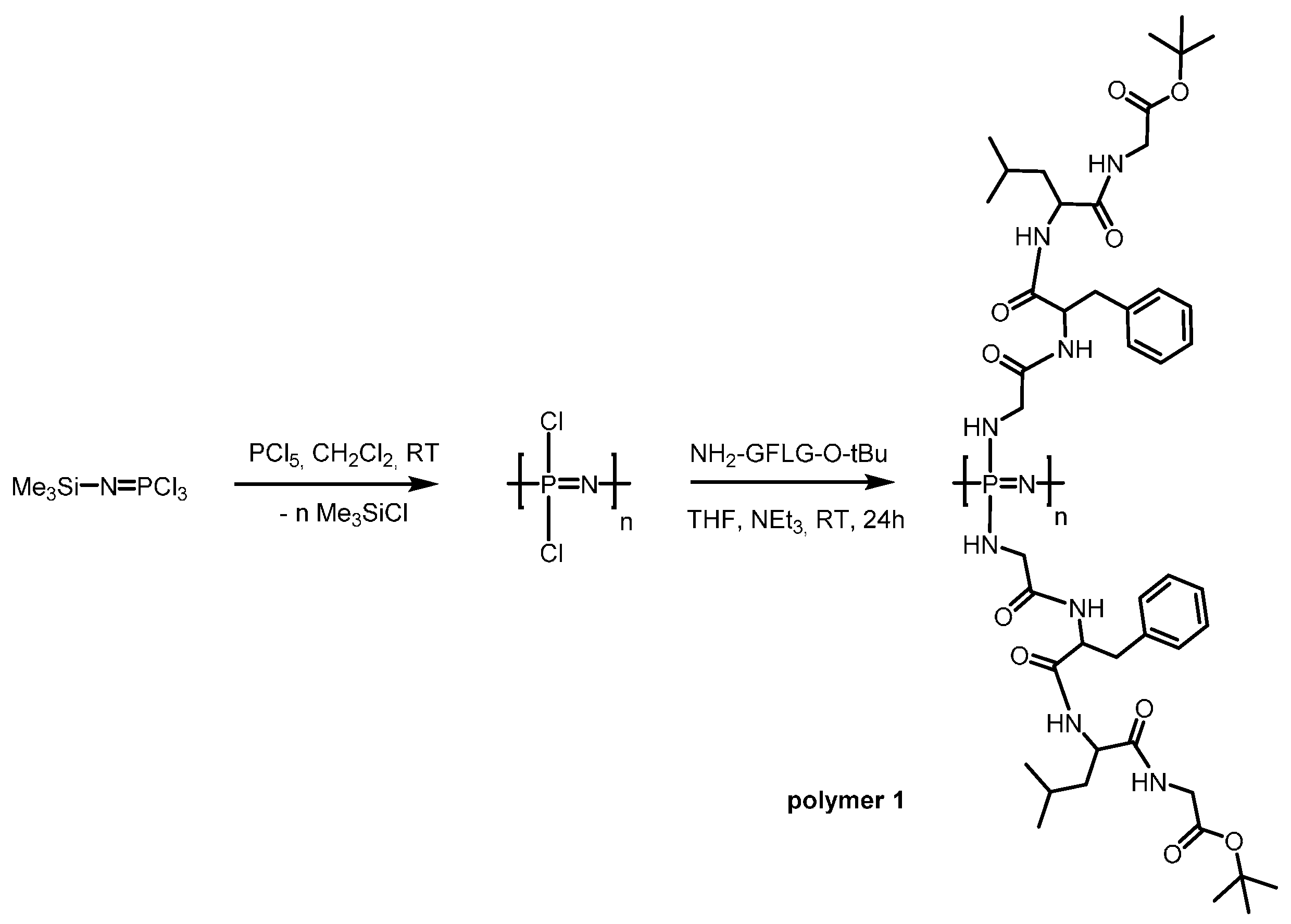
2.3. Polymerisation Procedure
2.4. Gly-Phe-Leu-Gly-Imiquimod
2.5. Gly-Jeffamine M-1000
2.6. Hydrolytic Degradation Study–Field Flow Fractionation
2.7. Hydrolytic Degradation Study—Dynamic Light Scattering
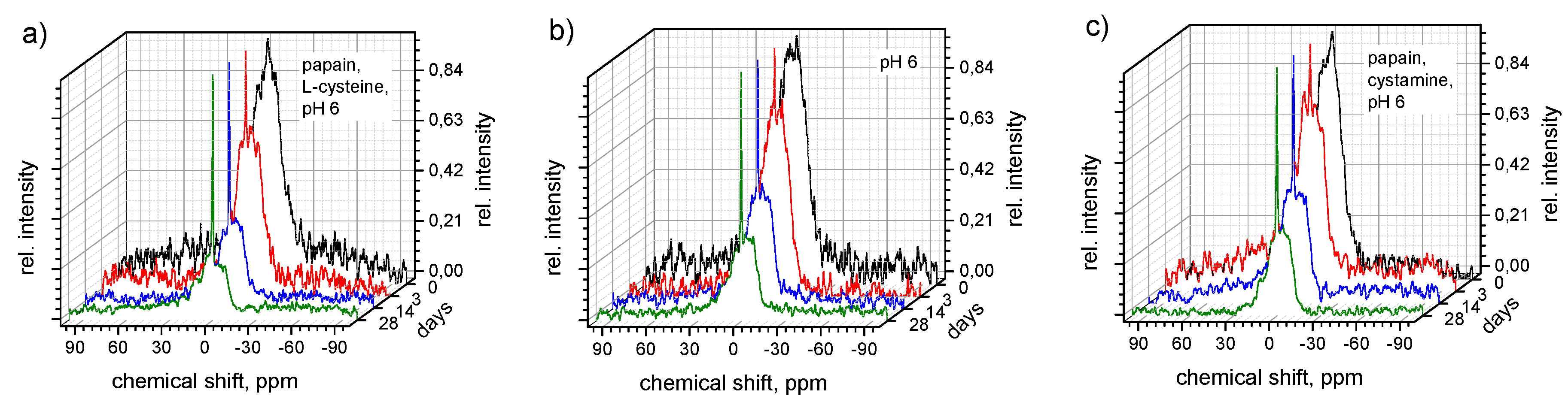
2.8. Enzymatic and Hydrolytic Degradation Study–NMR
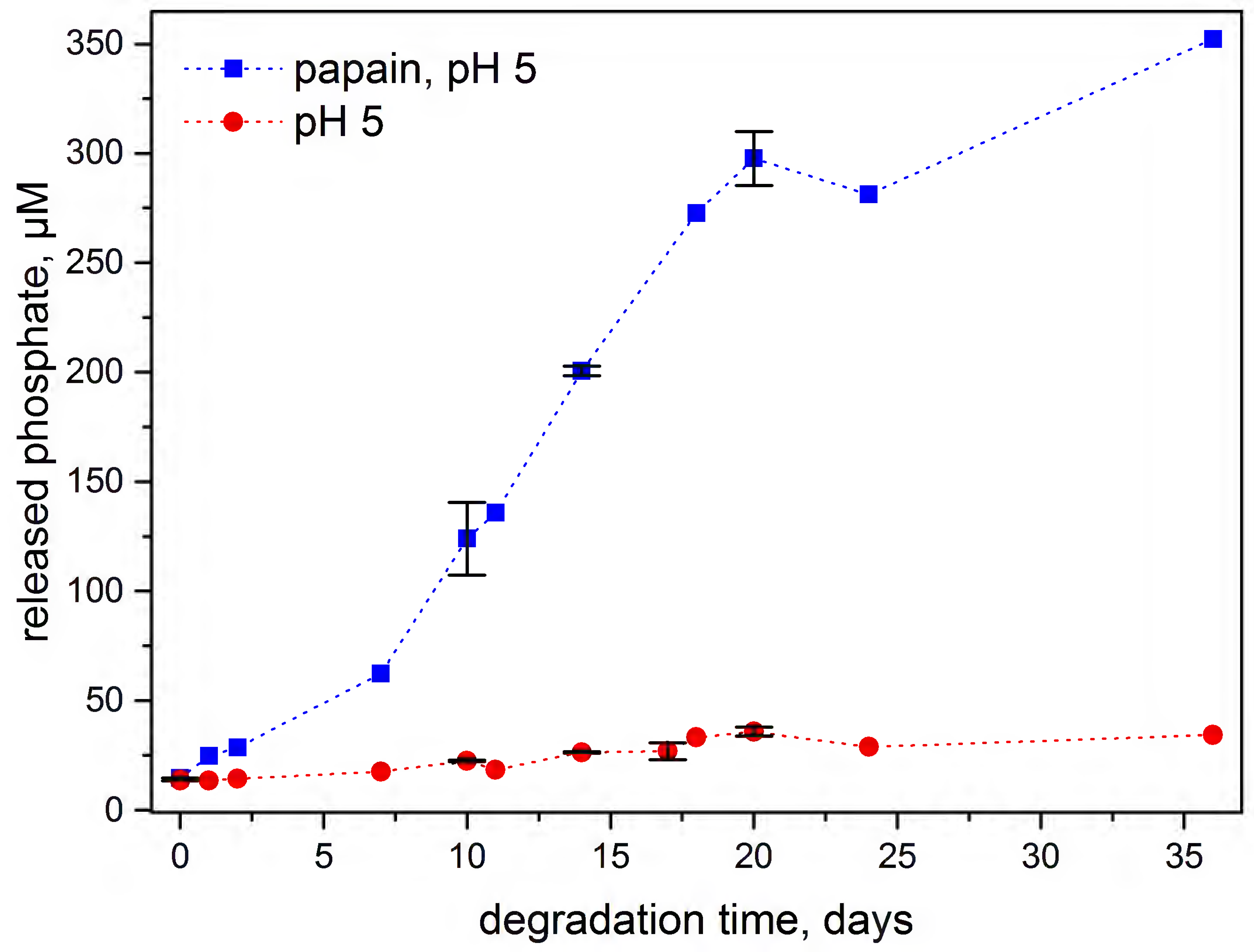
2.9. Degradation—Phosphate Determination
2.10. Drug Release—HPLC Measurements
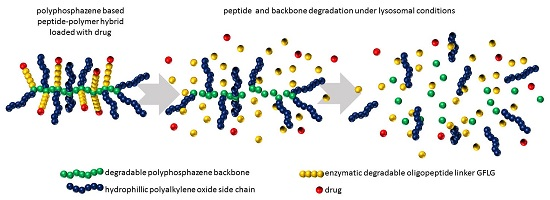
3. Results
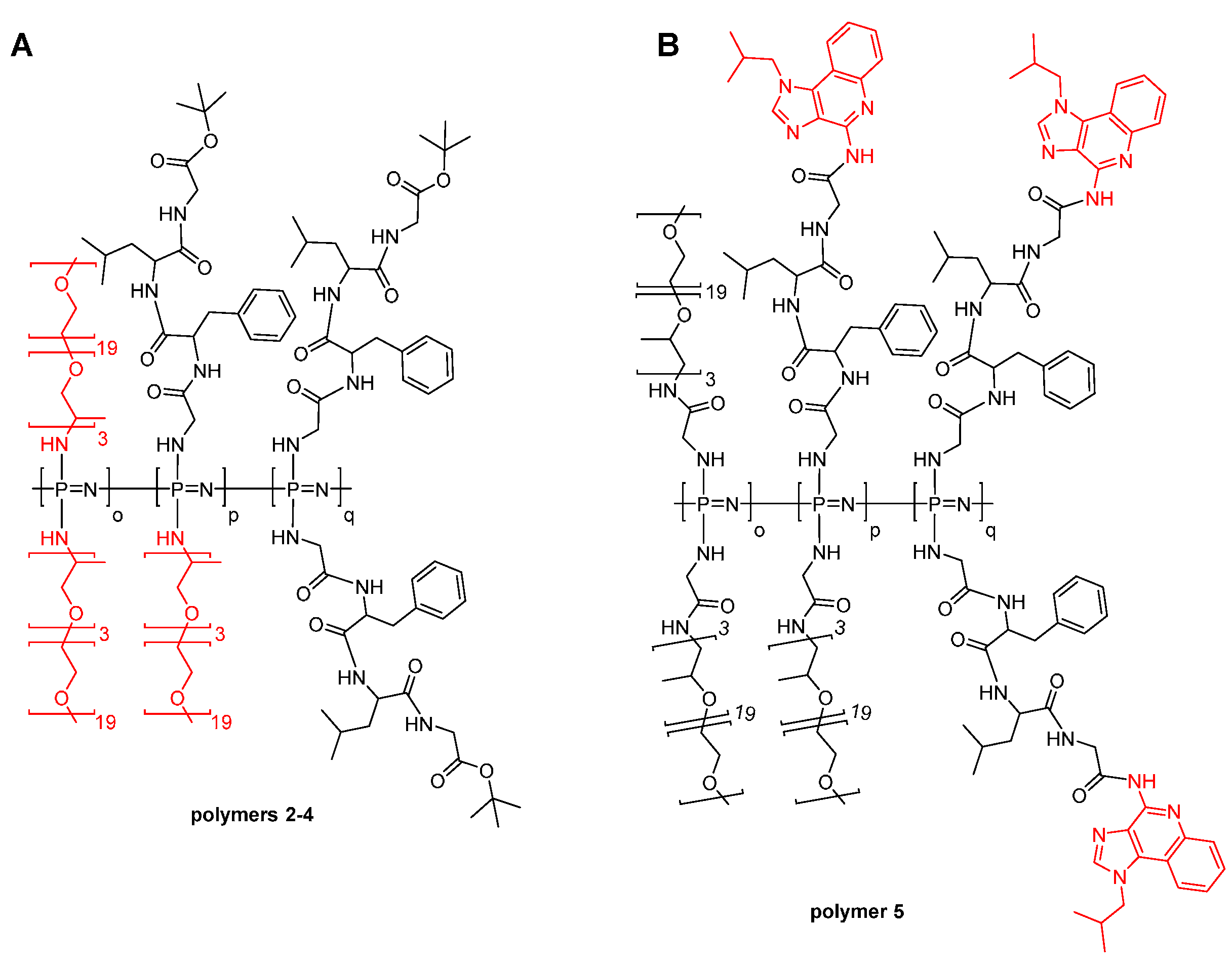
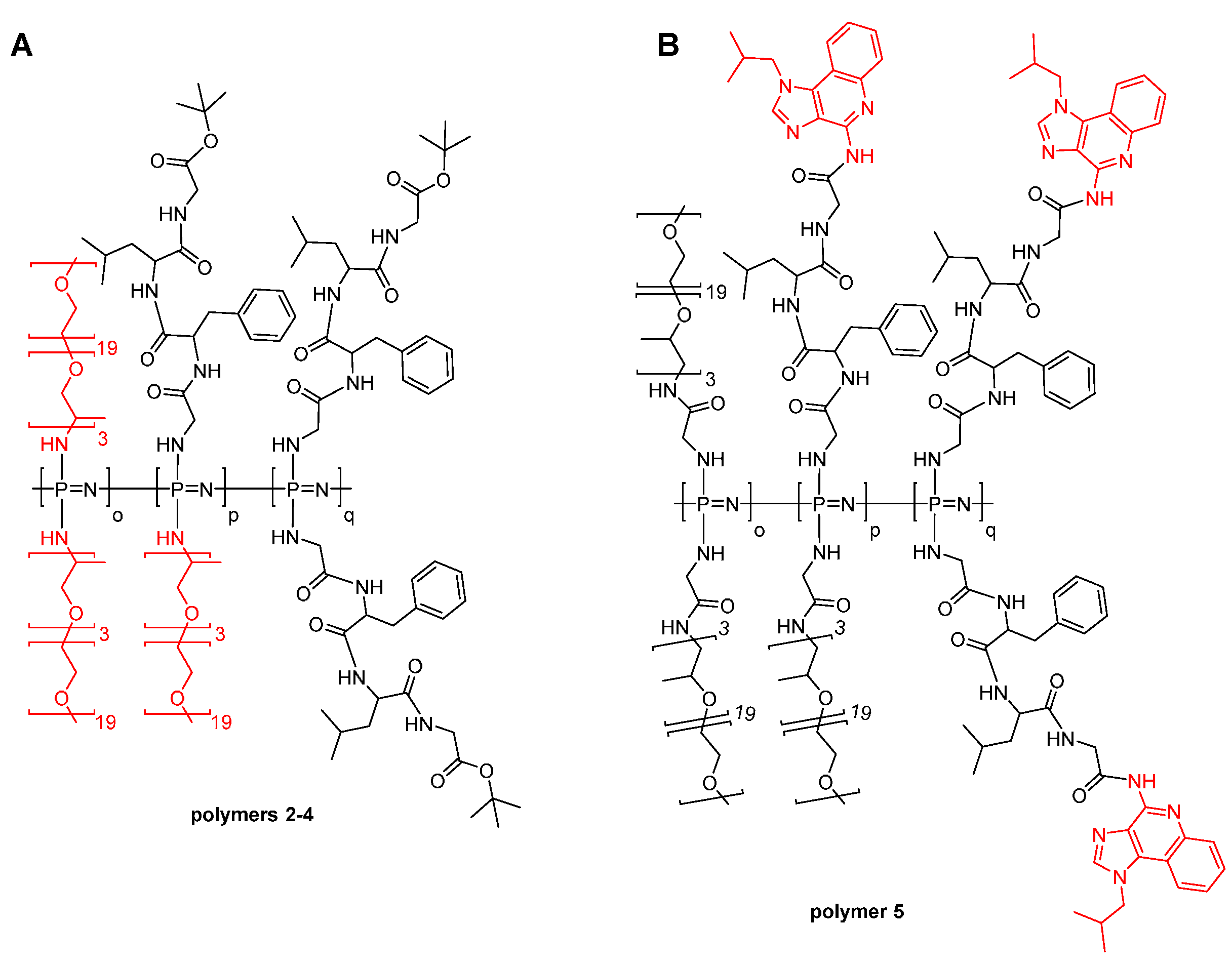
3.1. Synthesis
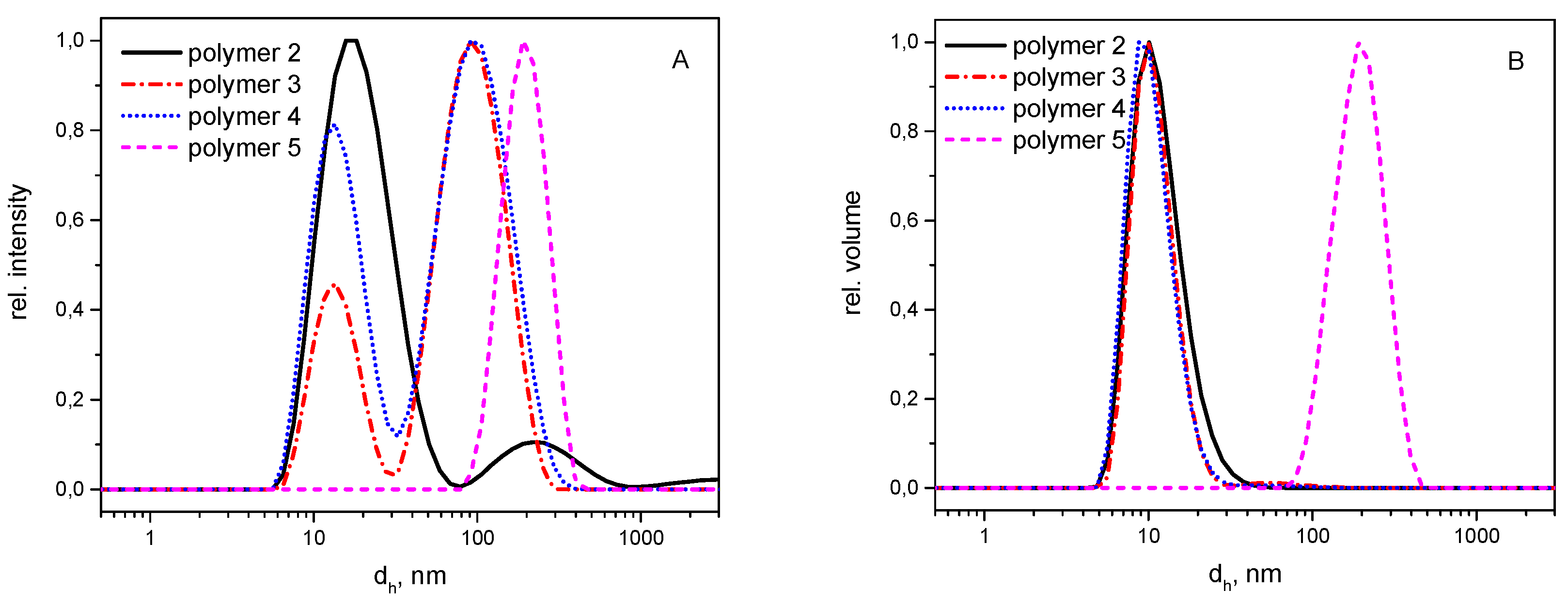
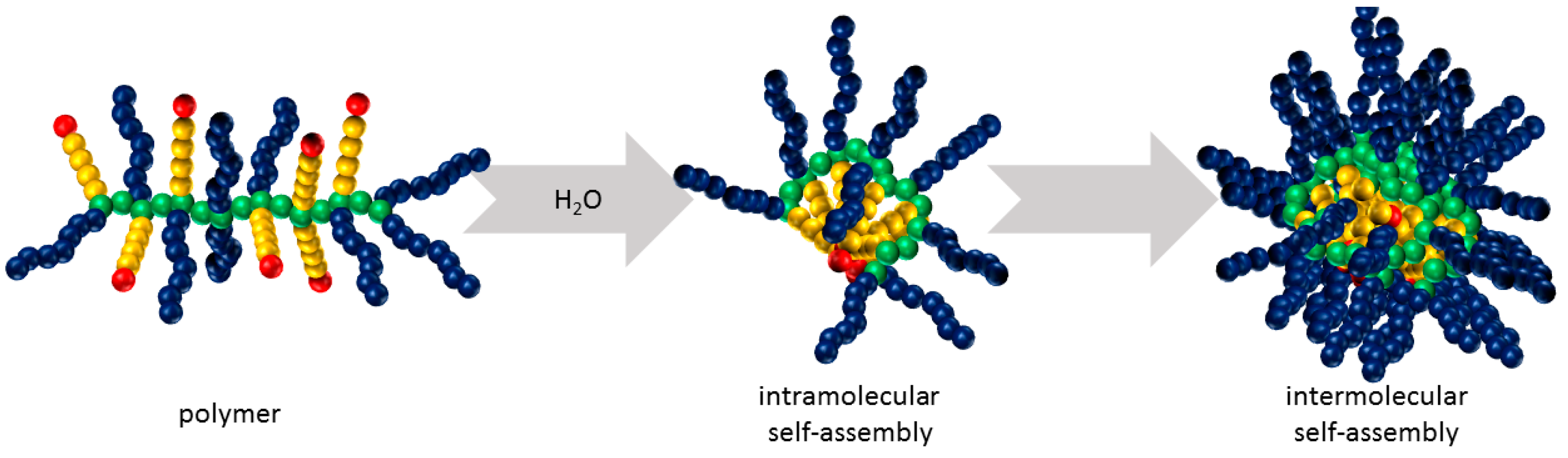
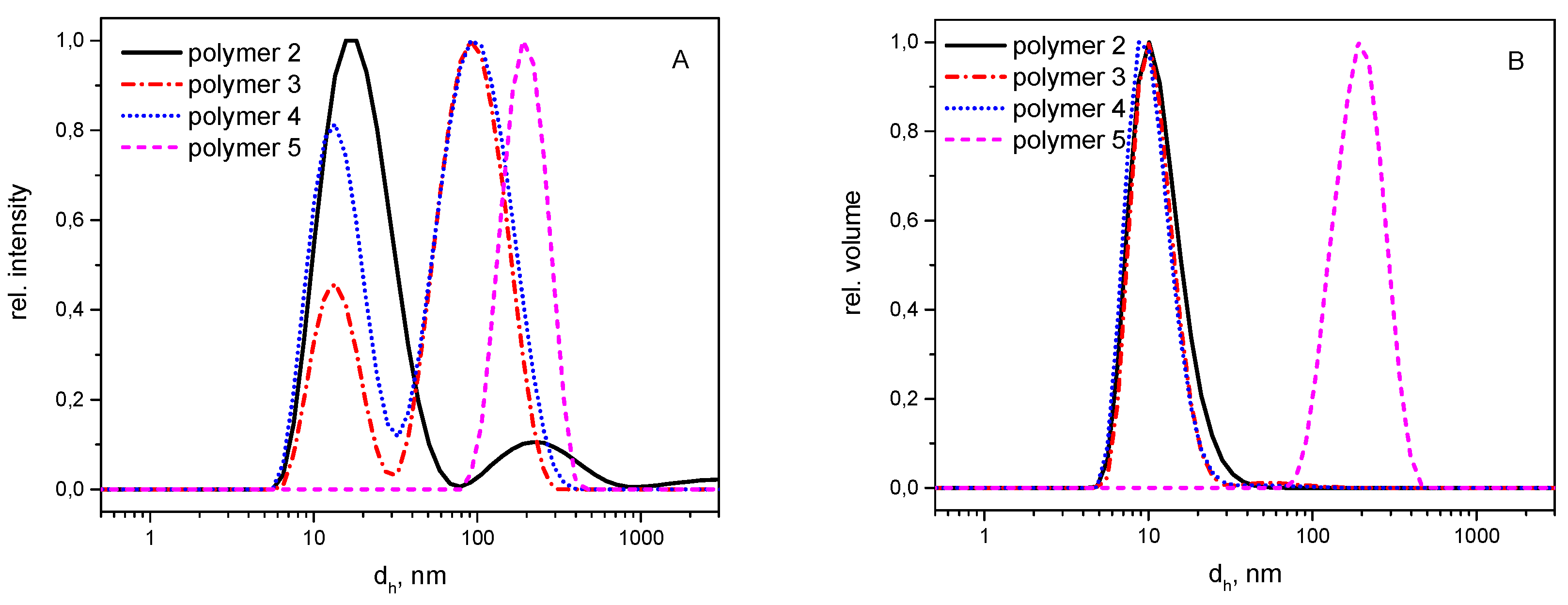
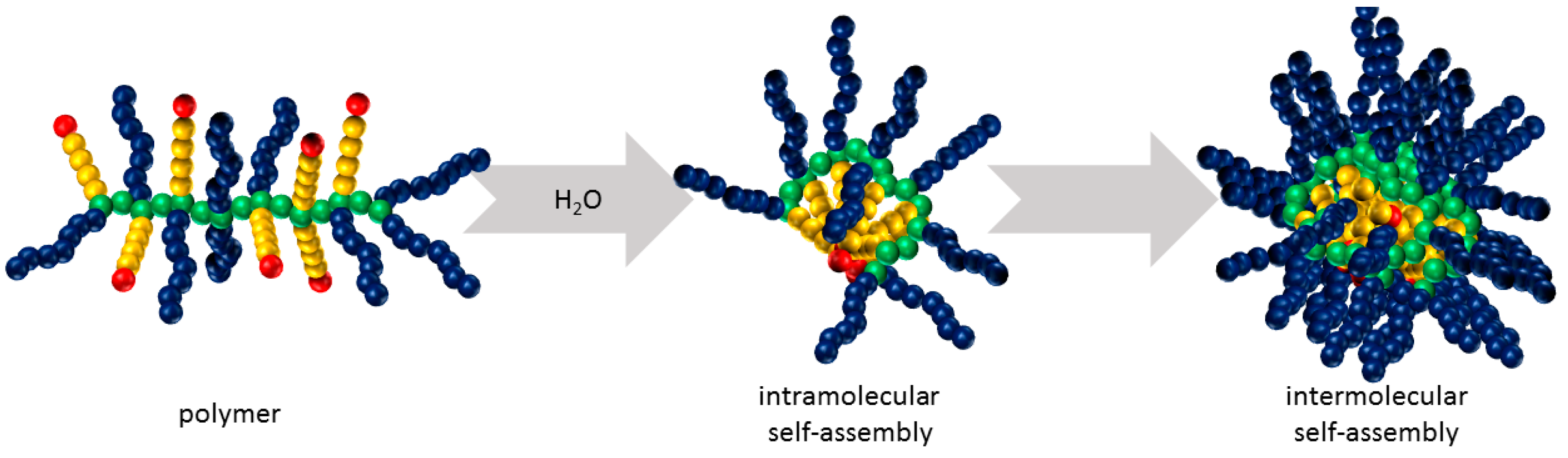
3.2. Self-Assembly
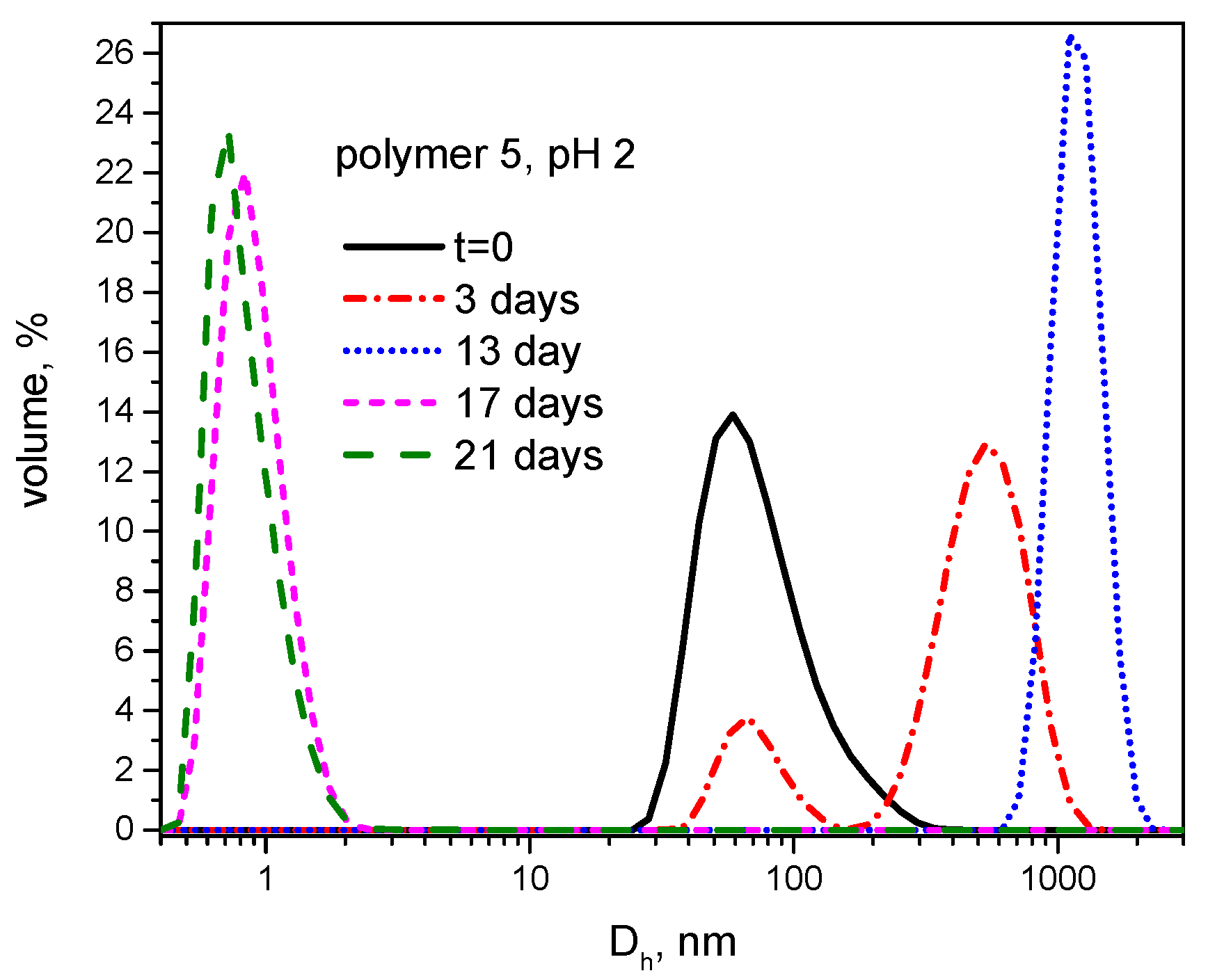
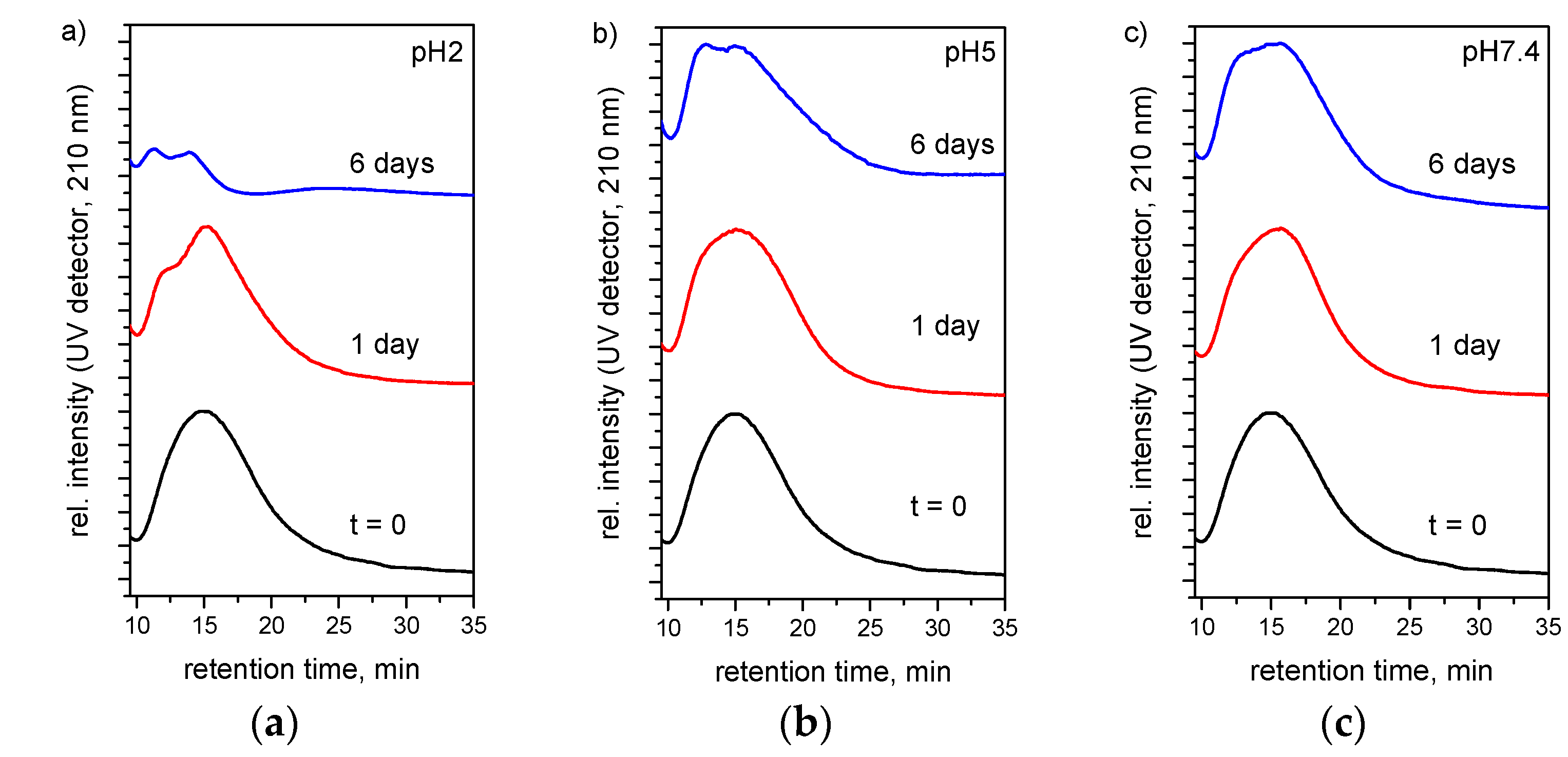
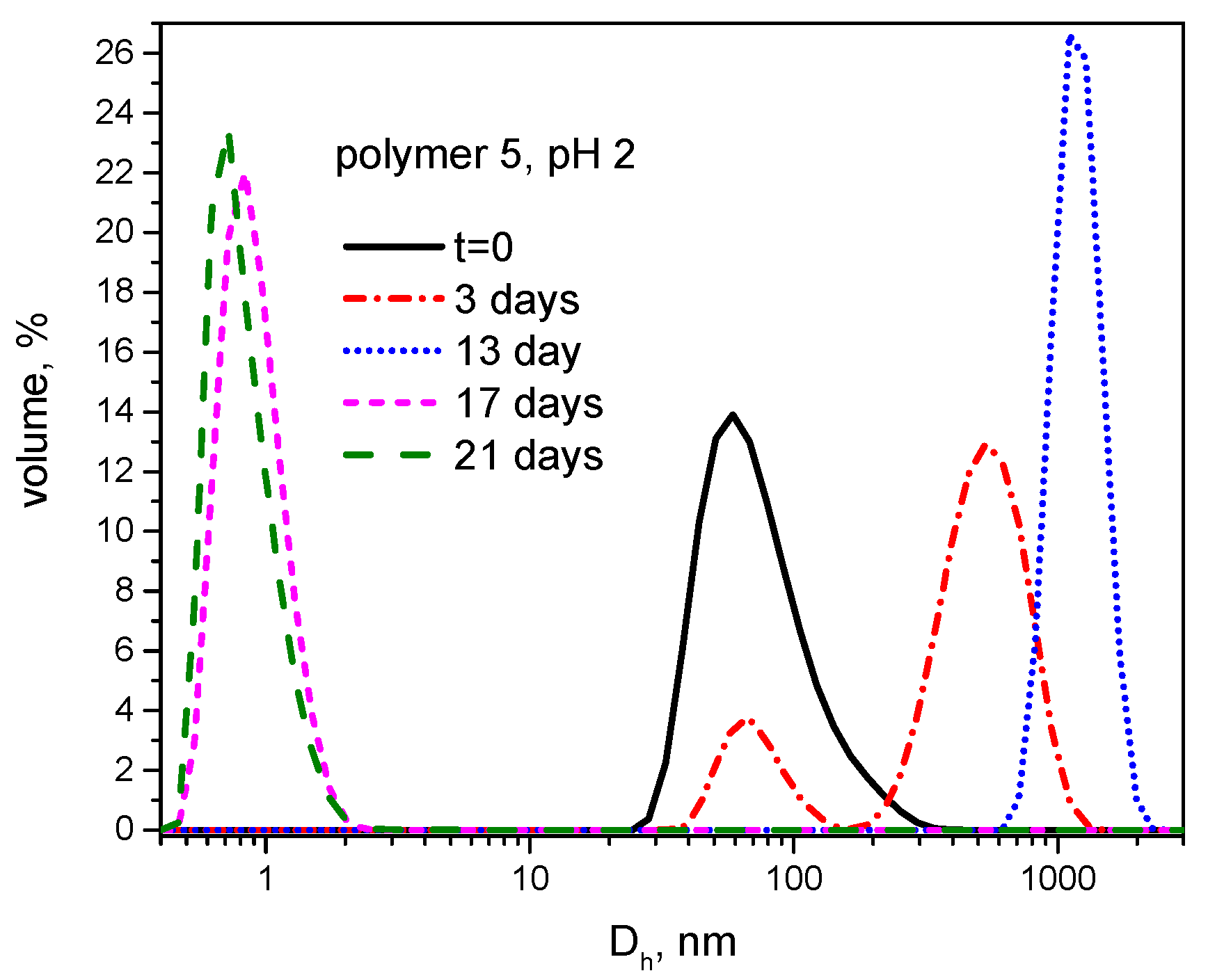
3.3. Hydrolytic Degradation
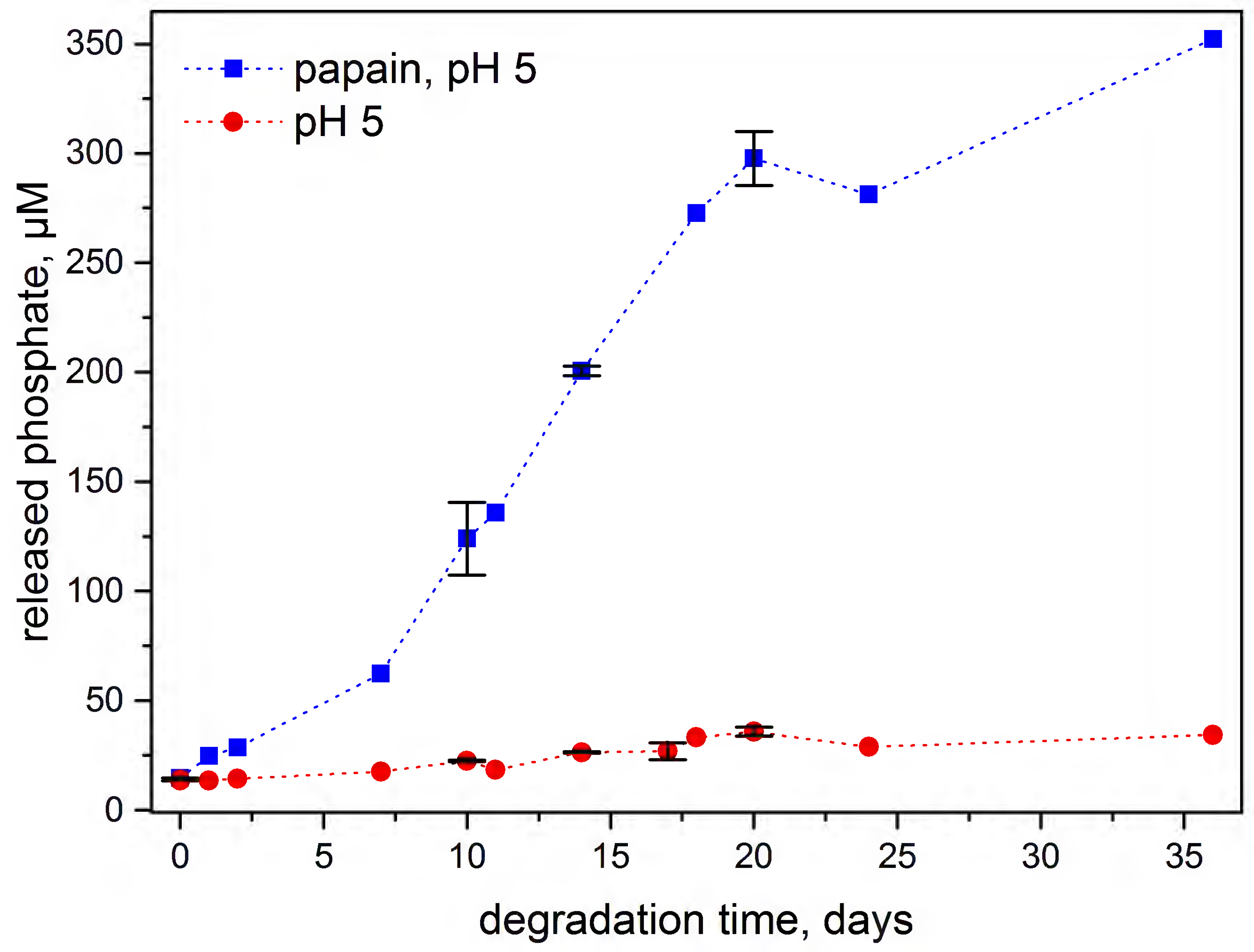
3.4. Enzymatic Degradation
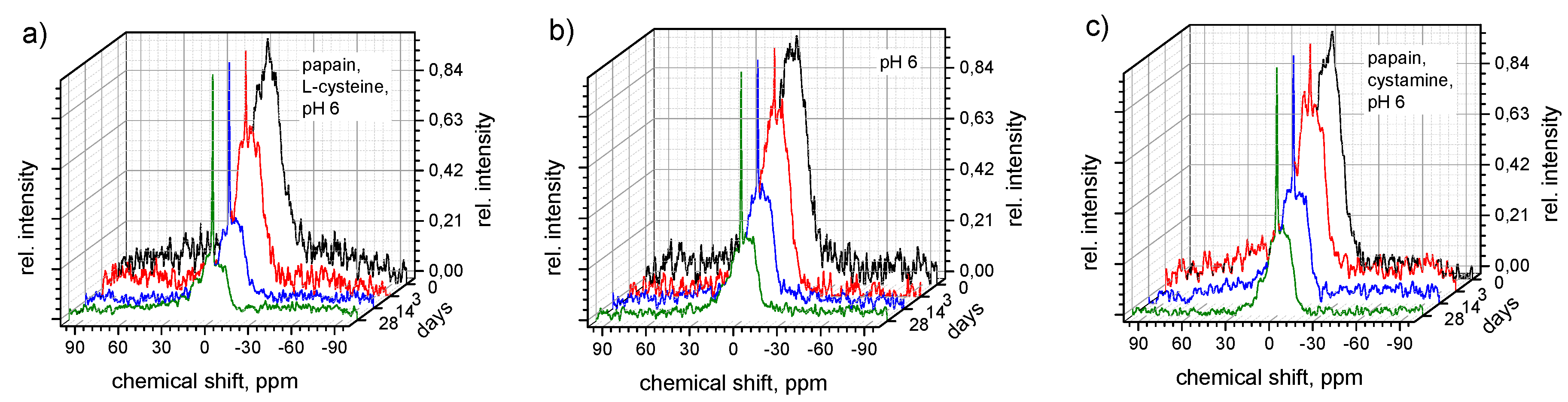
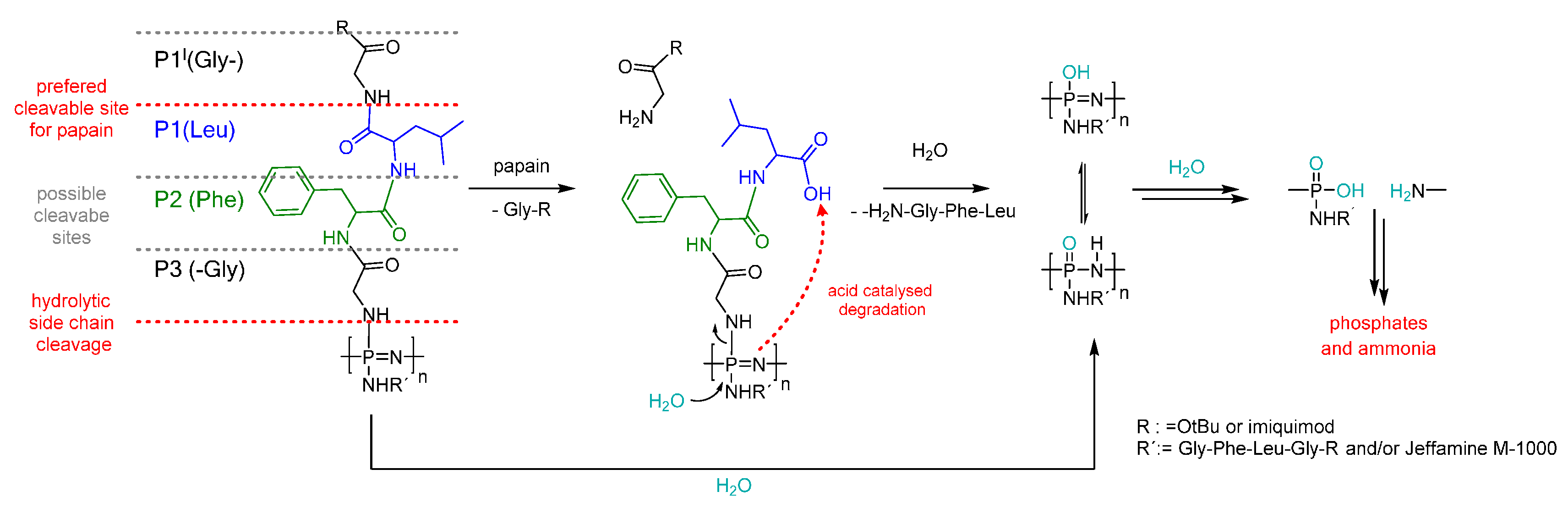
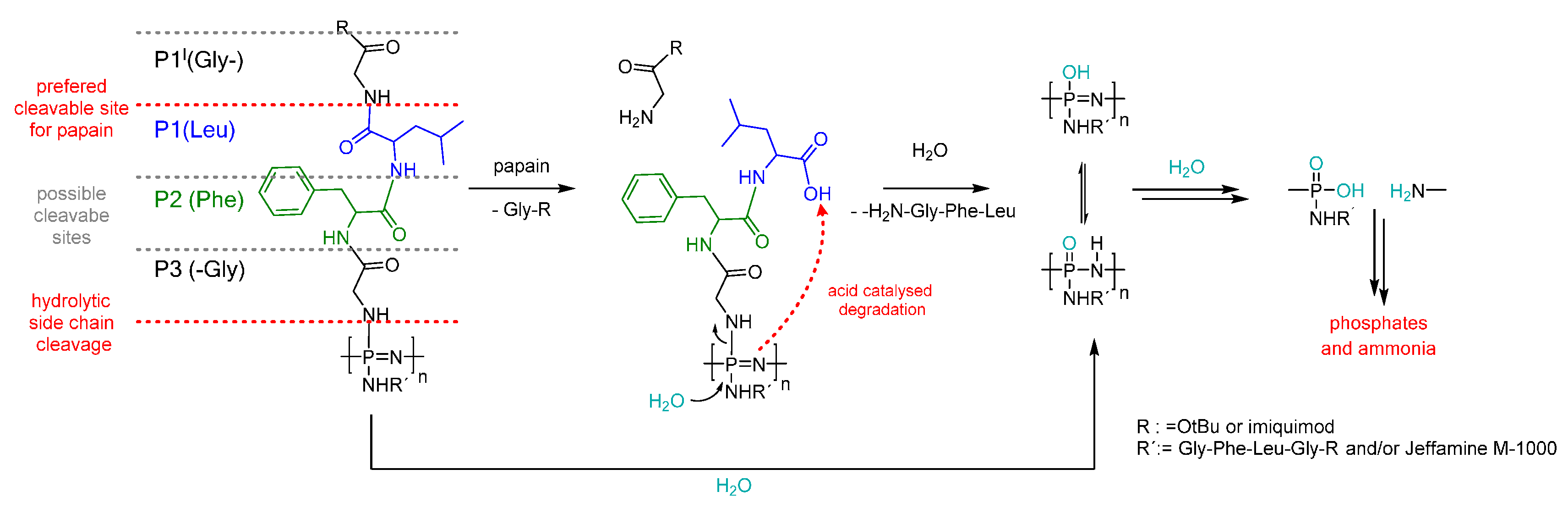
3.5. Degradation Mechanism
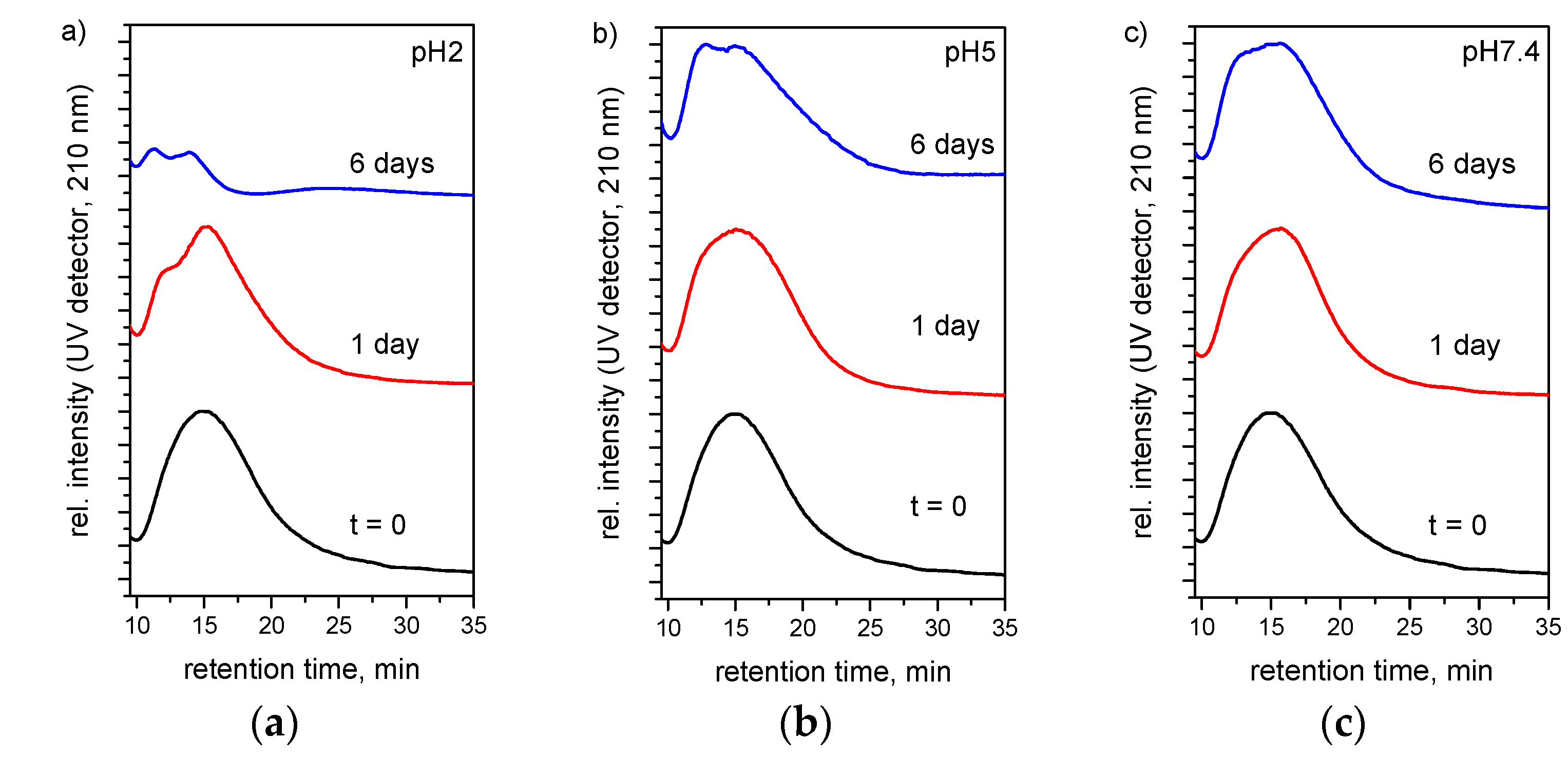
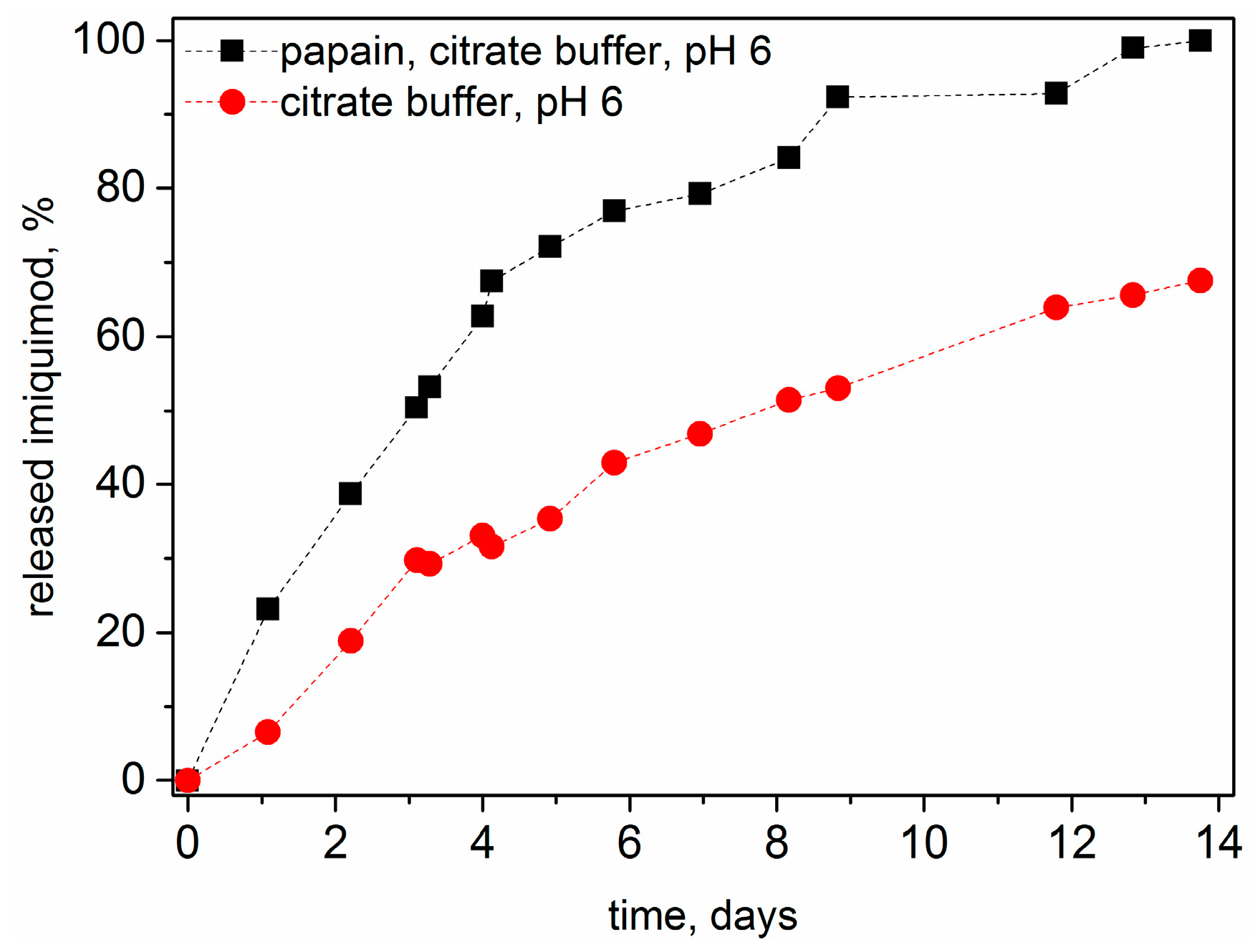
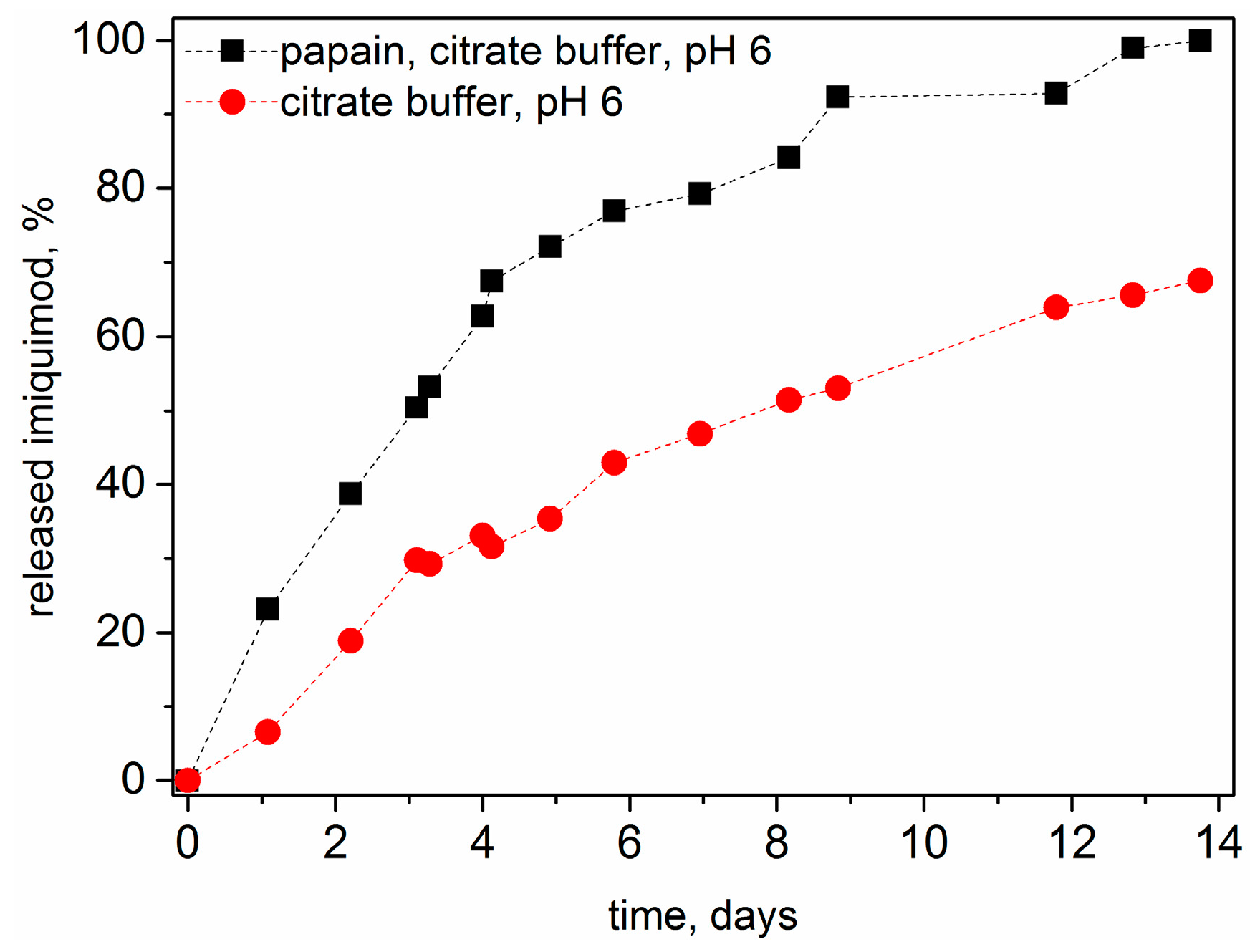
3.6. Drug Release
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| COMU | 1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DIEA | N,N-Diisopropylethylamine |
| DLS | dynamic light scattering |
| DMF | Dimethylformamide |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| FFF | field flow fractionation |
| Fmoc | 9-Fluorenylmethoxycarbonyl |
| PEG | polyethylene glycol |
| GFLG | Glycine-Phenylalanine-Leucine-Glycine |
| Gly | Glycine |
| GPC | gel permeation chromatography |
| HPLC | high performance liquid chromatography |
| HPMA | N-(2-hydroxypropyl)methacrylamide |
| Leu | Leucine |
| NMR | Nuclear magnetic resonance |
| PEG | polyethylene glycol |
| Phe | Phenylalanine |
| TAEA | Tris(2-aminoethyl)amine |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| UV–Vis | Ultraviolet–Visible |
References
- Duro-Castano, A.; Conejos-Sánchez, I.; Vicent, M. Peptide-based polymer therapeutics. Polymers 2014, 6, 515. [Google Scholar] [CrossRef]
- Chow, D.; Nunalee, M.L.; Lim, D.W.; Simnick, A.J.; Chilkoti, A. Peptide-based biopolymers in biomedicine and biotechnology. Mater. Sci. Eng. R R. 2008, 62, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.Y.; Panganiban, B.; Xu, T. Peptide-polymer conjugates: From fundamental science to application. Ann. Rev. Phys. Chem. 2013, 64, 631–657. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Puiggalí, J. Hybrid block copolymers constituted by peptides and synthetic polymers: An overview of synthetic approaches, supramolecular behavior and potential applications. Polymers 2013, 5, 188. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.P.; John, J.V.; Kim, I. Recent developments in polymer–block–polypeptide and protein–polymer bioconjugate hybrid materials. Eur. Polym. J. 2013, 49, 2925–2948. [Google Scholar] [CrossRef]
- Bacinello, D.; Garanger, E.; Taton, D.; Tam, K.C.; Lecommandoux, S. Tailored drug-release from multi-functional polymer-peptide hybrid vesicles. Eur. Polym. J. 2015, 62, 363–373. [Google Scholar] [CrossRef]
- Peters, D.; Kastantin, M.; Kotamraju, V.R.; Karmali, P.P.; Gujraty, K.; Tirrell, M.; Ruoslahti, E. Targeting atherosclerosis by using modular, multifunctional micelles. Proc. Natl. Acad. Sci. USA 2009, 106, 9815–9819. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J.; Kopečková, P.; Minko, T.; Lu, Z.-R. HPMA copolymer–anticancer drug conjugates: Design, activity, and mechanism of action. Eur. J. Pharm. Biopharm. 2000, 50, 61–81. [Google Scholar] [CrossRef]
- Veronese, F.M.; Schiavon, O.; Pasut, G.; Mendichi, R.; Andersson, L.; Tsirk, A.; Ford, J.; Wu, G.; Kneller, S.; Davies, J.; et al. Peg−doxorubicin conjugates: Influence of polymer structure on drug release, in vitro cytotoxicity, biodistribution, and antitumor activity. Bioconjugate Chem. 2005, 16, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur. J. Pharm. Biopharm. 2009, 71, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.J.; Kessler, J.A.; Stupp, S.I. Emerging peptide nanomedicine to regenerate tissues and organs. J. Intern. Med. 2010, 267, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, J.A.; Thomas, S.N.; Swartz, M.A. Materials engineering for immunomodulation. Nature 2009, 462, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Yang, J.; Kopečková, P.; Kopeček, J. Biodegradable multiblock poly[n-(2-hydroxypropyl)methacrylamide] via reversible addition-fragmentation chain transfer polymerization and click chemistry. Macromolecules 2011, 44, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Luo, K.; Pan, H.; Kopečková, P.; Kopeček, J. Synthesis of biodegradable multiblock copolymers by click coupling of raft-generated heterotelechelic polyhpma conjugates. React. Funct. Polym. 2011, 71, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Dvořák, M.; Kopečková, P.; Kopeček, J. High-molecular weight HPMA copolymer–adriamycin conjugates. J. Controll. Release 1999, 60, 321–332. [Google Scholar] [CrossRef]
- Lu, Z.-R.; Gao, S.-Q.; Kopečková, P.; Kopeček, J. Modification of cyclosporin a and conjugation of its derivative to hpma copolymers. Bioconjugate Chem. 2001, 12, 129–133. [Google Scholar] [CrossRef]
- Hersel, U.; Dahmen, C.; Kessler, H. Rgd modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials 2003, 24, 4385–4415. [Google Scholar] [CrossRef]
- Jun, Y.J.; Jadhav, V.B.; Min, J.H.; Cui, J.X.; Chae, S.W.; Choi, J.M.; Kim, I.-S.; Choi, S.-J.; Lee, H.J.; Sohn, Y.S. Stable and efficient delivery of docetaxel by micelle-encapsulation using a tripodal cyclotriphosphazene amphiphile. Int. J. Pharm. 2012, 422, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Andrianov, A.K. Polyphosphazenes for Biomedical Applications; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Teasdale, I.; Brüggemann, O. Polyphosphazenes for Medical Applications; Smithers Rapra: Shrewsbury, Shropshire, UK, 2014. [Google Scholar]
- Teasdale, I.; Brüggemann, O. Polyphosphazenes: Multifunctional, biodegradable vehicles for drug and gene delivery. Polymers 2013, 5, 161. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, V.; Lough, A.J.; Murray, M.; Manners, I. Probing the mechanism of the PCl5−initiated living cationic polymerization of the phosphoranimine Cl3p=NSiMe3 using model compound chemistry. J. Am. Chem. Soc. 2009, 131, 3658–3667. [Google Scholar] [CrossRef] [PubMed]
- Allcock, H.R.; Crane, C.A.; Morrissey, C.T.; Nelson, J.M.; Reeves, S.D.; Honeyman, C.H.; Manners, I. “Living” cationic polymerization of phosphoranimines as an ambient temperature route to polyphosphazenes with controlled molecular weights. Macromolecules 1996, 29, 7740–7747. [Google Scholar] [CrossRef]
- Wilfert, S.; Henke, H.; Schoefberger, W.; Brüggemann, O.; Teasdale, I. Chain-end-functionalized polyphosphazenes via a one-pot phosphine-mediated living polymerization. Macromol. Rapid Commun. 2014, 35, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Henke, H.; Wilfert, S.; Iturmendi, A.; Bruggemann, O.; Teasdale, I. Branched polyphosphazenes with controlled dimensions. J. Polym. Sci. A Polym. Chem. 2013, 51, 4467–4473. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Vicent, M.J. Do HPMA copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Adv. Drug Deliv. Rev. 2010, 62, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[(amino acid ester)phosphazenes] as substrates for the controlled release of small molecules. Biomaterials 1994, 15, 563–569. [Google Scholar] [CrossRef]
- Wilfert, S.; Iturmendi, A.; Schoefberger, W.; Kryeziu, K.; Heffeter, P.; Berger, W.; Brüggemann, O.; Teasdale, I. Water-soluble, biocompatible polyphosphazenes with controllable and pH-promoted degradation behavior. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Markovsky, E.; Baabur-Cohen, H.; Eldar-Boock, A.; Omer, L.; Tiram, G.; Ferber, S.; Ofek, P.; Polyak, D.; Scomparin, A.; Satchi-Fainaro, R. Administration, distribution, metabolism and elimination of polymer therapeutics. J. Controll. Release 2012, 161, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, R.; Duncan, R. Polymeric carriers: Preclinical safety and the regulatory implications for design and development of polymer therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, I.; Wilfert, S.; Nischang, I.; Brüggemann, O. Multifunctional and biodegradable polyphosphazenes for use as macromolecular anti-cancer drug carriers. Polym. Chem. 2011, 2, 828–834. [Google Scholar] [CrossRef]
- Zhong, Y.-J.; Shao, L.-H.; Li, Y.A.N. Cathepsin b-cleavable doxorubicin prodrugs for targeted cancer therapy. Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [PubMed]
- Bacinello, D.; Garanger, E.; Taton, D.; Tam, K.C.; Lecommandoux, S. Enzyme-degradable self-assembled nanostructures from polymer–peptide hybrids. Biomacromolecules 2014, 15, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- Musil, D.; Zucic, D.; Turk, D.; Engh, R.A.; Mayr, I.; Huber, R.; Popovic, T.; Turk, V.; Towatari, T.; Katunuma, N. The refined 2.15 a X-ray crystal structure of human liver cathepsin b: The structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [Google Scholar] [PubMed]
- Han, J.H.; Lee, J.; Jeon, S.J.; Choi, E.S.; Cho, S.D.; Kim, B.Y.; Kim, D.J.; Park, J.H. In vitro and in vivo growth inhibition of prostate cancer by the small molecule imiquimod. Int. J. Oncol. 2013, 42, 2087–2093. [Google Scholar] [PubMed]
- Greco, F.; Arif, I.; Botting, R.; Fante, C.; Quintieri, L.; Clementi, C.; Schiavon, O.; Pasut, G. Polysialic acid as a drug carrier: Evaluation of a new polysialic acid-epirubicin conjugate and its comparison against established drug carriers. Polym. Chem. 2013, 4, 1600–1609. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, J.; Kopeček, J. Selective inhibitory effect of hpma copolymer-cyclopamine conjugate on prostate cancer stem cells. Biomaterials 2012, 33, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.S.; Rosenbohm, C. Dry column vacuum chromatography. Synth.-Stuttg. 2001, 2001, 2431–2434. [Google Scholar] [CrossRef]
- Sheppeck, J.E.; Kar, H.; Hong, H. A convenient and scaleable procedure for removing the FMOC group in solution. Tetrahedron Lett. 2000, 41, 5329–5333. [Google Scholar] [CrossRef]
- Wang, B.; Rivard, E.; Manners, I. A new high-yield synthesis of Cl3PNSiMe3, a monomeric precursor for the controlled preparation of high molecular weight polyphosphazenes. Inorg. Chem. 2002, 41, 1690–1691. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, J.R.; Smith, E.L. Crystalline papain. I. Preparation, specificity, and activation. J. Biol. Chem. 1954, 207, 515–531. [Google Scholar] [PubMed]
- Andrianov, A.K.; Marin, A. Degradation of polyaminophosphazenes: Effects of hydrolytic environment and polymer processing. Biomacromolecules 2006, 7, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Song, S.-C.; Jin, J.-I.; Sohn, Y.S. A new class of biodegradable thermosensitive polymers. 2. Hydrolytic properties and salt effect on the lower critical solution temperature of poly(organophosphazenes) with methoxypoly(ethylene glycol) and amino acid esters as side groups. Macromolecules 1999, 32, 7820–7827. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[(amino acid ester)phosphazenes]: Synthesis, crystallinity, and hydrolytic sensitivity in solution and the solid state. Macromolecules 1994, 27, 1071–1075. [Google Scholar] [CrossRef]
- Jun, Y.J.; Park, M.K.; Jadhav, V.B.; Song, J.H.; Chae, S.W.; Lee, H.J.; Park, K.S.; Jeong, B.; Choy, J.H.; Sohn, Y.S. Tripodal amphiphiles tunable for self-assembly to polymersomes. J. Controll. Release 2010, 142, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Qiu, L.; Zhu, K. Novel polymersomes based on amphiphilic graft polyphosphazenes and their encapsulation of water-soluble anti-cancer drug. Polymer 2009, 50, 1173–1177. [Google Scholar] [CrossRef]
- Chen, C.; Qian, Y.-C.; Sun, C.-B.; Huang, X.-J. Self-assembly and morphological transitions of random amphiphilic poly([small β]-d-glucose-co-1-octyl) phosphazenes. Soft Matter 2015, 11, 6266–6274. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, X.-J.; Liu, Y.; Qian, Y.-C.; Xu, Z.-K. Synthesis and self-assembly of amphiphilic polyphosphazene with controllable composition via two step thiol-ene click reaction. Polymer 2014, 55, 833–839. [Google Scholar] [CrossRef]
- Jun, Y.J.; Toti, U.S.; Kim, H.Y.; Yu, J.Y.; Jeong, B.; Jun, M.J.; Sohn, Y.S. Thermoresponsive micelles from oligopeptide-grafted cyclotriphosphazenes. Angew. Chem. Int. Ed. 2006, 45, 6173–6176. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Toti, U.S.; Song, R.; Sohn, Y.S. A macromolecular prodrug of doxorubicin conjugated to a biodegradable cyclotriphosphazene bearing a tetrapeptide. Bioorganic Med. Chem. Lett. 2005, 15, 3576–3579. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | GFLG-OtBu, % a | Jeffamine M-1000, % a | Imiquimod loading, wt % b | Mw/Mn (GPC) c | Mn (GPC), kDa c | Dh (DLS), nm d |
|---|---|---|---|---|---|---|
| 1 | 100 | 0 | 0 | 1.3 | 6.84 | – f |
| 2 | 84 | 16 | 0 | 1.9 | 5.13 | 20,1 ± 1.6 |
| 3 | 57 | 43 | 0 | 1.9 | 4.05 | 14.2 ± 0.58 |
| 4 | 47 | 53 | 0 | 1.6 | 5.58 | 14.9 ± 0.58 |
| 5 | – e | – e | 2.4 | 1.6 | 6.20 | 201.6± 17.32 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linhardt, A.; König, M.; Schöfberger, W.; Brüggemann, O.; Andrianov, A.K.; Teasdale, I. Biodegradable Polyphosphazene Based Peptide-Polymer Hybrids. Polymers 2016, 8, 161. https://doi.org/10.3390/polym8040161
Linhardt A, König M, Schöfberger W, Brüggemann O, Andrianov AK, Teasdale I. Biodegradable Polyphosphazene Based Peptide-Polymer Hybrids. Polymers. 2016; 8(4):161. https://doi.org/10.3390/polym8040161
Chicago/Turabian StyleLinhardt, Anne, Michael König, Wolfgang Schöfberger, Oliver Brüggemann, Alexander K. Andrianov, and Ian Teasdale. 2016. "Biodegradable Polyphosphazene Based Peptide-Polymer Hybrids" Polymers 8, no. 4: 161. https://doi.org/10.3390/polym8040161