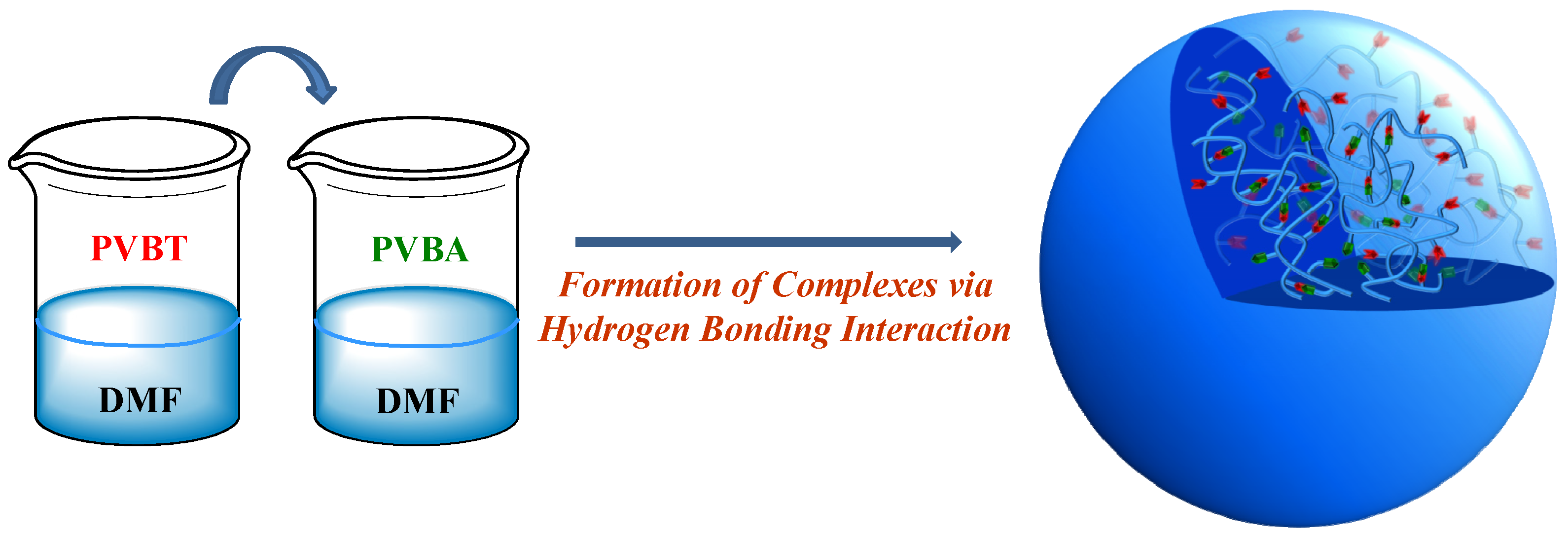
Thymine- and Adenine-Functionalized Polystyrene Form Self-Assembled Structures through Multiple Complementary Hydrogen Bonds


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Experimental Section
2.1. Materials
2.2. Poly(vinylbenzyl azide) (PVBN3)
2.3. Propargyl-Adenine (PA) [46]
2.4. Poly(4-vinylbenzyl triazolylmethyl methyladenine) (PVBA)
2.5. Propargyl-Thymine (PT) [26]
2.6. Poly(4-vinylbenzyl triazolylmethyl methylthymine) (PVBT)
2.7. PVBT/PVBA Blend Complexes
2.8. Complex Solutions
2.9. Characterization
3. Results and Discussion
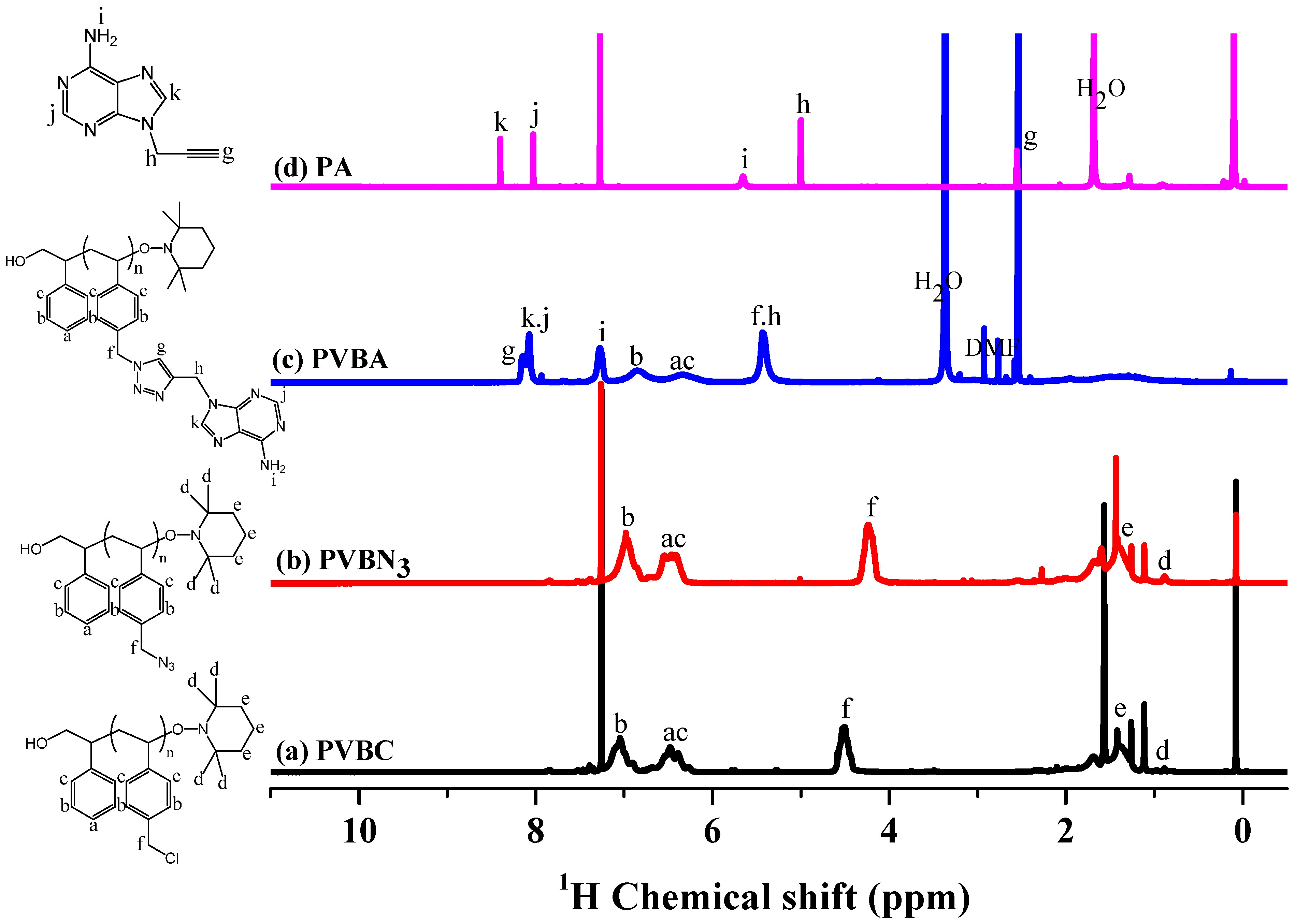
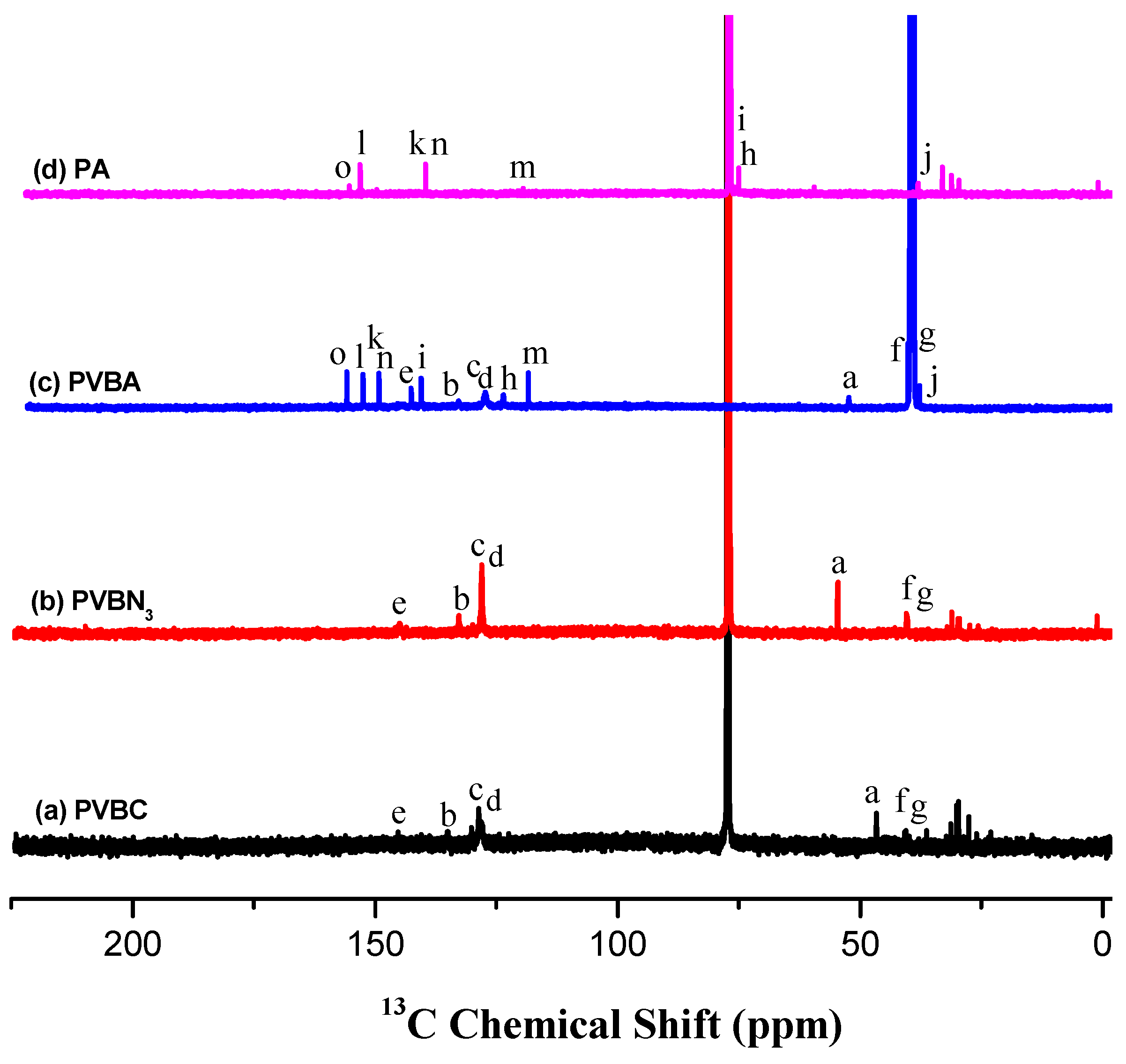
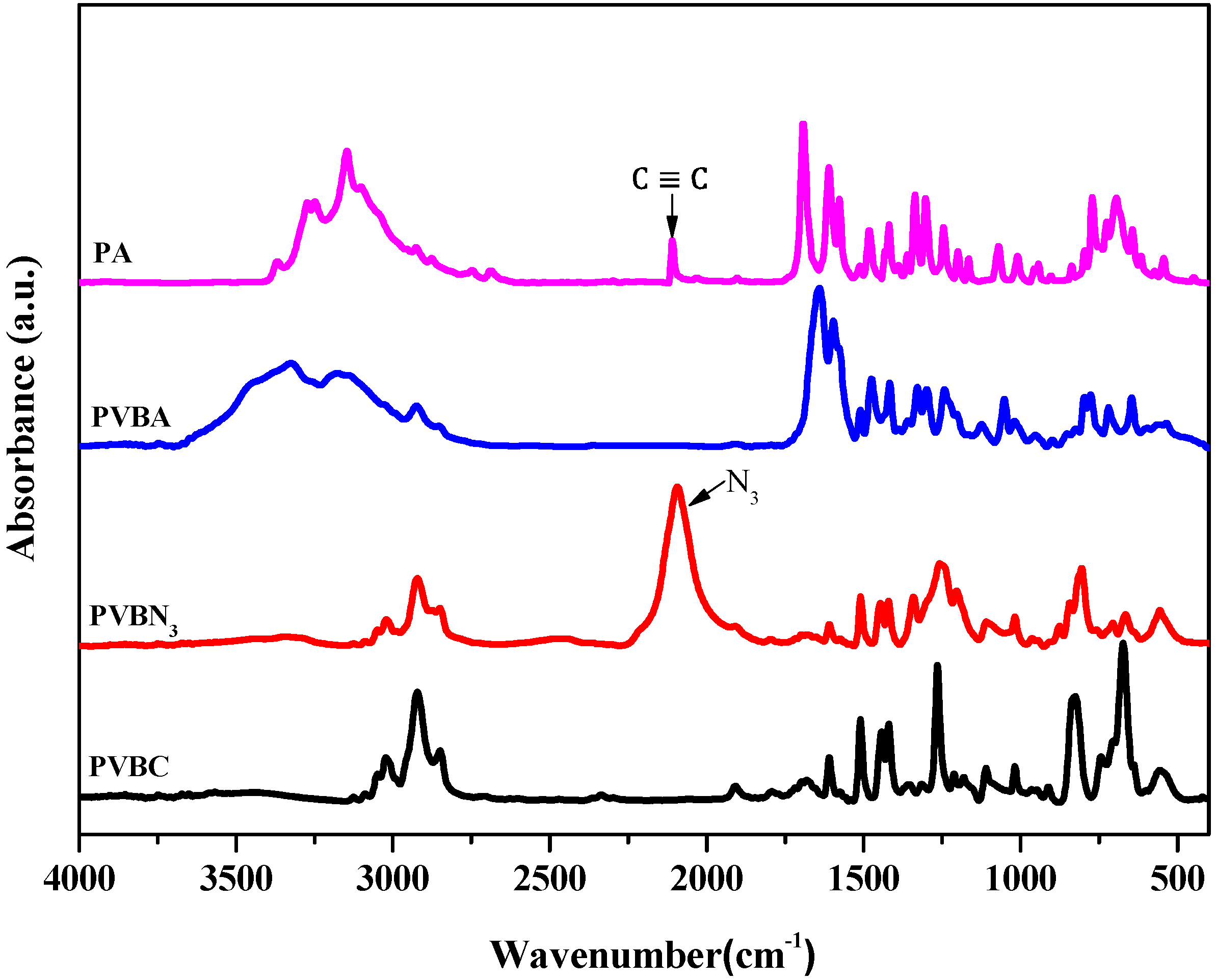
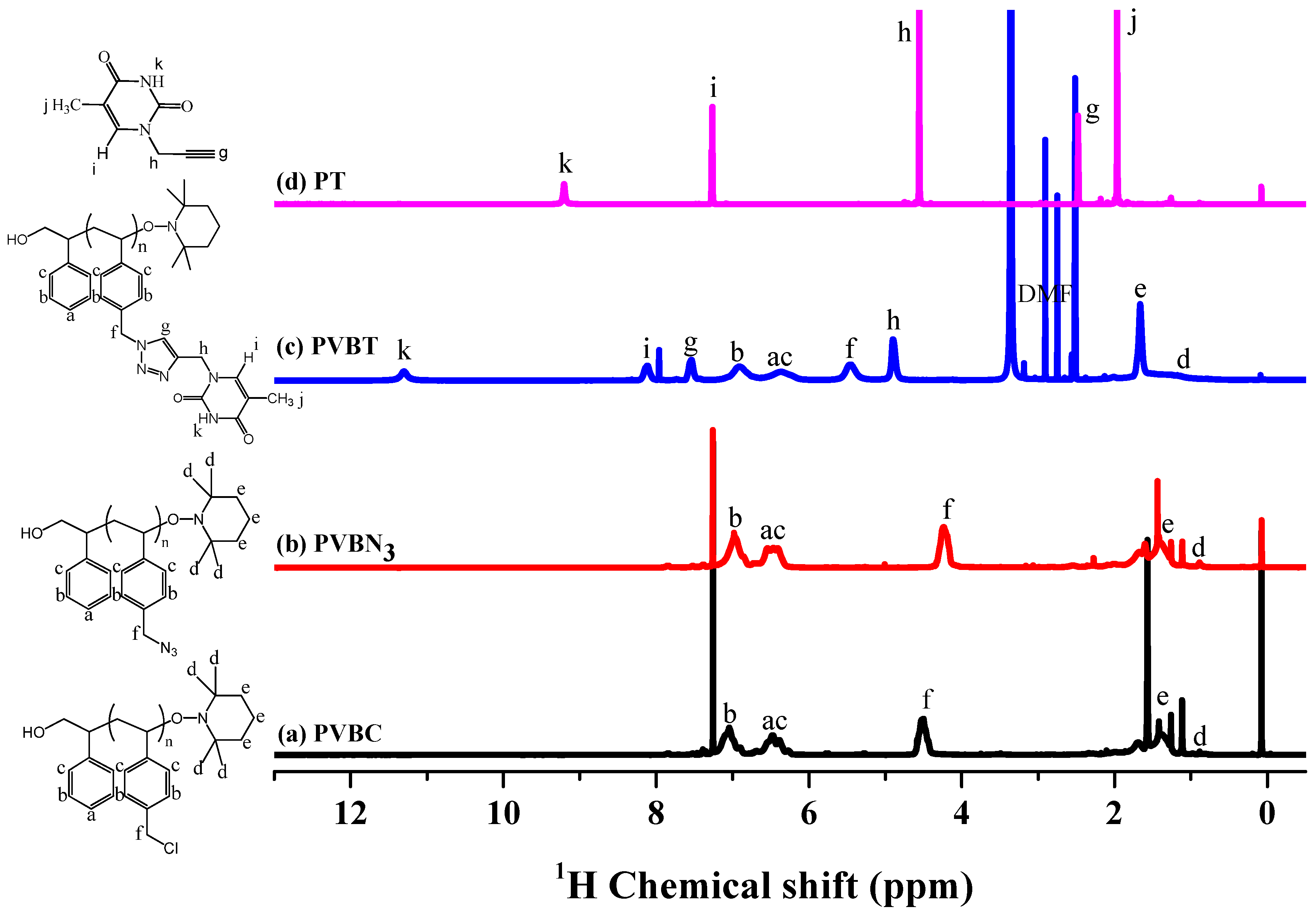
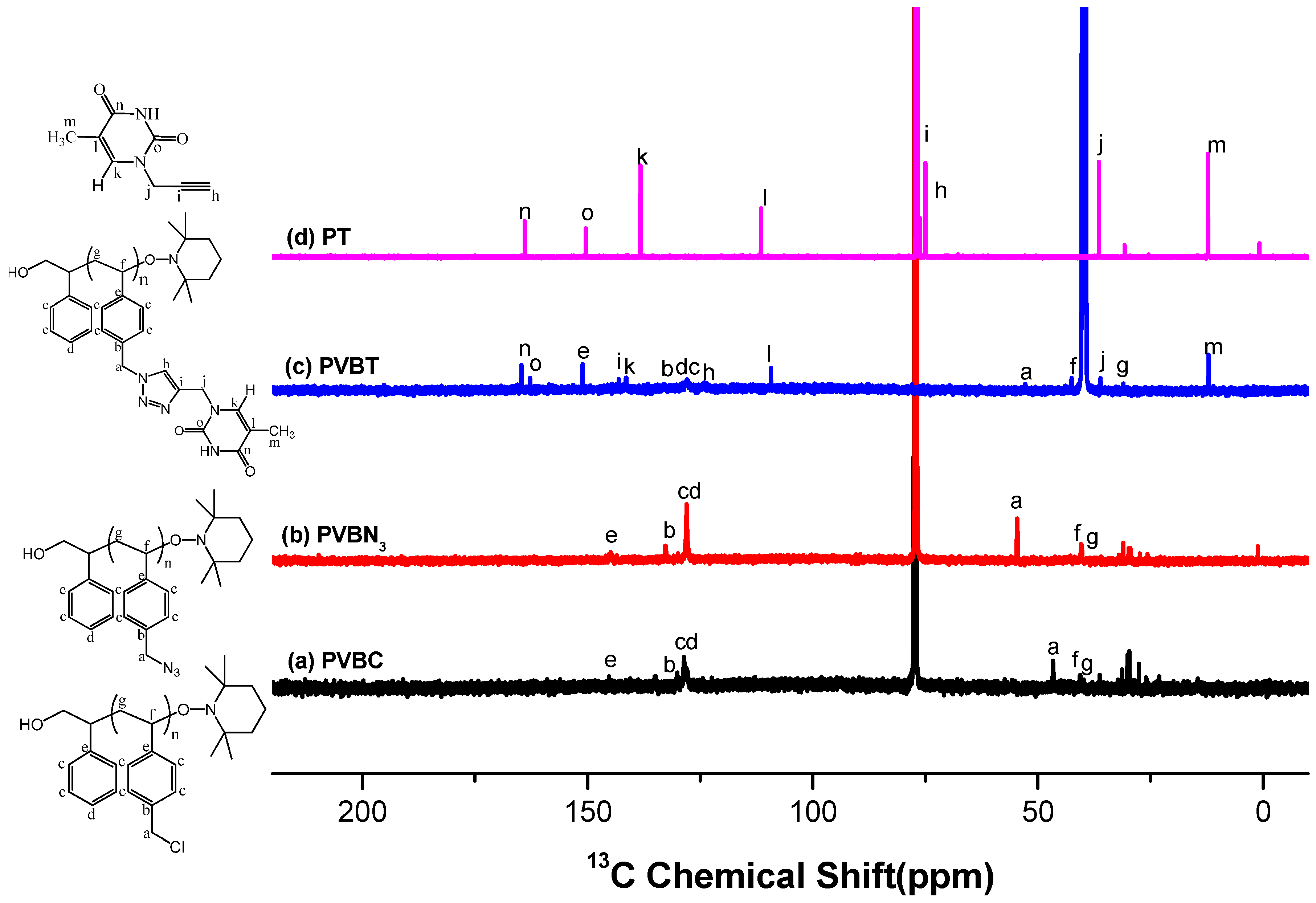
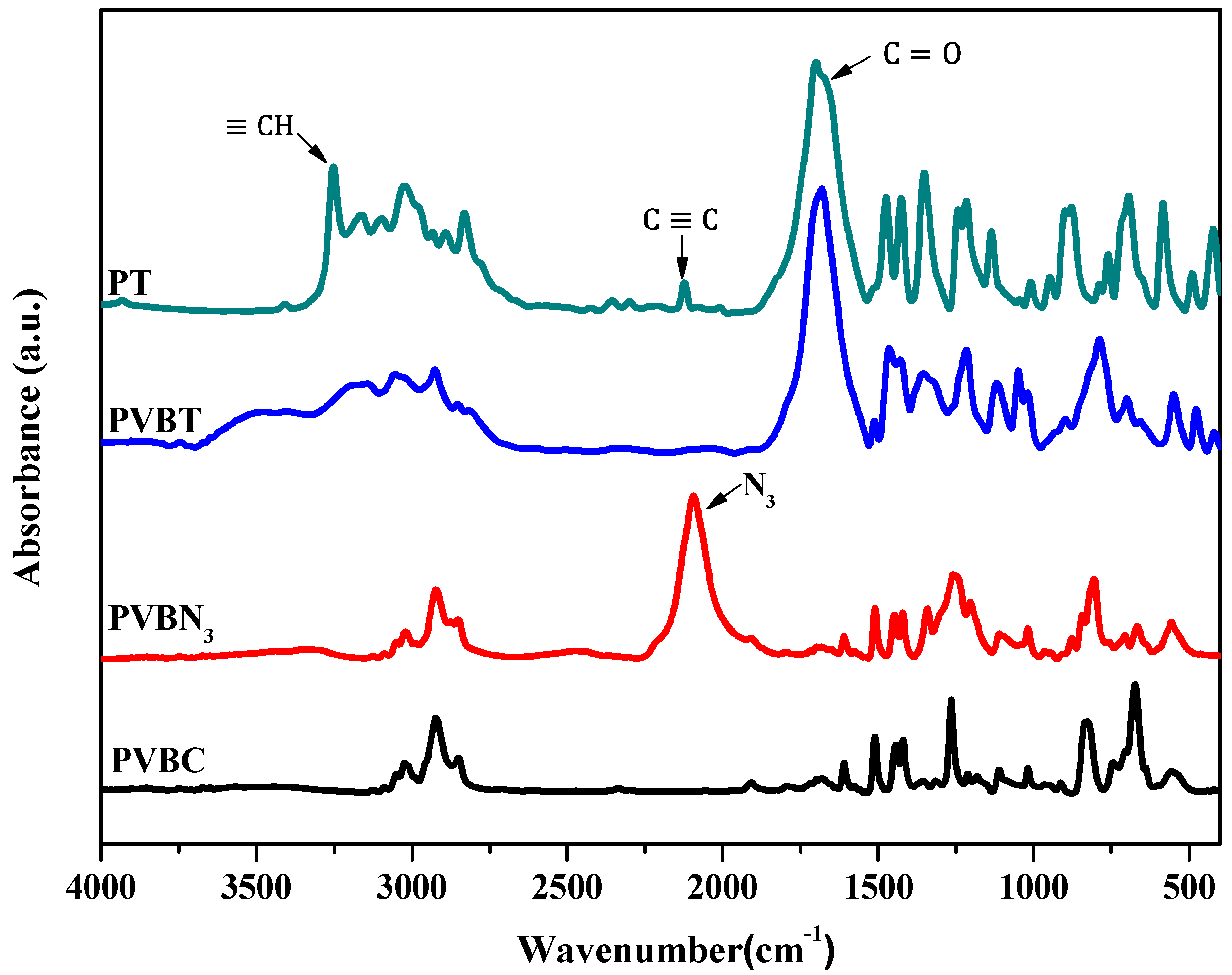
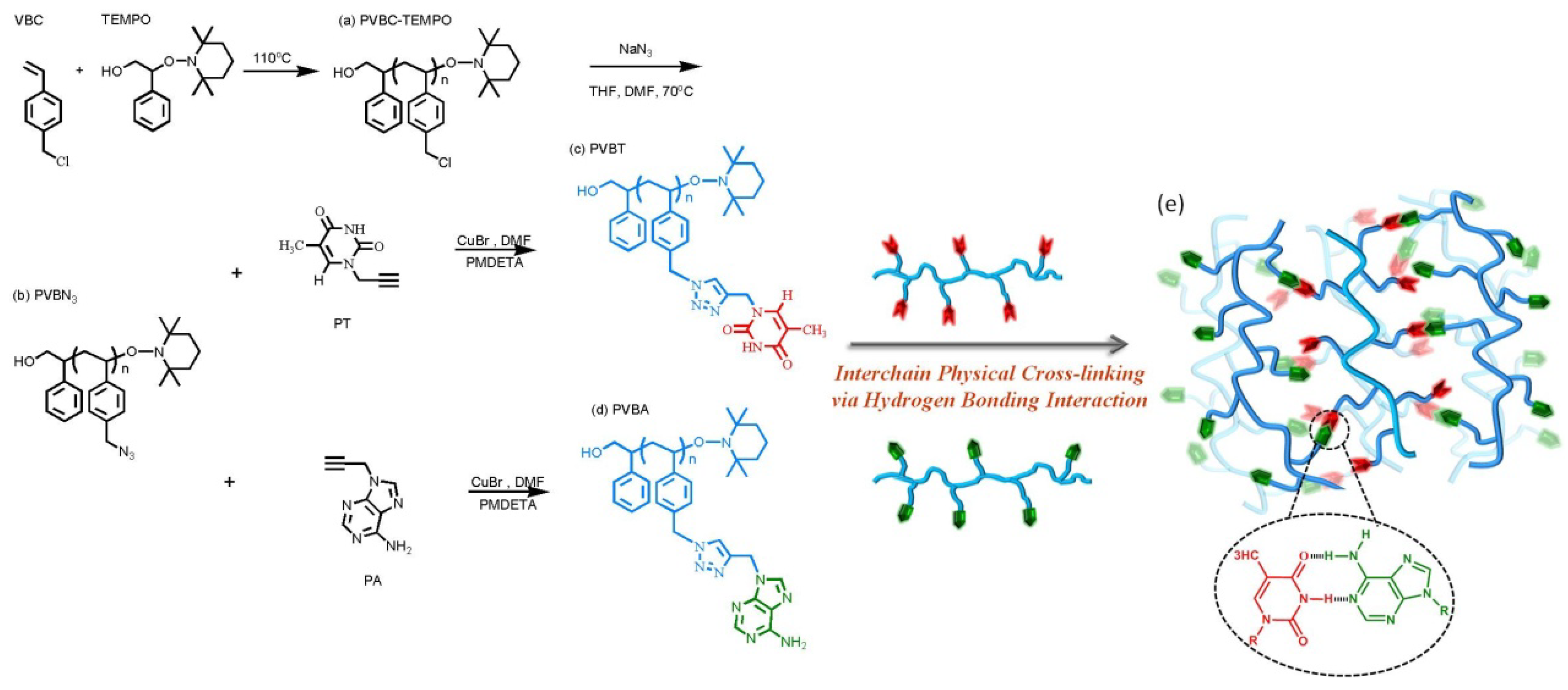
3.1. Synthesis of PVBA and PVBT
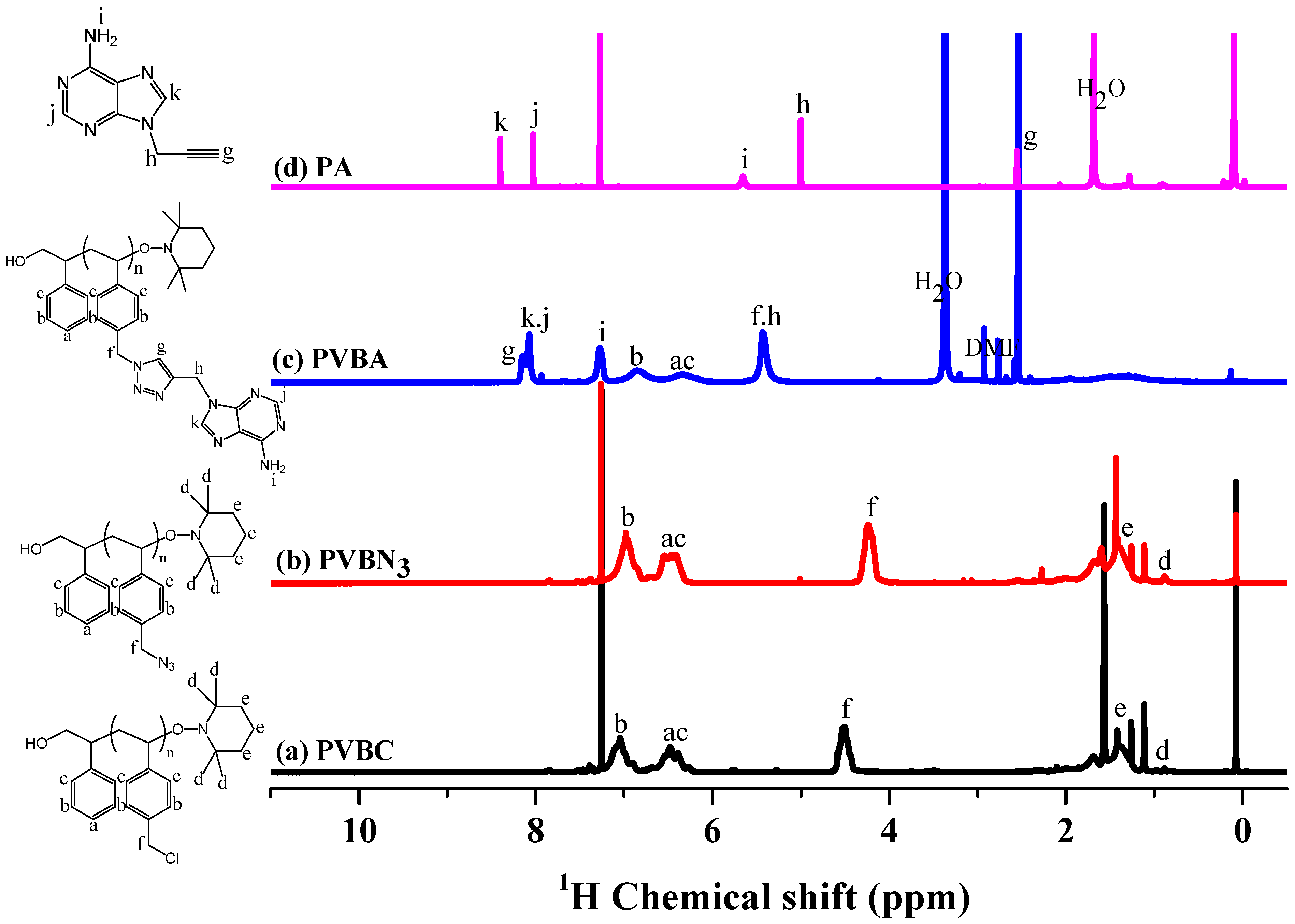
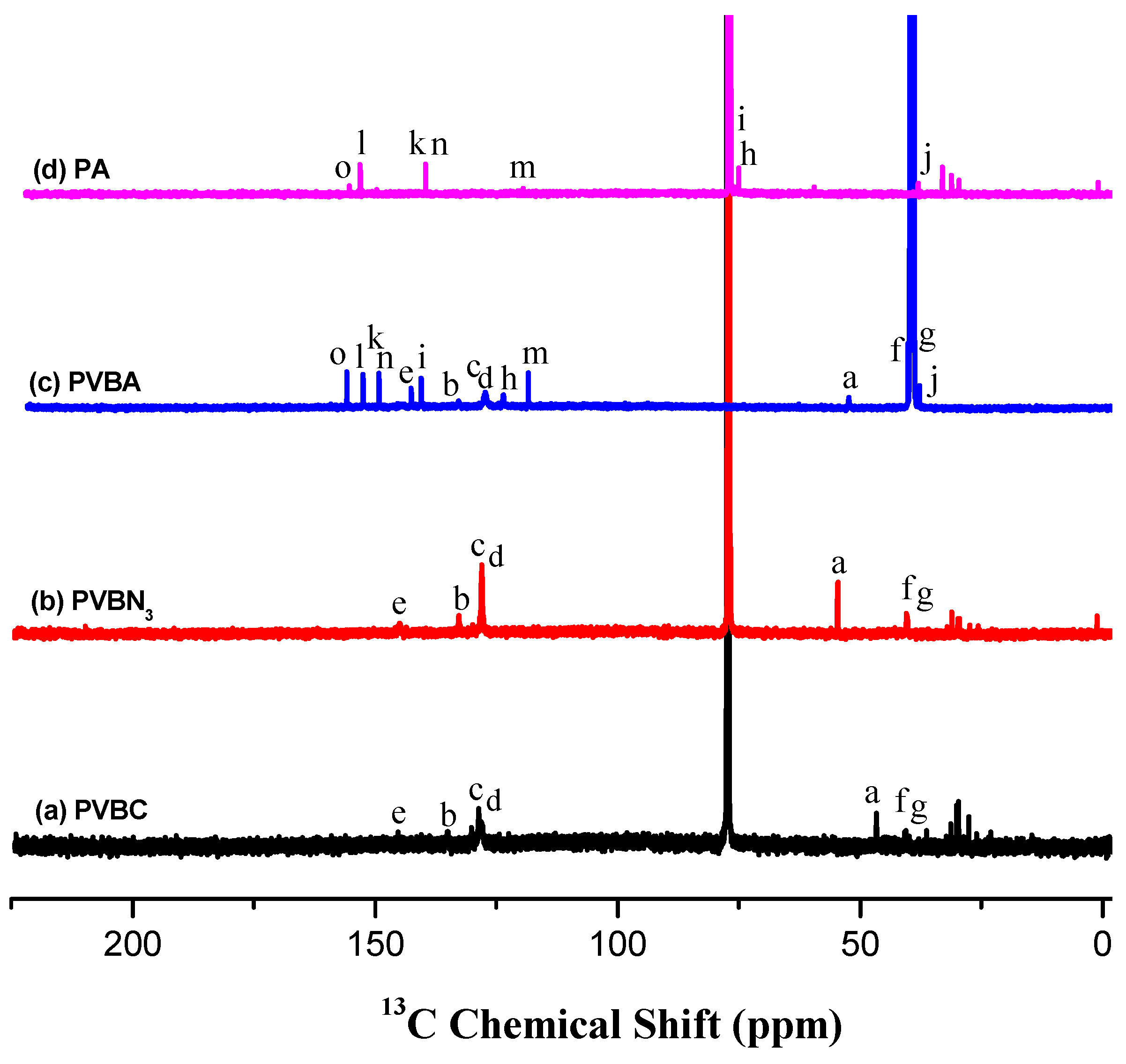
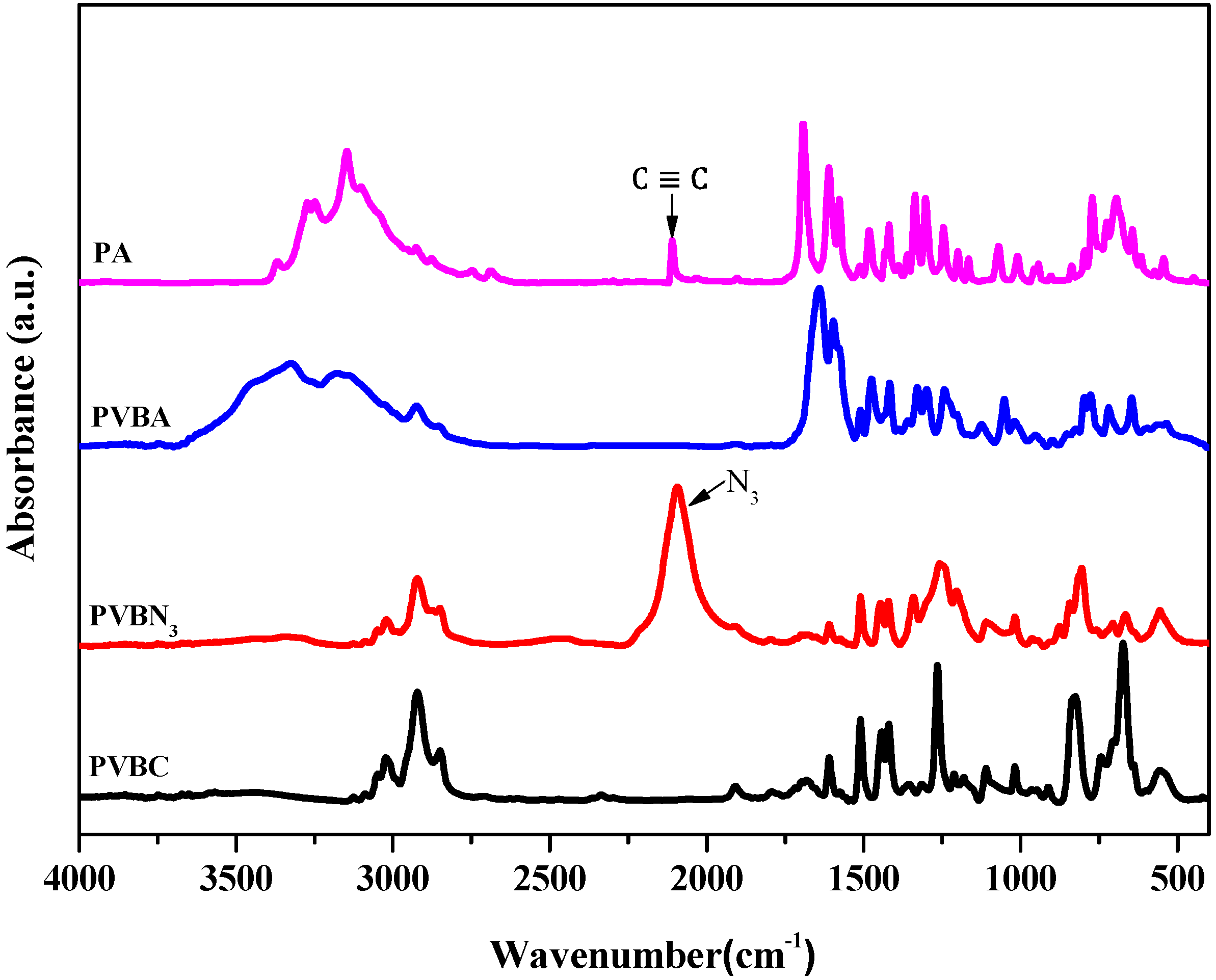


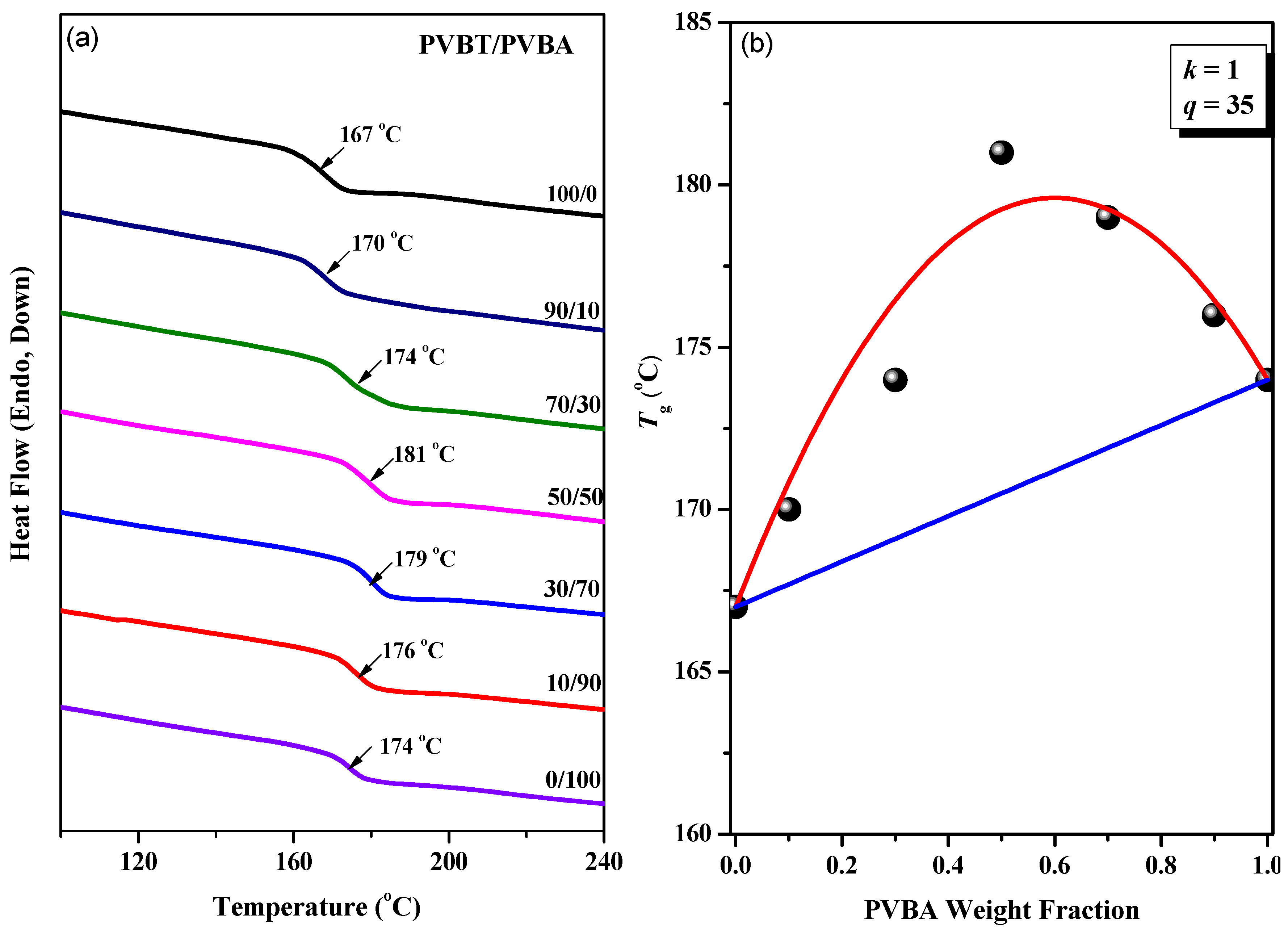
3.2. Thermal Analyses of PVBT/PVBA Blend Complexes
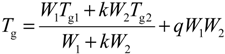

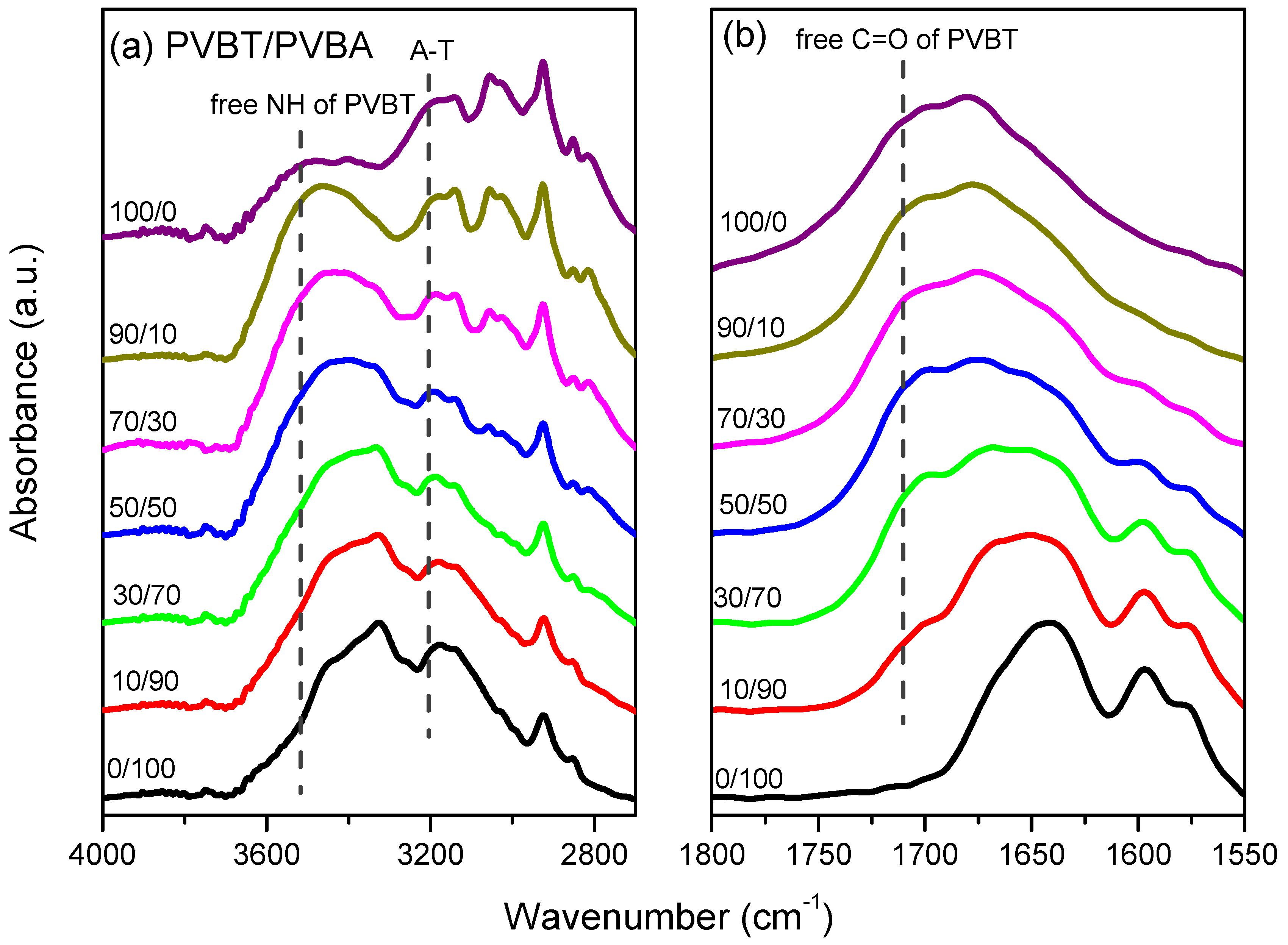
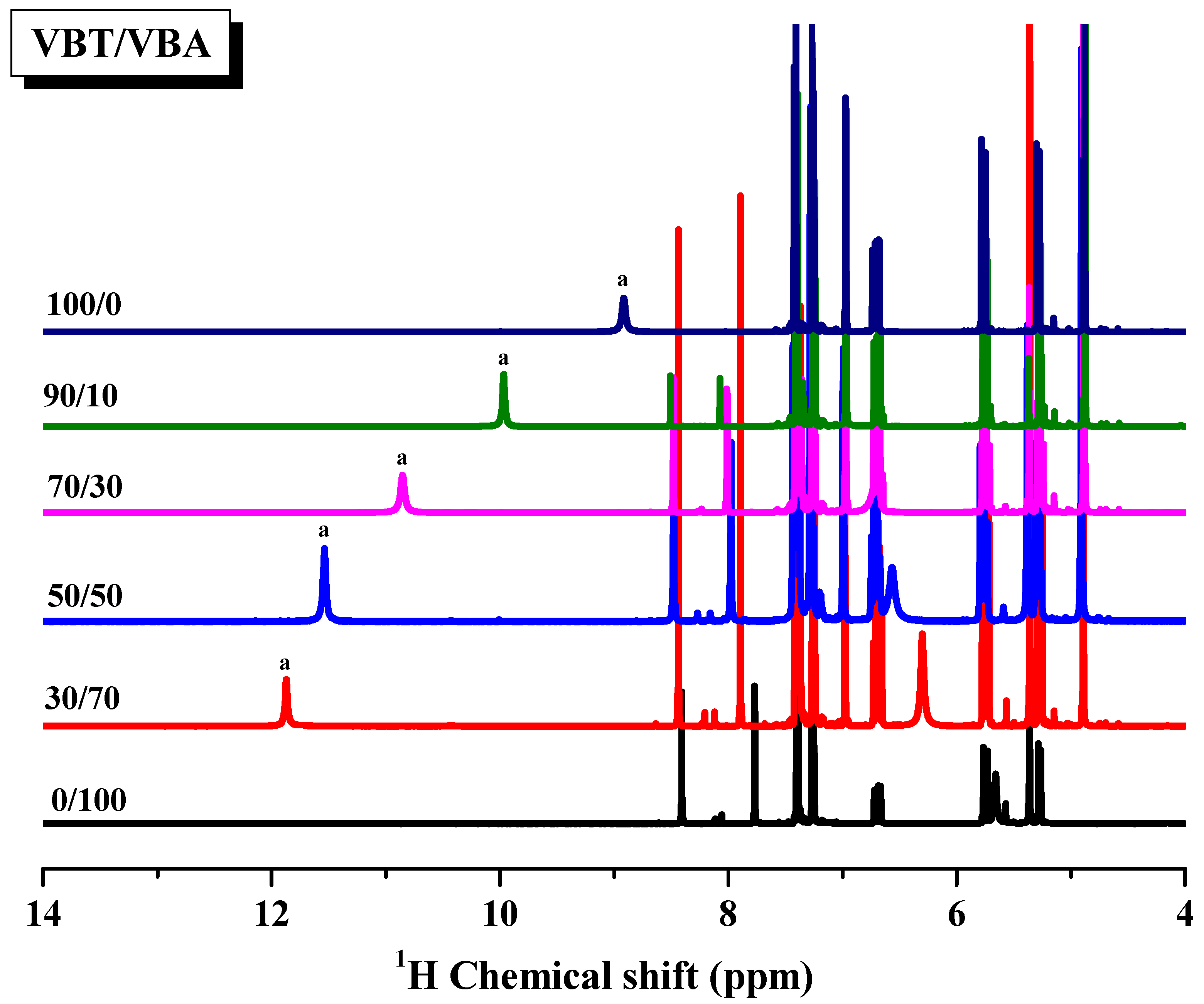
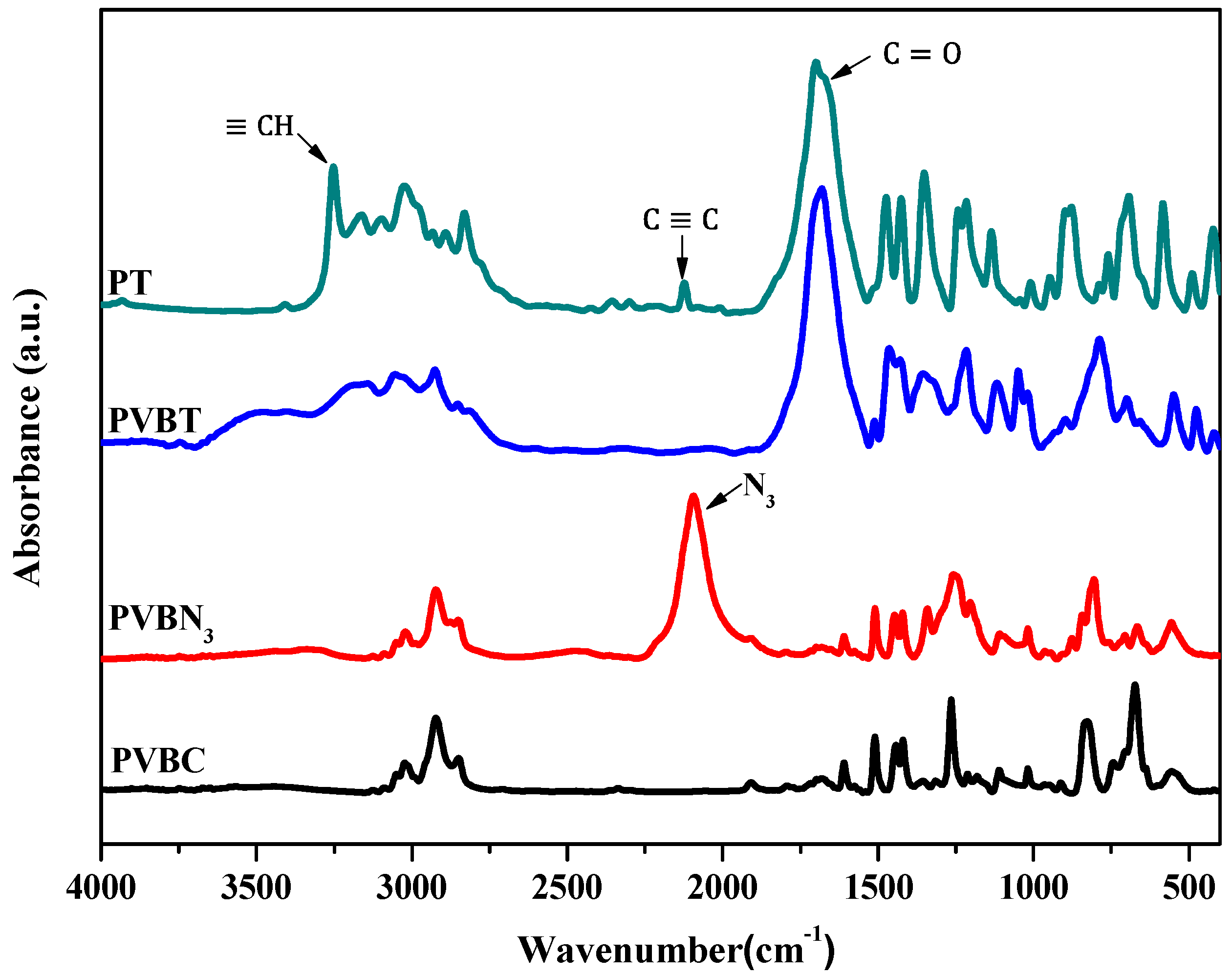
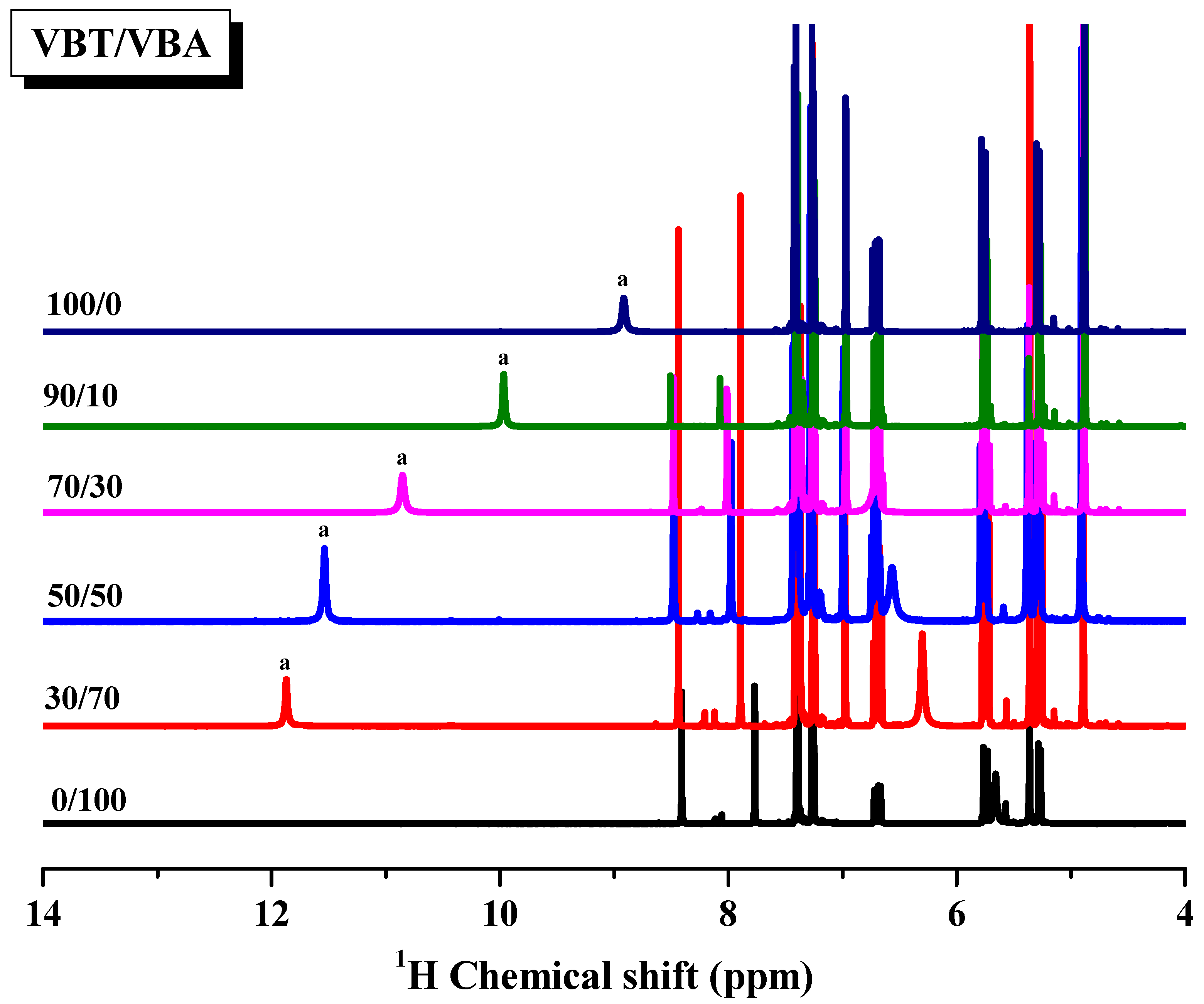
3.3. Multiple Hydrogen Bonding Interactions in PVBT/PVBA Blend Complexes

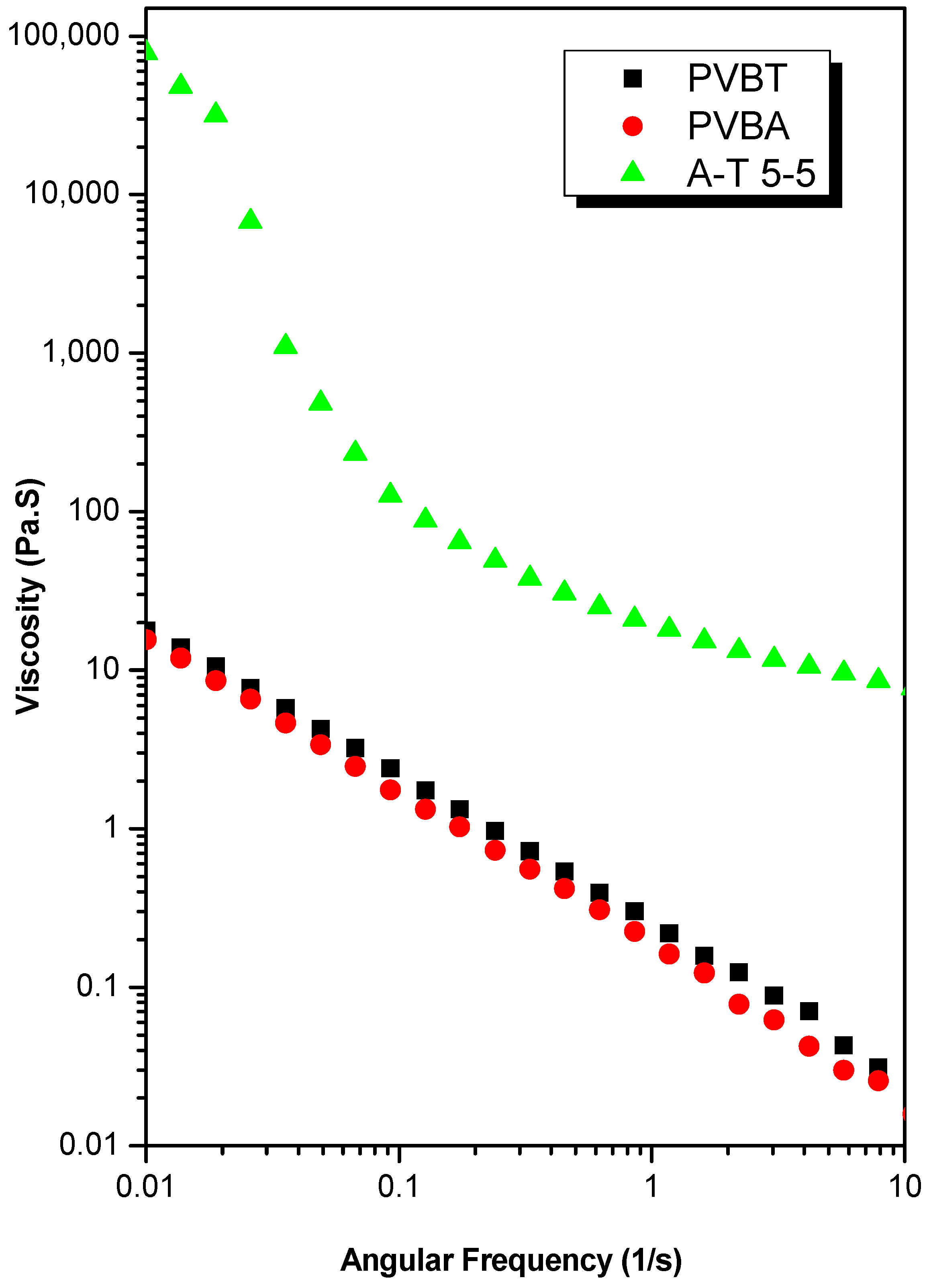
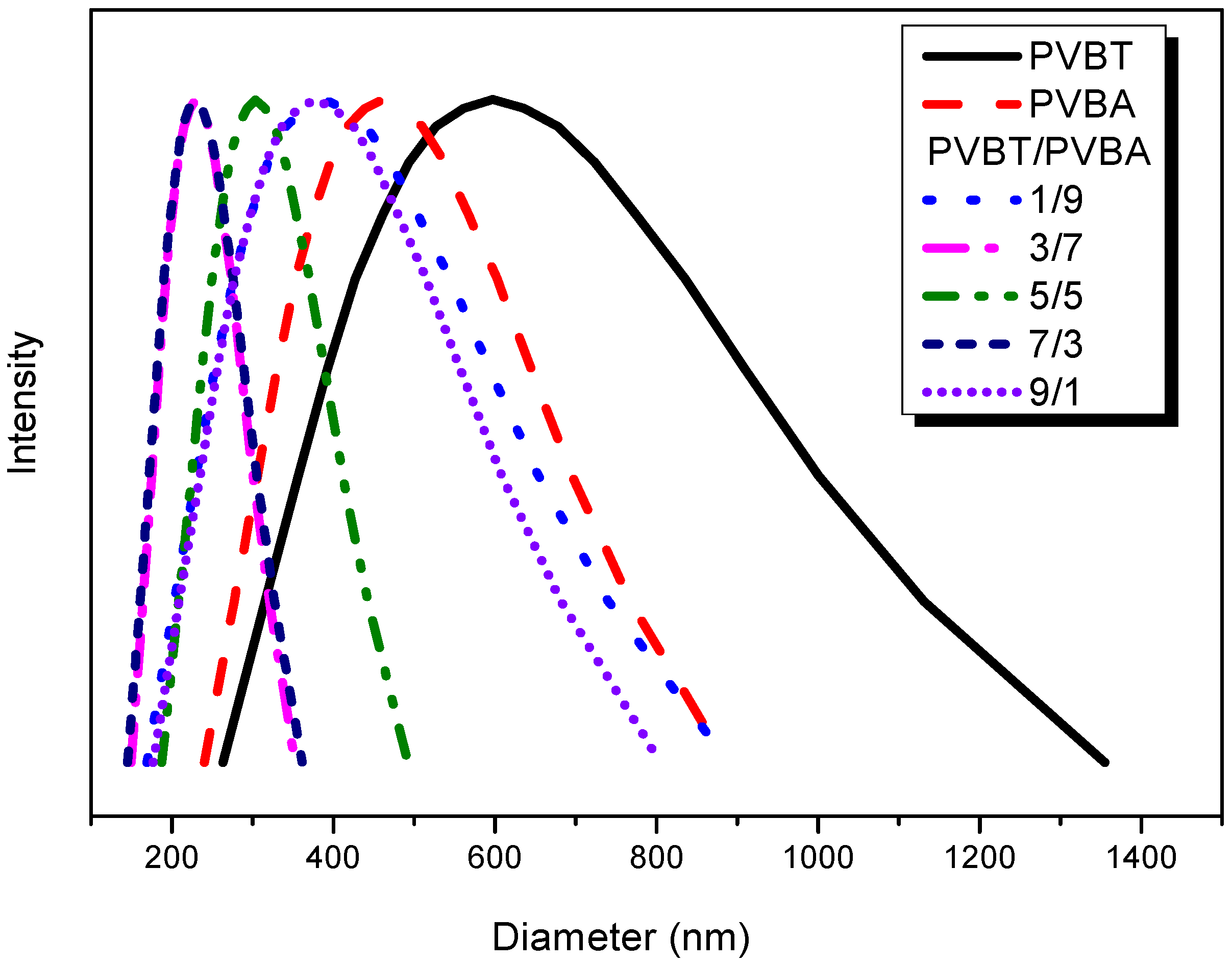
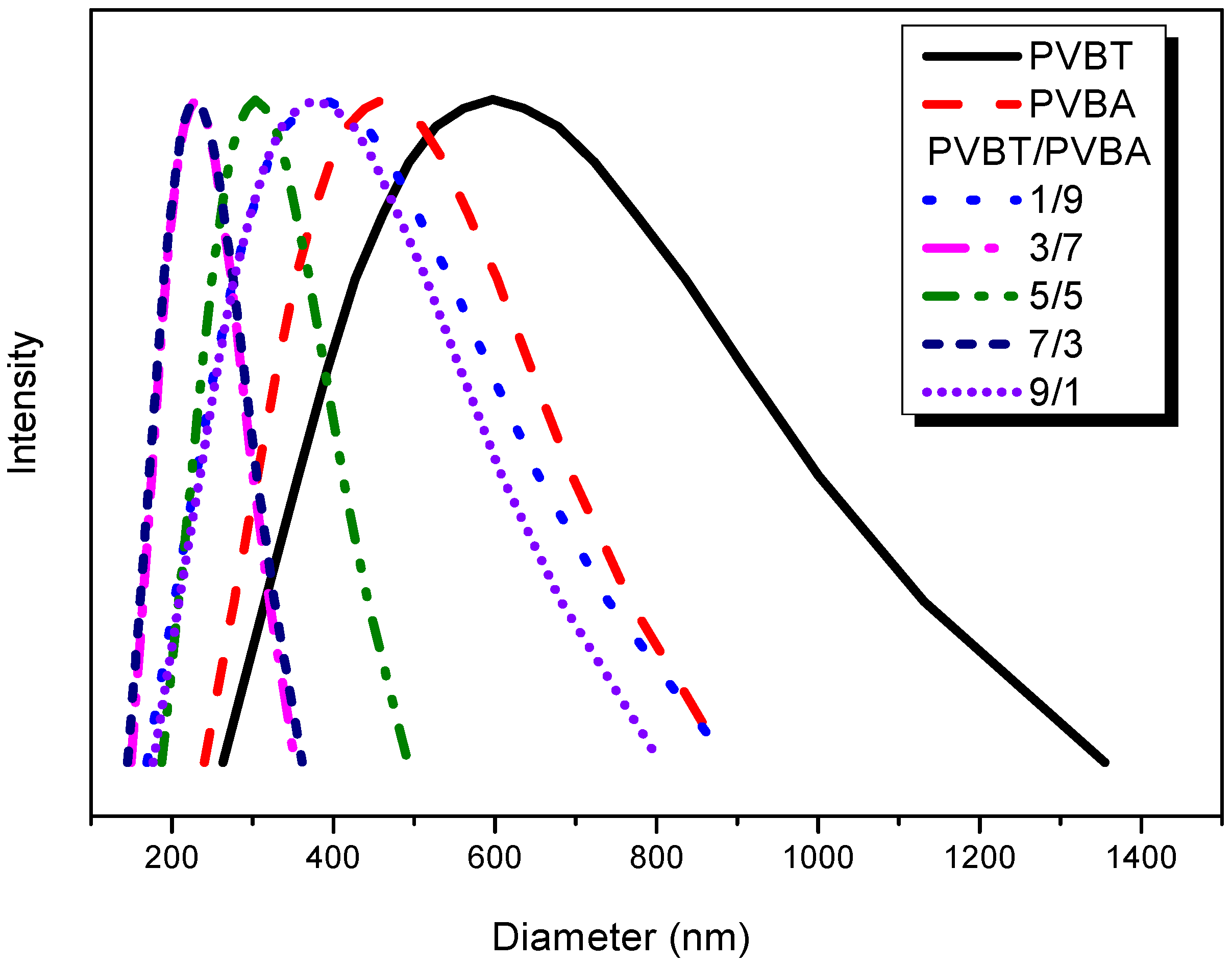
3.4. Viscosity, DLS, TEM and AFM Analyses of Supramolecular Structures of PVBT/PVBA Complexes



4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coleman, M.M.; Painter, P.C. Hydrogen bonded polymer blends. Prog. Polym. Sci. 1995, 20, 1–59. [Google Scholar] [CrossRef]
- He, Y.; Zhu, B.; Inoue, Y. Hydrogen bonds in polymer blends. Prog. Polym. Sci. 2004, 29, 1021–1051. [Google Scholar] [CrossRef]
- Kuo, S.W. Hydrogen-bonding in polymer blends. J. Polym. Res. 2008, 15, 459–486. [Google Scholar] [CrossRef]
- Kuo, S.W.; Chan, S.C.; Chang, F.C. Effect of hydrogen bonding strength on the microstructure and crystallization behavior of crystalline polymer blends. Macromolecules 2003, 36, 6653–6661. [Google Scholar] [CrossRef]
- Coleman, M.M.; Graf, J.F.; Painter, P.C. Specific Interactions and the Miscibility of Polymer Blends; Technomic Publishing: Lancaster, PA, USA, 1991. [Google Scholar]
- Kuo, S.W.; Chen, C.J. Using hydrogen-bonding interactions to control the peptide secondary structures and miscibility behavior of poly(l-glutamate)s with phenolic resin. Macromolecules 2011, 44, 7315–7326. [Google Scholar] [CrossRef]
- De Mefathi, M.V.; Frechet, J.M.J. Study of the compatibility of blends of polymers and copolymers containing styrene, 4-hydroxystyrene and 4-vinylpyridine. Polymer 1988, 29, 477–482. [Google Scholar] [CrossRef]
- Xiang, M.; Jiang, M.; Zhang, Y.; Wu, C. Intermacromolecular complexation due to specific interactions 4. The hydrogen-bonding complex of vinylphenol-containing copolymer and vinylpyridine-containing copolymer. Macromolecules 1997, 30, 2313–2319. [Google Scholar] [CrossRef]
- Wang, J.; Cheung, M.K.; Mi, Y. Miscibility in blends of poly(4-vinylpyridine)/poly(4-vinylphenol) as studied by 13C solid-state NMR. Polymer 2001, 42, 3087–3093. [Google Scholar] [CrossRef]
- Kuo, S.W.; Chang, F.C. Studies of miscibility behavior and hydrogen bonding in blends of poly(vinylphenol) and poly(vinylpyrrolidone). Macromolecules 2001, 34, 5224–5228. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tung, P.H.; Chang, F.C. Syntheses and the study of strongly hydrogen-bonded poly(vinylphenol-b-vinylpyridine) diblock copolymer through anionic polymerization. Macromolecules 2006, 39, 9388–9395. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, M.; Ning, F.L.; Chen, D.Y.; Liu, S.Y.; Duan, H.W. Block-copolymer-free strategy for preparing micelles and hollow spheres:self-assembly of poly(4-vinylpyridine) and modified polystyrene. Macromolecules 2002, 35, 5980–5989. [Google Scholar] [CrossRef]
- Chen, W.C.; Kuo, S.W.; Jeng, U.S.; Chang, F.C. Self-assembly through competitive interactions of miscible diblock copolymer/homopolymer blends: Poly(vinylphenol-b-methyl methacrylate)/poly(vinylpyrrolidone) blend. Macromolecules 2008, 41, 1401–1410. [Google Scholar] [CrossRef]
- Chen, W.C.; Kuo, S.W.; Lu, C.H.; Jeng, U.S.; Chang, F.C. Self-assembly structures through competitive interactions of crystalline-amorphous diblock copolymer/homopolymer blends: Poly(ε-caprolactone-b-4-vinyl pyridine)/poly(vinyl phenol). Macromolecules 2009, 42, 3580–3590. [Google Scholar] [CrossRef]
- Chen, W.C.; Kuo, S.W.; Chang, F.C. Self-assembly of an A–B diblock copolymer blended with a C homopolymer and a C–D diblock copolymer through hydrogen bonding interaction. Polymer 2010, 51, 4176–4184. [Google Scholar] [CrossRef]
- IIhan, F.; Gray, M.; Rotello, V.M. Reversible side chain modification through noncovalent interactions: “plug and play” polymers. Macromolecules 2001, 34, 2597–2601. [Google Scholar] [CrossRef]
- Sherrington, D.C.; Taskinen, K.A. Self-assembly in synthetic macromolecular systems via multiple hydrogen bonding interactions. Chem. Soc. Rev. 2001, 30, 83–93. [Google Scholar] [CrossRef]
- Feldman, K.E.; Kade, M.J.; de Greef, T.F.A.; Meijer, E.W.; Kramer, E.J.; Hawker, C.J. Polymers with multiple hydrogen-bonded end groups and their blends. Macromolecules 2008, 41, 4694–4700. [Google Scholar] [CrossRef]
- Sivakava, S.; Wu, J.; Campo, C.J.; Mather, P.T.; Rowan, S.J. Liquid-crystalline supramolecular polymers formed through complementary nucleobase-pair interactions. Chem. A Eur. J. 2005, 12, 446–456. [Google Scholar]
- Altintas, O.; Schulze-Suenninghausen, D.; Luy, B.; Barner-Kowollik, C. Facile preparation of supramolecular H-shaped (Ter)polymers via multiple hydrogen bonding. ACS Macro Lett. 2013, 2, 211–216. [Google Scholar] [CrossRef]
- Kuo, S.W.; Cheng, R.S. DNA-Like interactions enhance the miscibility of supramolecular polymer blends. Polymer 2009, 50, 177–188. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tsai, H.T. Complementary multiple hydrogen-bonding interactions increase the glass transition temperatures to PMMA copolymer mixtures. Macromolecules 2009, 42, 4701–4711. [Google Scholar] [CrossRef]
- Kuo, S.W.; Hsu, C.H. Miscibility enhancement of supramolecular polymer blends through complementary multiple hydrogen bonding interactions. Polym. Int. 2010, 59, 998–1005. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tsai, H.T. Self-complementary multiple hydrogen bonding interactions increase the glass transition temperatures to supramolecular poly(methyl methacrylate) copolymers. J. Appl. Polym. Sci. 2012, 123, 3275–3282. [Google Scholar] [CrossRef]
- Hu, W.H.; Huang, K.W.; Kuo, S.W. Heteronucleobase-functionalized benzoxazine: Synthesis, thermal property and multiple hydrogen bonding interactions. Polym. Chem. 2012, 3, 1546–1554. [Google Scholar] [CrossRef]
- Wu, Y.C.; Kuo, S.W. Complementary multiple hydrogen bonding interactions mediate the self-assembly of supramolecular structures from thymine-containing block copolymers and hexadecyladenine. Polym. Chem. 2012, 3, 3100–3111. [Google Scholar] [CrossRef]
- Wang, J.H.; Altukhov, O.; Cheng, C.C.; Chang, F.C.; Kuo, S.W. Supramolecular structures of uracil-functionalized PEG with multi-diamidopyridine POSS through complementary hydrogen bonding interactions. Soft Matter 2013, 9, 5196–5206. [Google Scholar]
- Huang, K.W.; Wu, Y.R.; Kuo, S.W. From random coil to helical structure induced by carbon nanotube through supramolecular interactions. Macromolecules Rapid Commun. 2013, 34, 1530–1536. [Google Scholar] [CrossRef]
- Park, T.; Zimmerman, S.C. Formation of a miscible supramolecular polymer blend through self-assembly mediated by a quadruply hydrogen-bonded heterocomplex. J. Am. Chem. Soc. 2006, 128, 11582–11590. [Google Scholar] [CrossRef]
- Mather, B.D.; Baker, M.B.; Beyer, F.L.; Berg, M.A.G.; Green, M.D.; Long, T.E. Supramolecular triblock copolymers containing complementary nucleobase nolecular recognition. Macromolecules 2007, 40, 6834–6845. [Google Scholar] [CrossRef]
- Nair, K.P.; Breedveld, V.; Weck, M. Complementary hydrogen-bonded thermoreversible polymer networks with tunable properties. Macromolecules 2008, 41, 3429–3438. [Google Scholar] [CrossRef]
- Mather, B.D.; Lizotte, J.R.; Long, T.E. Synthesis of chain end functionalized multiple hydrogen bonded polystyrenes and poly(alkyl acrylates) using controlled radical polymerization. Macromolecules 2004, 37, 9331–9337. [Google Scholar] [CrossRef]
- Bazzi, H.S.; Sleiman, H.F. Adenine-containing block copolymers via ring-opening metathesis polymerization: synthesis and self-assembly into rod morphologies. Macromolecules 2002, 35, 9617–9620. [Google Scholar] [CrossRef]
- Park, T.; Zimmerman, S.C. Interplay of fidelity, binding strength, and structure in supramolecular polymers. J. Am. Chem. Soc. 2006, 128, 14236–14237. [Google Scholar] [CrossRef]
- Smith, J.R.; Walter, T. Nucleic acid models. Prog. Polym. Sci. 1996, 21, 209–253. [Google Scholar] [CrossRef]
- Watson, J.D.; Berry, A. DNA: The Secret of Life; Knopf: NewYork, NY, USA, 2003. [Google Scholar]
- Lutz, J.F.; Thunemann, A.F.; Rurack, K. DNA-like “melting” of adenine- and thymine-tunctionalized synthetic copolymers. Macromolecules 2005, 38, 8124–8126. [Google Scholar] [CrossRef]
- Lutz, J.F.; Thunemann, A.F.; Nehbring, R. Preparation by controlled radical polymerization and self-assembly via base-recognition of synthetic polymers bearing complementary nucleobases. J. Polym. Sci. Polym. Chem. 2005, 43, 4805–4818. [Google Scholar]
- Lutz, J.F.; Pfeifer, S.; Chanana, M.; Thunemann, A.F.; Bienert, R. H-Bonding-directed self-assembly of synthetic copolymers containing nucleobases: organization and colloidal fusion in a noncompetitive solvent. Langmuir 2006, 22, 7411–7415. [Google Scholar] [CrossRef]
- Yamauchi, K.; Kanomata, A.; Inoue, T.; Long, T.E. Thermoreversible polyesters consisting of multiple hydrogen bonding (MHB). Macromolecules 2004, 37, 3519–3522. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization; Wiley Publication: New York, NY, USA, 1991; p. 470. [Google Scholar]
- Overberger, C.C.; Inaki, Y. Graft copolymers containing nucleic acid bases and l-amino acids. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 1739–1958. [Google Scholar] [CrossRef]
- Inaki, Y.; Sakuma, Y.; Suda, Y.; Takemoto, K. Functional monomers and polymers. XCVII. Synthesis of oligomer models of polyethyleneimine derivatives containing pendant thymine bases. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 1917–1933. [Google Scholar] [CrossRef]
- Cheng, C.C.; Huang, C.F.; Yen, Y.C.; Chang, F.C. A “plug and play” polymer through biocomplementary hydrogen bonding. J. Polym. Sci. A Polym. Chem. 2008, 46, 6416–6424. [Google Scholar] [CrossRef]
- Lu, C.H.; Huang, C.F.; Kuo, S.W.; Chang, F.C. Synthesis and characterization of poly(ε-caprolactone-b-4-vinylpyridine): Initiation, polymerization, solution morphology, and gold metalation. Macromolecules 2009, 42, 1067–1078. [Google Scholar] [CrossRef]
- Wu, Y.C.; Kuo, S.W. Self-Assembly supramolecular structure through complementary multiple hydrogen bonding of heteronucleobase-multifunctionalized polyhedral oligomeric silsesquioxane (POSS) complexes. J. Mater. Chem. 2012, 22, 2982–2991. [Google Scholar] [CrossRef]
- Kwei, T. The effect of hydrogen bonding on the glass transition temperatures of polymer mixtures. J. Polym. Sci. Polym. Lett. Ed. 1984, 22, 307–313. [Google Scholar] [CrossRef]
- Wang, J.H.; Altukhov, O.; Cheng, C.C.; Chang, F.C.; Kuo, S.W. Investigating the Effect of Uracil-Functionalized Poly(ethylene glycol) on the Thermal Properties, Interactions, and Conductivity Behavior with LiAsF6 Salt. Polymers 2013, 5, 937–953. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Multiple morphologies of “Crew-Cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers. Science 1995, 268, 1728–1731. [Google Scholar]
- Wang, X.; Chen, J.; Hong, K.; Mays, J.W. Well-defined polyisoprene-b-poly(acrylic acid)/polystyrene-b-polyisoprene-b-poly(acrylic acid) block copolymers: synthesis and their self-assembled hierarchical structures in aqueous media. ACS Macro Lett. 2012, 1, 743–747. [Google Scholar] [CrossRef]
- Guo, M.; Jiang, M. Non-covalently connected micelles (NCCMs): The origins and development of a new concept. Soft Matter 2009, 5, 495–500. [Google Scholar] [CrossRef]
- Wang, D.; Su, Y.; Jin, C.; Zhu, B.; Pan, Y.; Zhu, L.; Liu, J.; Tu, C.; Yan, D.; Zhu, X. Supramolecular copolymer micelles based on the complementary multiple hydrogen bonds of nucleobases for drug delivery. Biomacromolecules 2011, 12, 1370–1379. [Google Scholar] [CrossRef]
- Wang, D.; Huan, X.; Zhu, L.; Liu, J.; Qiu, F.; Yan, D.; Zhu, X. Salt/pH dual-responsive supramolecular brush copolymer micelles with molecular recognition of nucleobases for drug delivery. RSC Adv. 2012, 2, 11953–11962. [Google Scholar] [CrossRef]
- Wang, D.; Chen, H.; Su, Y.; Qiu, F.; Zhu, L.; Huan, X.; Zhu, B.; Yan, D.; Guo, F.; Zhu, X. Supramolecular amphiphilic multiarm hyperbranched copolymer: Synthesis, self-assembly and drug delivery applications. Polym. Chem. 2013, 4, 85–94. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, Y.-S.; Wu, Y.-C.; Kuo, S.-W. Thymine- and Adenine-Functionalized Polystyrene Form Self-Assembled Structures through Multiple Complementary Hydrogen Bonds. Polymers 2014, 6, 1827-1845. https://doi.org/10.3390/polym6061827
Wu Y-S, Wu Y-C, Kuo S-W. Thymine- and Adenine-Functionalized Polystyrene Form Self-Assembled Structures through Multiple Complementary Hydrogen Bonds. Polymers. 2014; 6(6):1827-1845. https://doi.org/10.3390/polym6061827
Chicago/Turabian StyleWu, Yu-Shian, Yi-Chen Wu, and Shiao-Wei Kuo. 2014. "Thymine- and Adenine-Functionalized Polystyrene Form Self-Assembled Structures through Multiple Complementary Hydrogen Bonds" Polymers 6, no. 6: 1827-1845. https://doi.org/10.3390/polym6061827