Peptide-Based Polymer Therapeutics

Abstract
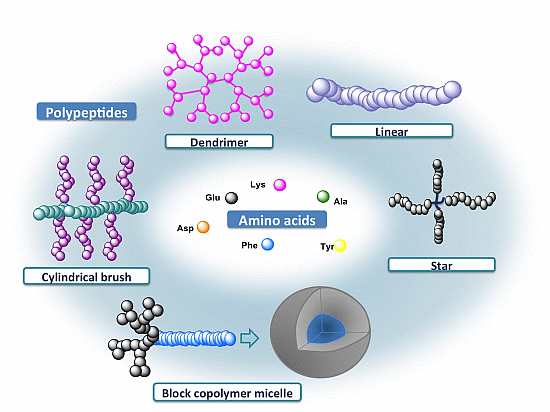
:
1. Introduction
2. Polypeptide Design and Synthesis
2.1. Recombinant DNA Expression of Proteins
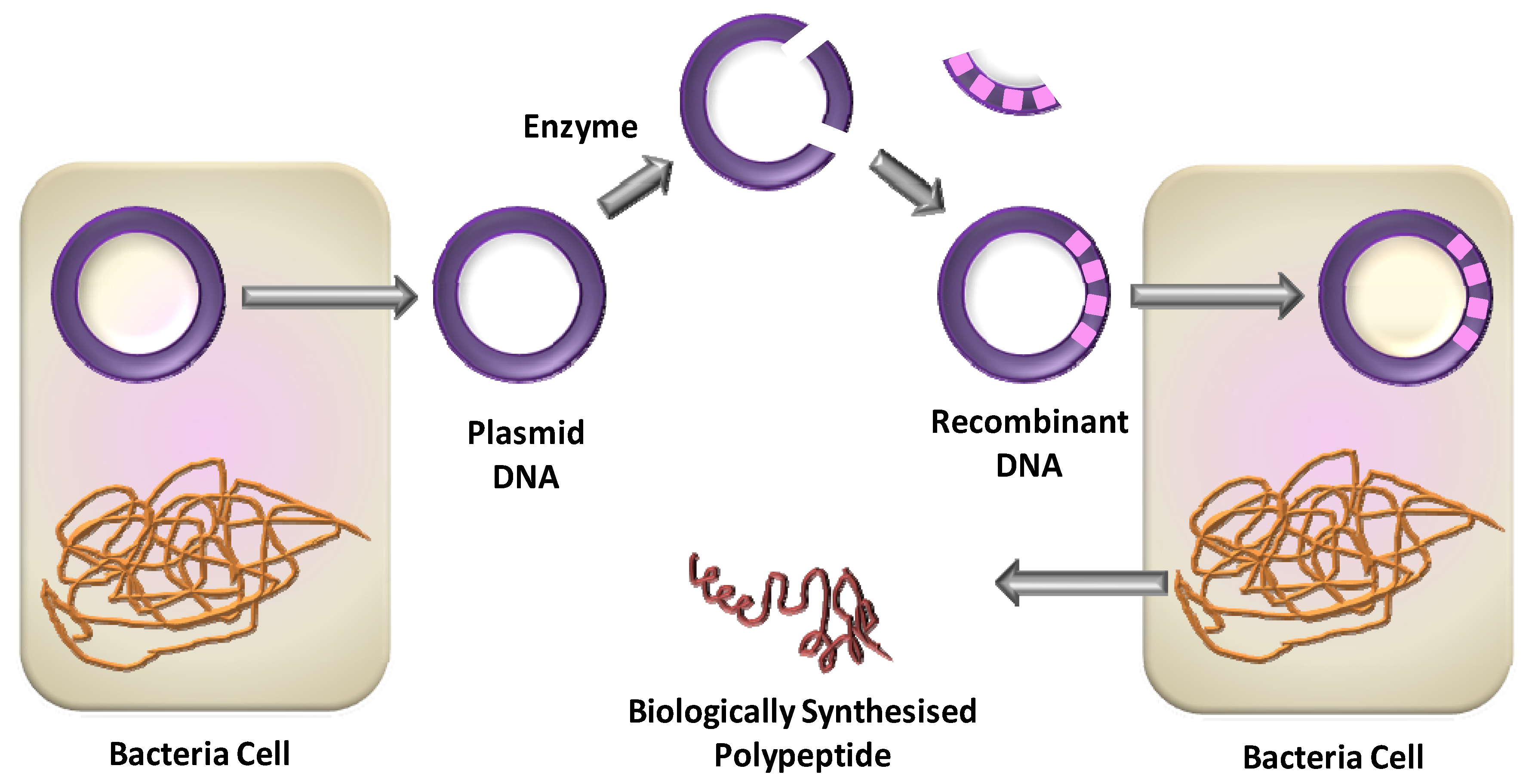
- Polypeptides produced by genetically encoded synthesis have a defined sequence, stereochemistry, and Mw based on a genetic template.
- Once optimised, transformed cells can provide a continuous supply of the polypeptide.
- If the polypeptide has a particular secondary or tertiary structure, the in vivo folding machinery of the cell can assist to ensure the correct conformation.
- The technique is, in general, time- and effort- consuming, mainly due to the synthesis optimisation of the gene (especially if large Mw are desired); and to the optimisation of the expression levels in the host cell.
- Multidomain proteins are much more challenging to express than proteins smaller than 30 kDa.
- Incompatibility between protein and bacteria leads to toxicity, reducing protein production.
2.2. Synthetic Approaches
- Unnatural amino acids can be used to produce a vast number of novel architectures with different properties and structure-function relationships.
- It enables the combination of polypeptides with other synthetic polymers such as poly(ethylene glycol) (PEG).
- Easier and faster methodology compared with the genetically encoded techniques.
- Higher yields and large scale synthesis.
2.2.1. Solid-Phase Peptide Synthesis (SPPS)

- It allows the synthesis of natural peptides that are difficult to express in bacteria, the incorporation of unnatural amino acids, peptide/protein backbone modification, and the synthesis of d-polypeptides, which consist of d-amino acids.
- Total control over peptide composition is achieved.
- The possibility to perform wash cycles after each reaction, removing reagents in excess with all of the growing peptide of interest remaining covalently attached to the insoluble resin.
- The process can be automated using a peptide synthesiser.
- On the other hand, some drawbacks must also be taken into account:
- Large, complex polypeptides/proteins cannot be prepared, due to a low coupling efficiency as the length of the peptide increases.
- High purity is not usually obtained when large polypeptides are synthesised. Significant quantities of resin-bound by-products can be often detected.
- It is necessary to use an excess of amino acids and coupling reagents to ensure the maximum coupling efficiency.
- Solubility of protected peptide segments is sometimes challenging.
- Complete deprotection of long peptides can be difficult to accomplish.
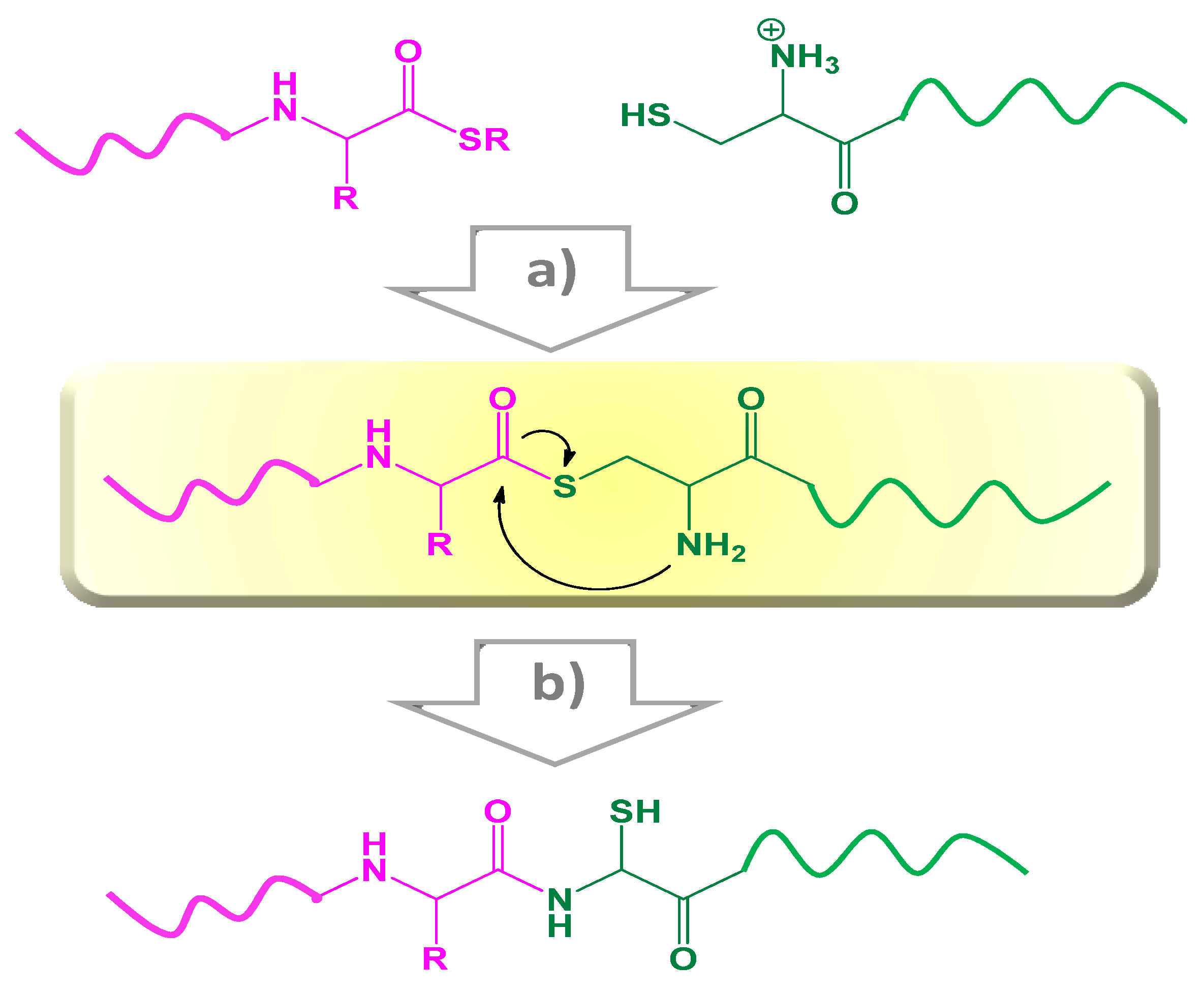
2.2.2. Native Chemical Ligation (NCL)
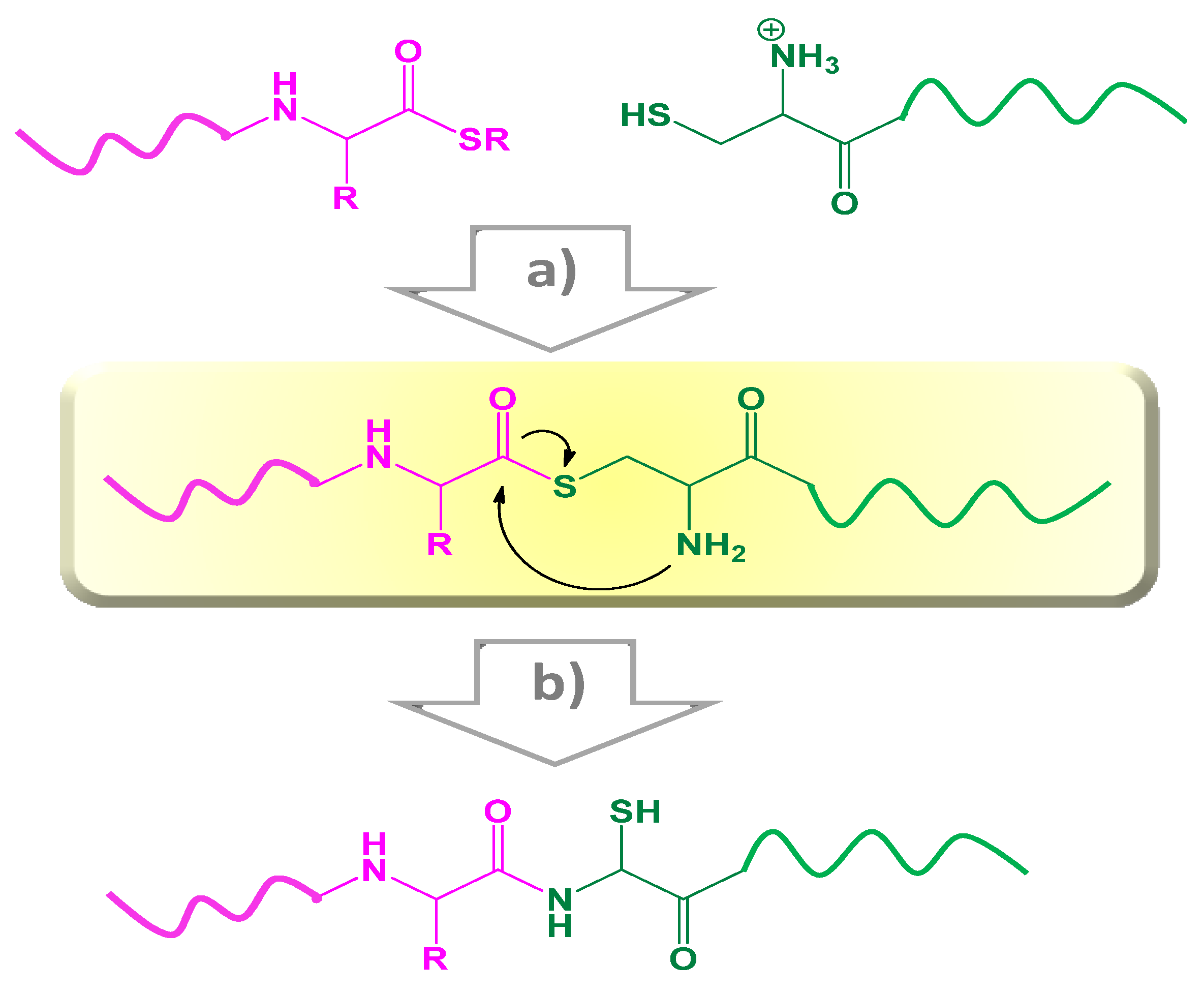
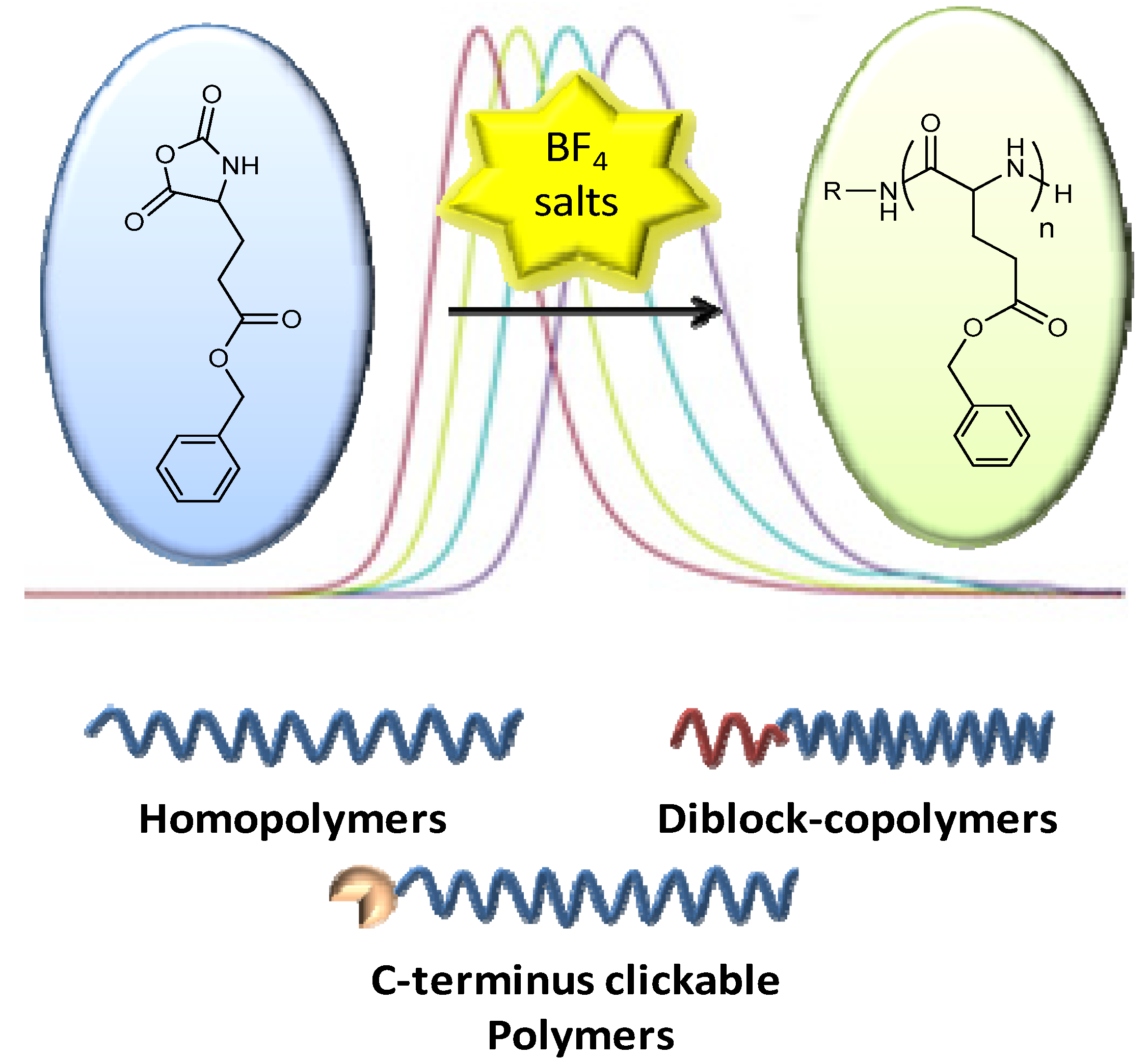
2.2.3. Ring-Opening Polymerisation (ROP) of α-Amino Acid N-Carboxyanhydrides (NCAs)

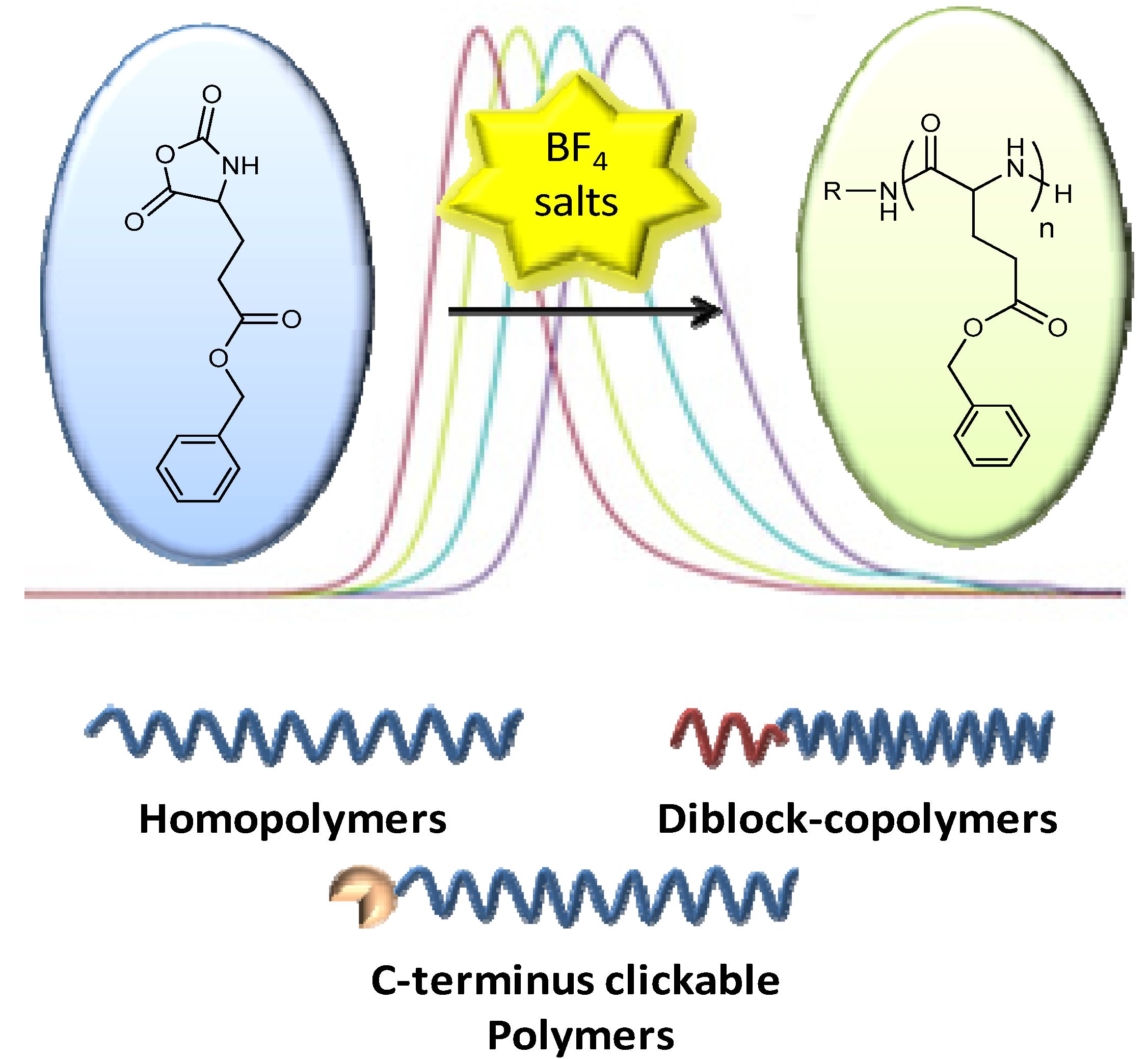
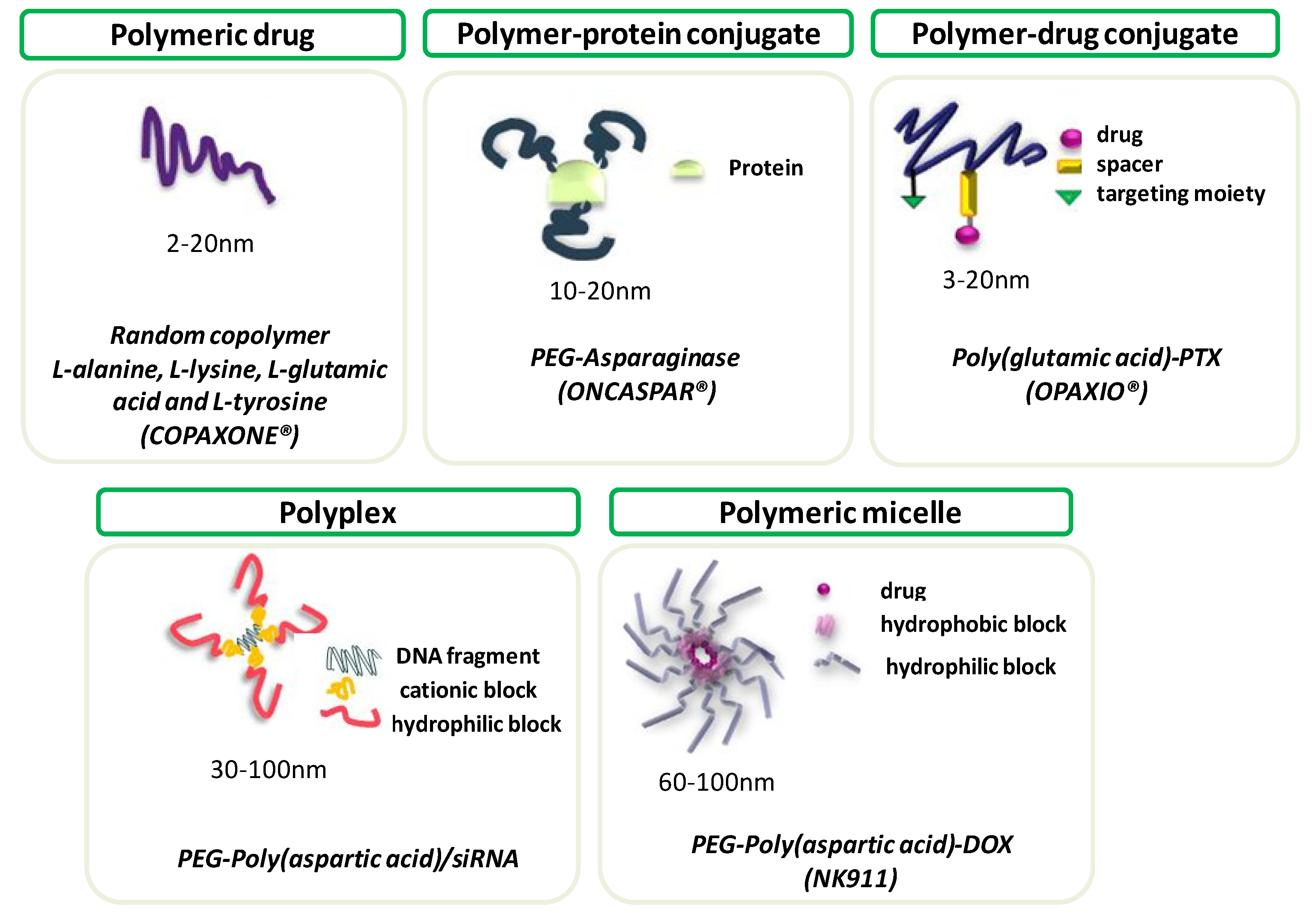
3. Polypeptides as Polymer Therapeutics
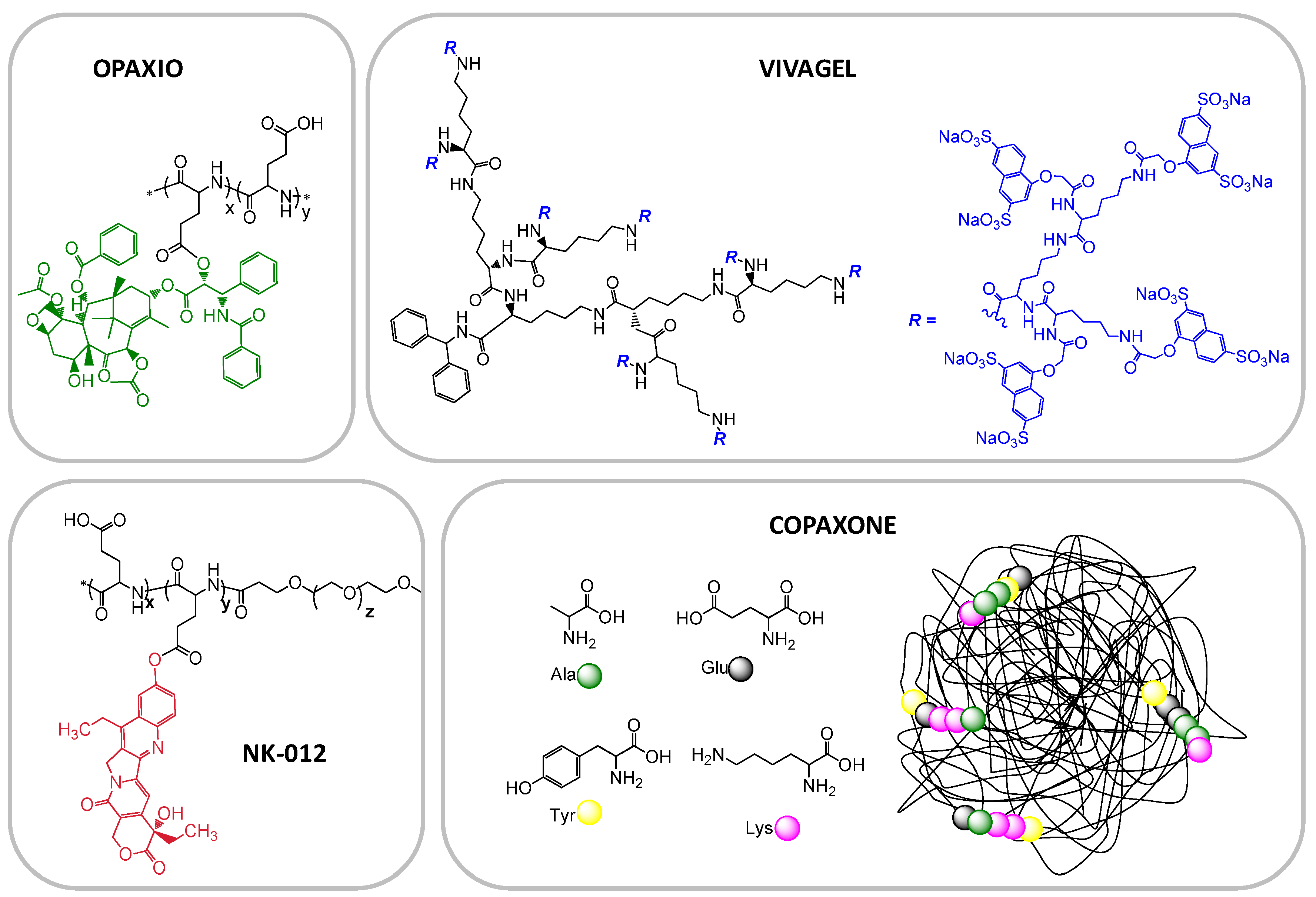
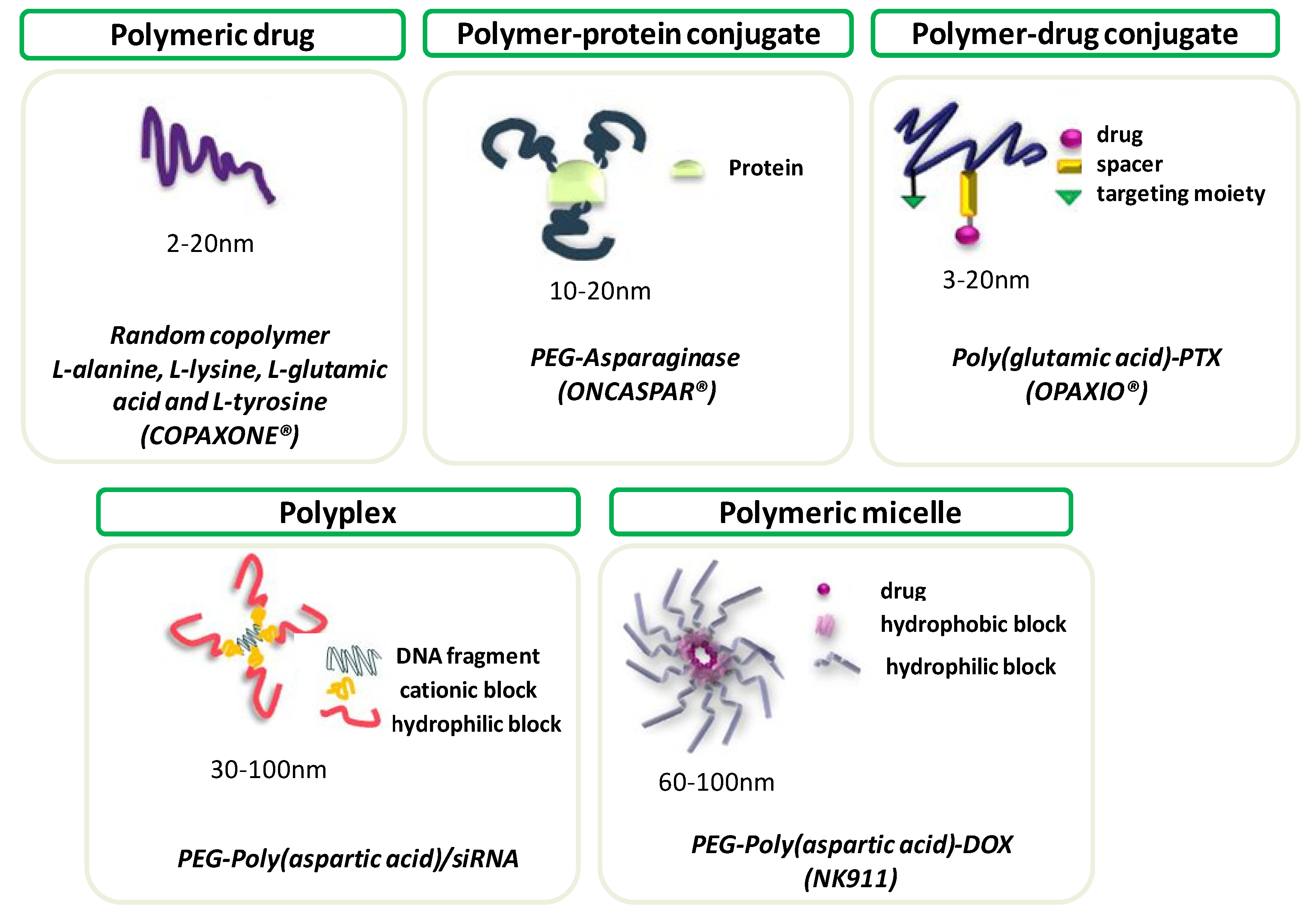
3.1. Drug and Gene Delivery
3.1.1. Drug Delivery
3.1.2. Gene Delivery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Technology | Indication | Stage | Info Source |
|---|---|---|---|---|
| Drug Delivery | ||||
| Polymer-Drug Conjugates | ||||
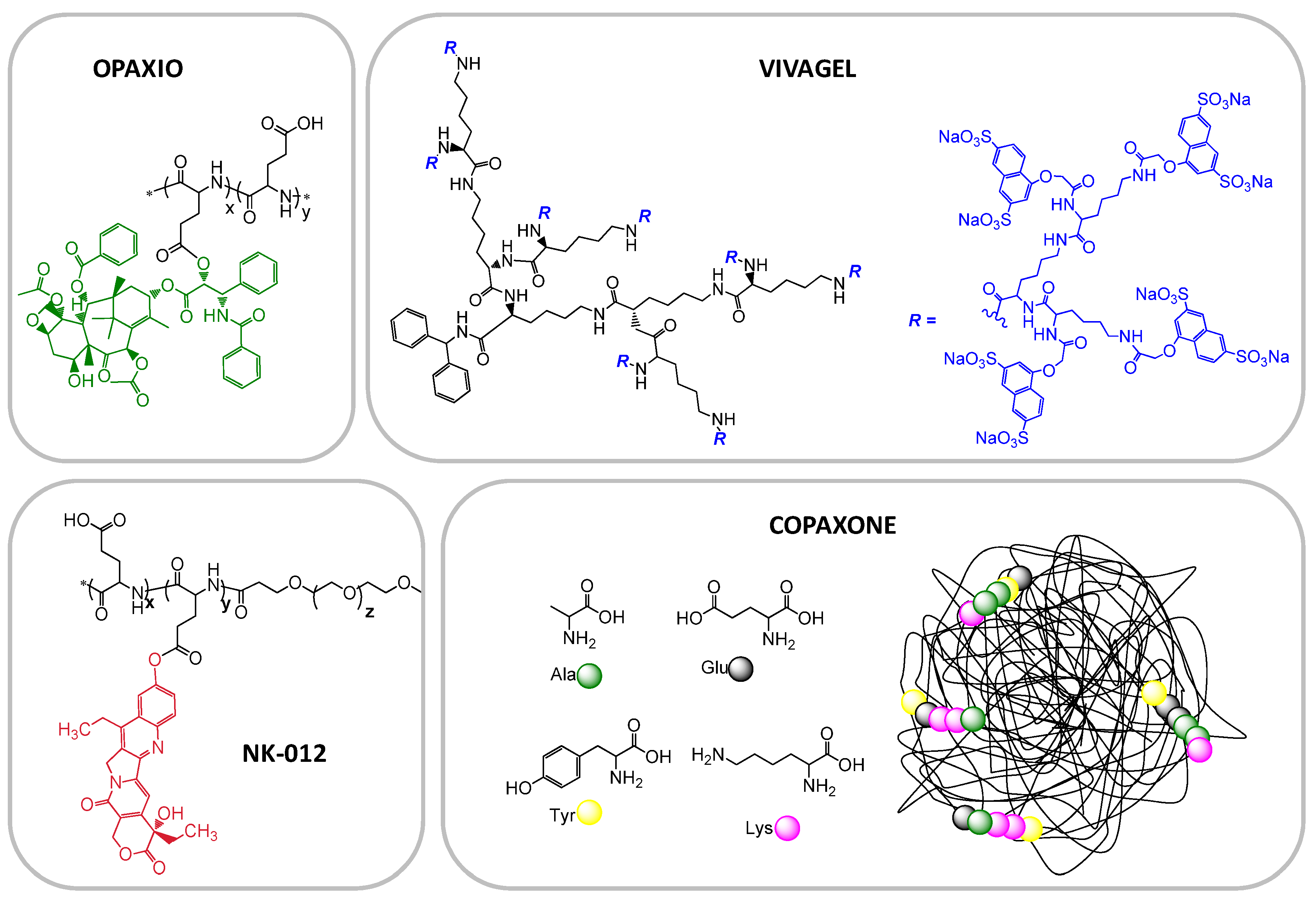
| CT-2103; Xyotax; Opaxio | Poly glutamic acid (PGA)-Paclitaxel | Cancer—NSCLC, ovarian, various, other cancers and combinations | Phase III, | Cell Therapeutics Inc. |
| CT-2106 | Poly glutamic acid (PGA)-Camptothecin | Cancer -colorectal and ovarian | Phase II | Cell Therapeutics Inc. |
| – | Poly glutamic acid (PGA)-Paclitaxel-RGD | Cancer-glioblastoma, breast cancer, pancreatic adenocarcinoma | Preclinical | [91] |
| – | Poly glutamic acid (PGA)-N,N-dimethylsphinosine (DMSP) | Cancer-Breast adenocarcinoma | Early stage | [95] |
| – | Poly glutamic acid (PGA)-Apaf 1 Inhibitor | Regenerative medicine. Regeneration in the course of inflammation-induced tissue injury | Early stage | [99] |
| – | Poly glutamic acid (PGA)-d-Penicillamine and idarubicin | Cancer-non-small cell lung cancer | Preclinical | [103] |
| – | Block copolymer lysine 8-elastin-like polypeptide (K8-ELP)-Geldanamycin | Cancer therapy in general | Early stage | [127] |
| – | Elastin-like polypeptide-Penetrating peptide-oncogen inhibitor peptide H1: Pen-ELP-H1 | Cancer therapy in general | Early stage | [108] |
| – | ELP-Doxorubicin, and Pen-ELP-Doxorubicin | Cancer therapy in general | Early stage | [109,110,111] |
| Polymeric Drugs | ||||
| Copaxone | Glu, Ala, Tyr copolymer | multiple sclerosis | Marketed | Teva |
| – | Poly(Arg-Gly-Asp) | Cancer: lung metastasis | Preclinical | [115] |
| Vivagel | Lysine-based dendrimer | Microbiocide | Phase II/III | StarPharma |
| Polymeric Micelles | ||||
| NK911 | mPEG-poly(aspartic acid)-doxorubicin | Solid tumors | Phase I | Nippon Kayaku Co |
| NK012 | PEG-poly(glutamic acid)-SN38 | SCLC and triple negative breast cancer | Phase II | Nippon Kayaku Co |
| NK105 | mPEG-poly(aspartic acid) PTX | Recurrent or meta-static breast cancer | Phase III | NanoCarrier Co.-Nippon Kayaku Co |
| NC-6004 | mPEG-poly(glutamic acid) with cysplatin | Locally advanced or metastatic pancreatic cancer | Phase I/II | NanoCarrier Co. TOUDAI |
| NC-6301 | mPEG-poly(aspartic acid)-docetaxel | Cancer therapy | Preclinical | NanoCarrier Co. TOUDAI |
| NC-4016 | mPEG-poly(glutamic acid) with oxaliplatin | Cancer therapy | Phase I | NanoCarrier Co. TOUDAI |
| NC-6300/K-912 | mPEG-poly(aspartate-hydrazide)-epirubicin | Cancer therapy | Phase I | NanoCarrier Co. TOUDAI |
| – | mPEG-poly(aspartate hidrazide)-Dox and Wortmannin | Cancer therapy | Early stage | [127] |
| Polymer-Protein conjugates | ||||
| Zinostatin stimalmer® | Styrene maleic anhydride-neocarzinostatin, (SMANCS) | Cancer- hepatocellular carcinoma | Marketed | Yamanouchia Japan |
| Oncaspar® | PEG-asparaginase | Cancer -acute lymphocytic leukaemia (ALL) | Marketed | Enzon |
| Peg-intron® | PEG-Interferon alpha 2b | Hepatitis C | Marketed | Schering-Plough |
| Pegasys® | PEG-Interferon alpha 2a | Hepatitis C | Marketed | Roche |
| Neulasta™ | PEG-hrGCSF | Chemotherapy-induced neutropenia | Marketed | Amgen |
| Adagen® | PEG-adenosine deaminase | Severe combined immune deficiency syndrome | Marketed | Enzon |
| Somavert® | PEG-HGH antagonist | Acromegalia | Marketed | Pfizer |
| Mircera® | PEG-EPO (polyethylene glycol-epoetin beta) | Treatment of anemia associated with chronic kidney disease | Marketed | Roche |
| KrystexxaTM (pegloticase) | PEG-uricase | Chronic gout | Marketed | Savient farmaceuticals |
| Cimzia (certolizumab pegol) | PEG-anti-TNF Fab | Rheumatoid arthritis, Crohn’s disease | Marketed | UCB |
| ADI-PEG 20 | PEG–arginine deaminase | Cancer-hepatocellular carcinoma, melanoma | Phase I/II | Phoenix Pharmalogics-Polaris Group |
| Hemospan® MP4OX | PEG-haemoglobin | Delivery of O2 in post surgery and trauma patients | Phase I/II | Sangart |
| CDP 791 | PEG-anti VEGFR-2 Fab | Cancer-NSCLC | Phase II | UCB Pharma |
| ADI-PEG 20 | PEG-arginine deaminase | Cancer-hepatocellular carcinoma, melanoma | Phase I/II | Phoenix Pharmalogics-Polaris Group |
| Gen Delivery | ||||
| – | PEG-poly(lysine) block copolymer-siRNA | Nephrin protein silencing for renal diseases | Preclinical | [145] |
| – | Poly(phenylalanine)coPoly(ethylene imine) multiarm copolymer-DNA | DNA delivery | Early stages | [146] |
| – | Poly(lysine-co-phenylalanine) | DNA/siRNA delivery | Early stages | [147] |
| – | Poly(lysine) dendritic scaffold with HIV tat protein sequences | DNA/siRNA delivery | Early stages | [148] |
| – | Poly(lysine) dendrimers containing lipidated units | DNA/siRNA delivery | Early stages | [149] |
| – | Poly(lysine) dendrimers containing Cell Penetrating Peptides (CPPs) | DNA/siRNA delivery | Early stages | [150,151] |
| – | Poly(lysine-co-hystidine) dendrimers-DNA | DNA delivery | Preclinical | [152,153,154] |
| Imaging and monitoring | ||||
| Gadomer 17 | Poly(lysine) dendrimer-DOTA-Gd | MRI contrast agent | Retired from clinics due to non adequate PK profile | [155,156] |
| – | Poly(lysine) dendrimer-RGD-DTPA-Gd | MRI contrast agent | Preclinical | [157] |
| – | Poly(glutamic acid)-DTPA-Gd | MRI contrast agent | Preclinical | [158] |
| – | Poly(glutamic acid)-DTPA-Gd-NIR813 | MRI contrast agent + fluorescence agent | Preclinical | [159] |
| Other applications | ||||
| – | Dendrimeric Peptides with microbial surface recognition tetra and octapeptides | Antimicrobial agents | Early studies | [160] |
| – | Dendrimeric peptide (RW)4D | Antimicrobial agents | Early studies | [161] |
| – | Branched His-Lys dendrimers | Antimicrobial agents | Early studies | [162] |
| – | Peptide dendrimers based on His-Ser and diaminopropionic acid (Dap) | Enzyme mimic applications | Early studies | [163,164,165,166] |
| – | MAPs (Multiple Antigenic peptides), poly(lysine dendrimers) | Peptide antigens and inmunomodulating molecules to be used as vaccines | Early studies | [167] |
3.2. Molecular Imaging and Theranostics
3.3. Other Roles of Polypeptides in Polymer Therapeutics
4. Polypeptide-Based Complex Architectures
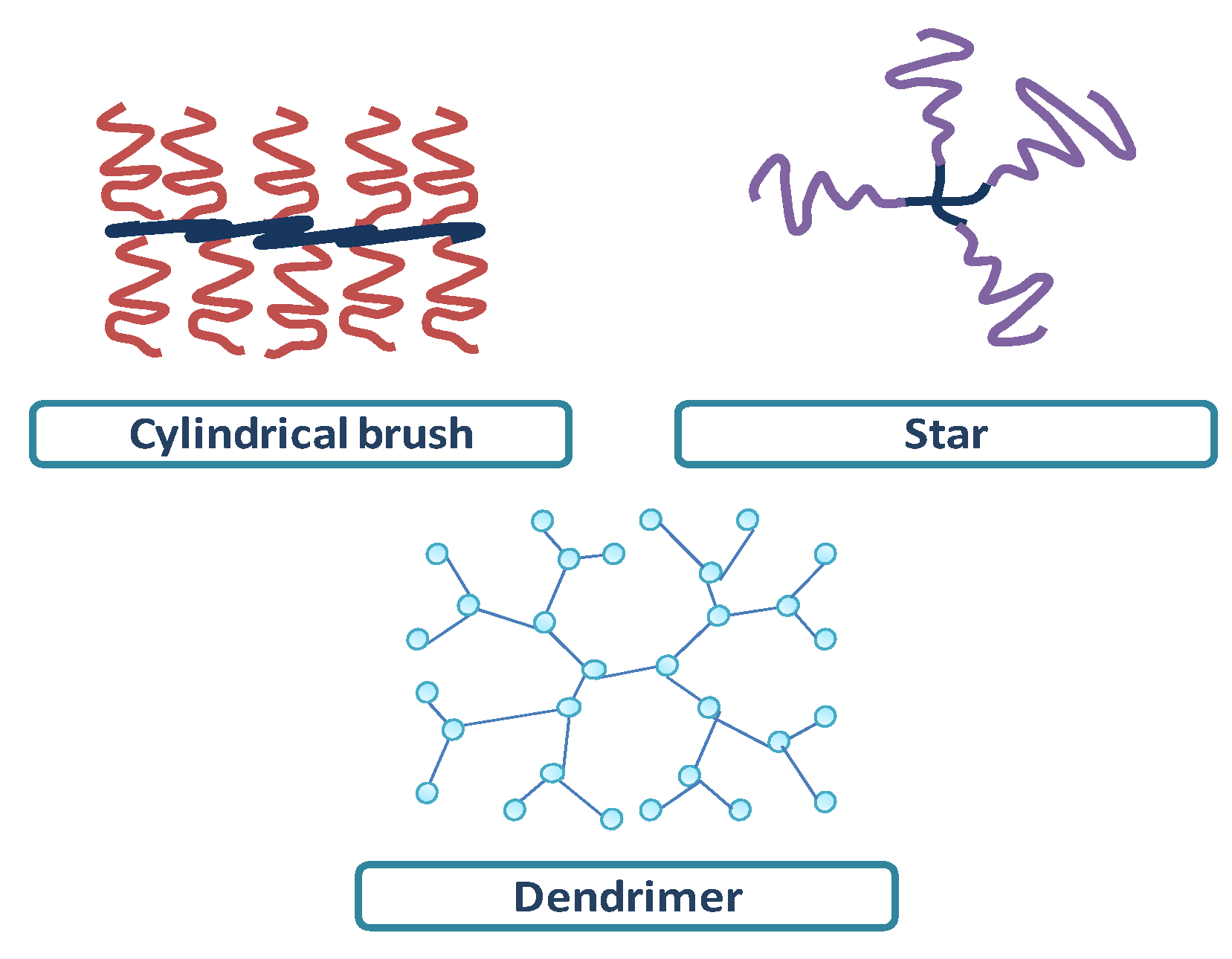
4.1. Star and Branched Polypeptides
4.2. Cylindrical Brushes
4.3. Dendrimers
5. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Nanomedicine 2020. Contribution of Nanomedicine to Horizon. Available online: http://www.etp-nanomedicine.eu/public/press-documents/publications/etpn-publications/etpn-white-paper-H2020 (accessed on 27 December 2013).
- Nitta, S.K.; Numata, K. Biopolymer-based nanoparticles for drug/gene delivery and tissue engineering. Int. J. Mol. Sci. 2013, 14, 1629–1654. [Google Scholar] [CrossRef]
- Duncan, R. The dawning era of polymer therapeutics. Natl. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Duncan, R. Polymer therapeutics as nanomedicines: New perspectives. Curr. Opin. Biotechnol. 2011, 22, 492–501. [Google Scholar] [CrossRef]
- Donaruma, L.G. Synthetic biologically active polymers. Prog. Polym. Sci. 1975, 4, 1–25. [Google Scholar] [CrossRef]
- Seymour, L. Review: Synthetic polymers with intrinsinc anticancer activity. J. Bioact. Comp. Polym. 1991, 6, 178–216. [Google Scholar]
- Ringsdorf, H. Structure and properties of pharmacologically active polymers. J. Polym. Sci. 1975, 51, 135–153. [Google Scholar]
- Vicent, M.J.; Ringsdorf, H.; Duncan, R. Polymer therapeutics: Clinical applications and challenges for development. Adv. Drug Deliv. Rev. 2009, 61, 1117–1120. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Felgner, P.L.; Seymour, L.W. Self-Assembling Complexes for Gene Delivery. From Laboratory to Clinical Trial; Wiley: Chichester, UK, 1998. [Google Scholar]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Natl. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef]
- Masayuli, Y.; Mizue, M.; Noriko, Y.; Teruo, O.; Yasuhisa, S. Polymer micelles as novel drug carrier: Adriamycin-conjugated poly(ethylene glycol)-(poly(aspartic acid) block copolymer. J. Control. Release 1990, 11, 269–278. [Google Scholar] [CrossRef]
- Matsumura, Y.; Kataoka, K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009, 100, 572–579. [Google Scholar] [CrossRef]
- Haag, R.; Kratz, F. Polymer therapeutics: Concepts and applications. Angew. Chem. Int. Ed. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Duncan, R.; Vicent, M.J. Polymer therapeutics-prospects for 21st century: The end of the beginning. Adv. Drug Deliv. Rev. 2013, 65, 60–70. [Google Scholar] [CrossRef]
- Gaspar, R.; Duncan, R. Polymeric carriers: Preclinical safety and the regulatory implications for design and development of polymer therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 1220–1231. [Google Scholar] [CrossRef]
- Vicent, M.J.; Duncan, R. Polymer conjugates: Nanosized medicines for treating cancer. Trend Biotechnol. 2006, 24, 39–47. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Maeda, H.; Fang, J.; Inutsuka, T.; Kitamoto, Y. Vascular permeability enhancement in solid tumor: Various factors, mechanisms involved and its implications. Int. Immunopharmacol. 2003, 3, 319–328. [Google Scholar] [CrossRef]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef]
- Gaspar, R.; Duncan, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef]
- Duncan, R.; Ringsdorf, H.; Satchi-Fainaro, R. Polymer therapeutics—Polymers as drugs, drug and protein conjugates and gene delivery systems: Past, present and future opportunities. Adv. Polym. Sci. 2006, 192, 1–8. [Google Scholar] [CrossRef]
- Alconcel, S.N.S.; Baas, A.S.; Maynard, H.D. FDA-approved poly(ethylene glycol)-protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Barz, M.; Luxenhofer, R.; Zentel, R.; Vicent, M.J. Overcoming the PEG-addiction: Well-defined alternatives to PEG, from structure-property relationships to better defined therapeutics. Polym. Chem. 2011, 2, 1900–1918. [Google Scholar] [CrossRef]
- Lavasanifar, A.; Samuel, J.; Kwon, G.S. Poly(ethylene oxide)-block-poly(l-amino acid) micelles for drug delivery. Adv. Drug Deliv. Rev. 2002, 54, 169–190. [Google Scholar] [CrossRef]
- Canal, F.; Sanchis, J.; Vicent, M.J. Polymer-drug conjugates as nano-sized medicines. Curr. Opin. Biotech. 2011, 22, 894–900. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Polymerization of Peptide Polymers for Biomaterial Applications. Available online: http://cdn.intechopen.com/pdfs/42100/InTech-Polymerization_of_peptide_polymers_for_biomaterial_applications.pdf (accessed on 27 December 2013).
- Johnson, R.P.; John, J.V.; Kim, I. Recent developments in polymer–block–polypeptide and protein–polymer bioconjugate hybrid materials. Eur. Polym. J. 2013, 49, 2925–2948. [Google Scholar] [CrossRef]
- Morell, M.; Puiggali, J. Hybrid block copolymers constituted by peptides and synthetic polymers: An overview of synthetic approaches, supramolecular behavior and potential applications. Polymers 2013, 5, 188–224. [Google Scholar] [CrossRef]
- Chow, D.; Nunalee, M.L.; Lim, D.W.; Simnick, A.J.; Chilkoti, A. Peptide-based biopolymers in biomedicine and biotechnology. Mater. Sci. Eng. R Rep. Rev. J. 2008, 62, 125–155. [Google Scholar] [CrossRef]
- Svenson, S. Clinical translation of nanomedicines. Curr. Opin. Solid State Mater. Sci. 2012, 16, 287–294. [Google Scholar] [CrossRef]
- Shaffer, S.A.; Baker-Lee, C.; Kennedy, J.; Lai, M.S.; de Vries, P.; Buhler, K.; Singer, J.W. In vitro and in vivo metabolism of paclitaxel poliglumex: Identification of metabolites and active proteases. Cancer Chemother. Pharmacol. 2007, 59, 537–548. [Google Scholar] [CrossRef]
- Hardwicke, J.; Moseley, R.; Stephens, P.; Harding, K.; Duncan, R.; Thomas, D.W. Bioresponsive dextrin-rhEGF conjugates: In vitro evaluation in models relevant to its proposed used as treatment for chronic wounds. Mol. Pharm. 2010, 7, 699–707. [Google Scholar] [CrossRef]
- XMT-1001. Available online: http://www.mersana.com/product-pipeline/xmt-1001.php (accessed on 27 December 2013).
- Markovsky, E.; Baabur-Cohen, H.; Eldar-Boock, A.; Omer, L.; Tiram, G.; Ferber, S.; Ofek, P.; Polyak, D.; Scomparin, A.; Satchi-Fainaro, R. Administration, distribution, metabolism and elimination of polymer therapeutics. J. Control. Release 2012, 161, 446–460. [Google Scholar] [CrossRef]
- Thomas, C.M.; Lutz, J.F. Precision synthesis of biodegradable polymers. Angew. Chem. Int. Ed. 2011, 50, 9244–9246. [Google Scholar] [CrossRef]
- Qiu, L.Y.; Bae, Y.H. Polymer architecture and drug delivery. Pharm. Res. 2006, 23, 1–30. [Google Scholar]
- Seymour, L.W.; Miyamoto, Y.; Maeda, H.; Brereton, M.; Strohalm, J.; Ulbrich, K; Duncan, R. Influence of molecular weight on passive tumour accumulation of a soluble macromolecular drug carrier. Eur. J. Cancer 1995, 31, 766–770. [Google Scholar] [CrossRef]
- Sun, J.; Zuckermann, R.N. Peptoid polymers: A highly designable bionspired material. ACS Nano 2013, 7, 4715–4732. [Google Scholar] [CrossRef]
- Luxenhofer, R.; Fetsch, C.; Grossmann, A. Polypeptoids: A perfect match for molecular definition and macromolecular engineering? J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2731–2752. [Google Scholar] [CrossRef]
- Kidchob, T.; Kimura, S.; Imanishi, Y. Amphiphilic poly(Ala)-b-poly(Sar) microspheres loaded with hydrophobic drug. J. Control. Release 1998, 51, 241–248. [Google Scholar] [CrossRef]
- Birke, A.; Huesmann, D.; Kelsch, A.; Weilbächer, M.; Xie, J.; Bros, M.; Bopp, T.; Becker, C.; Landfester, K.; Barz, M. Polypeptoid-block-polypeptide copolymers: Synthesis, characterization, and application of amphiphilic block copolypept(o)ides in drug formulations and miniemulsion techniques. Biomacromolecules 2014, 2014. [Google Scholar] [CrossRef]
- Sanhai, W.R.; Sakamoto, J.H.; Canady, R.; Ferrari, M. Seven challenges for nanomedicine. Nat.Nanotechnol. 2008, 3, 242–244. [Google Scholar] [CrossRef]
- Sakamoto, J.H.; van de Ven, A.L.; Godin, B.; Blanco, E.; Serda, R.E.; Grattoni, A.; Ziemys, A.; Bouamrani, A.; Hu, T.; Ranganathan, S.I.; et al. Enabling individualized therapy through nanotechnology. Pharmacol. Res. 2010, 62, 57–89. [Google Scholar]
- Merrifield, R.B. Solid phase peptide synthesis I. Synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Bayer, E. Towards the chemical synthesis of proteins. Angew. Chem. Int. Ed. 1991, 30, 113–129. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar]
- Deming, T.J. Living polymerization of α-amino acid-N-carboxyanhydrides. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3011–3018. [Google Scholar] [CrossRef]
- Kricheldorf, H. Polypeptides and 100 years of chemistry of alpha-amino acid N-carboxyanhydrides. Angew. Chem. Int. Ed. 2006, 45, 5752–5784. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of well-defined polypeptide-based materials via the ring-opening polymerization of alpha-amino acid N-carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar] [CrossRef]
- Conejos-Sanchez, I.; Duro-Castano, A.; Birke, A.; Barz, M.; Vicent, M.J. A controlled and versatile NCA polymerization method for the synthesis of polypeptides. Polym. Chem. 2013, 4, 3182–3186. [Google Scholar] [CrossRef]
- Barz, M.; Duro-Castano, A.; Vicent, M.J. A versatile post-polymerization modification method for polyglutamic acid: Synthesis of orthogonal reactive polyglutamates and their use in “click chemistry”. Polym. Chem. 2013, 4, 2989–2994. [Google Scholar] [CrossRef]
- Rhodes, A.J. Three-Dimensional Polypeptide Architectures through Tandem Catalysis and Click Chemistry. Ph.D Thesis, University of California, Los Angeles, CA, USA, 2013. [Google Scholar]
- Cappello, J.; Crissman, J.; Dorman, M.; Mikolajczak, M.; Textor, G.; Marquet, M.; Ferrari, F. Genetic engineering of structural protein polymers. Biotechnol. Prog. 1990, 6, 198–202. [Google Scholar] [CrossRef]
- Chilkoti, A.; Dreher, M.R.; Meyer, D.E. Design of thermally responsive, recombinant polypeptide carriers for targeted drug delivery. Adv. Drug Deliv. Rev. 2002, 54, 1093–1111. [Google Scholar] [CrossRef]
- Elion, E.A.; Marina, P.; Yu, L. Constructing recombinant DNA molecules by PCR. Curr. Protoc. Mol. Biol. 2007, 3. [Google Scholar] [CrossRef]
- Kim, E.; George, A.; Evans, M.; Conticello, V.P. Cotranslational incorporation of a structurally diverse series of proline analogues in an escherichia coli expression system. ChemBioChem 2004, 5, 928–936. [Google Scholar] [CrossRef]
- Xie, J.; Schultz, P.G. A chemical toolkit for proteins—An expanded genetic code. Natl. Rev. Mol. Cell. Biol. 2006, 7, 775–782. [Google Scholar] [CrossRef]
- Connor, R.E.; Tirrell, D.A. Non-canonical amino acids in protein polymer design. J. Macromol. Sci. Part C Polym. Rev. 2007, 47, 9–28. [Google Scholar]
- Merrifield, R.B. Solid-phase peptide synthesis. Ad. Enzymol. Relat. Areas Mol. Biol. 1969, 32, 221–296. [Google Scholar]
- Yan, L.Z.; Dawson, P.E. Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization. J. Am. Chem. Soc. 2001, 123, 526–533. [Google Scholar] [CrossRef]
- Canne, L.E.; Botti, P.; Simon, R.J.; Chen, Y.; Dennis, E.A.; Kent, S.B.H. Chemical protein synthesis by solid phase ligation of unprotected peptide segments. J. Am. Chem. Soc. 1999, 121, 8720–8727. [Google Scholar]
- Schnolzer, M.; Kent, S.B. Constructing proteins by dovetailing unprotected synthetic peptides: Backbone-engineered HIV protease. Science 1992, 256, 221–225. [Google Scholar]
- Dawson, P.E.; Churchill, M.J.; Ghadiri, M.R.; Kent, B.H. Modulation of reactivity in native chemical ligation through the use of thiol additives. J. Am. Chem. Soc. 1997, 119, 4325–4329. [Google Scholar]
- Tam, J.P.; Lu, Y.A.; Liu, C.F.; Shao, J. Peptide synthesis using unprotected peptides through orthogonal coupling methods. Proc. Natl. Acad. Sci. USA 1995, 92, 12485–12489. [Google Scholar]
- Lu, W.; Qasim, M.A.; Kent, S.B.H. Comparative total syntheses of turkey ovomucoid third domain by both stepwise solid phase peptide synthesis and native chemical ligation. J. Am. Chem. Soc. 1996, 118, 8518–8523. [Google Scholar] [CrossRef]
- Kent, S.B.H.; Bark, S.J.; Canne, L.E.; Dawson, P.E.; Fitzgerald, M.C.; Hackeng, T.; Lu, W.; Muir, T. Total Chemical Synthesis of Proteins as a Probe of Structure and Function. Peptides 1996. In Proceedings of the Twenty-Fourth European Peptide Symposium, Edinburgh, UK, 8–13 September 1996.
- Deming, T.J. Methodologies for preparation of synthetic block copolypeptides: Materials with future promise in drug delivery. Adv. Drug Deliv. Rev. 2002, 54, 1145–1155. [Google Scholar] [CrossRef]
- Wang, X.; Kim, H.J.; Wong, C.; Vepari, C.; Matsumoto, A.; Kaplan, D.L. Fibrous proteins and tissue engineering. Mater. Today 2006, 9, 44–53. [Google Scholar]
- Santos, S.D.; Chandravarkar, A.; Mandal, B.; Mimna, R.; Murat, K.; Saucède, L.; Tella, P.; Tuchscherer, G.; Mutter, M. Switch-peptides: Controlling self-assembly of amyloid β-derived peptides in vitro by consecutive triggering of acyl migrations. J. Am. Chem. Soc. 2005, 127, 11888–11889. [Google Scholar] [CrossRef]
- Mart, R.J.; Osborne, R.D.; Stevens, M.M.; Ulijn, R.V. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006, 2, 822–835. [Google Scholar] [CrossRef]
- Leuchs, H. Fiber die Glycin-carbonsauren. Chem. Food Sci. 1906, 39, 857–861. [Google Scholar]
- Leuchs, H.; Manasse, W. Über die Isomerie der Carbäthoxyl-glycyl glycinester. Ber. Dtsch. Chem. Ges. 1907, 40. [Google Scholar] [CrossRef]
- Leuchs, H.; Geiger, W. Über die Anhydride von α-Amino-N-carbonsäuren und die von α-Aminosäuren. Ber. Dtsch. Chem. Ges. 1908, 41. [Google Scholar] [CrossRef]
- Deming, T.J. Facile synthesis of block copolypeptides of defined architecture. Nature 1997, 390, 386–389. [Google Scholar] [CrossRef]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Well-defined linear multiblock and branched polypeptides by linking chemistry. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 4670–4673. [Google Scholar] [CrossRef]
- Dimitrov, I.; Schlaad, H. Synthesis of nearly monodisperse polystyrene-polypeptide block copolymers via polymerisation of N-carboxyanhydrides. Chem. Commun. 2003, 2003, 2944–2945. [Google Scholar] [CrossRef]
- Vayaboury, W.; Giani, O.; Collet, H.; Commeyras, A.; Schué, F. Synthesis of N-protected-l-lysine and alpha-benzyl-l-glutamate N-carboxyanhydrides (NCA) by carbamoylation and nitrosation. Amino Acids 2004, 27, 161–167. [Google Scholar] [CrossRef]
- Lu, H.; Cheng, J. Hexamethyldisilazane-mediated controlled polymerization of alpha-amino acid N-carboxyanhydrides. J. Am. Chem. Soc. 2007, 129, 14114–14115. [Google Scholar] [CrossRef]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Living polypeptides. Biomacromolecules 2004, 5, 1653–1656. [Google Scholar] [CrossRef]
- Vayaboury, W.; Giani, O.; Cottet, H.; Deratani, A.; Schué, F. Living polymerization of α-amino acid N-carboxyanhydrides (NCA) upon decreasing the reaction temperature. Macromol. Rapid Commun. 2004, 25, 1221–1224. [Google Scholar] [CrossRef]
- Vicent, M.J.; Barz, M.; Canal, F.; Conejos-Sánchez, I.; Castaño, A.D. Controled Ynthesis Od Polyglutamates with Low Polydispersity and Versatile Architectures. PCT/ES2012/070740; 23 October 2012. [Google Scholar]
- Polypeptide Therapeutic Solutions. Available online: http://www.pts-polymers.com (accessed on 27 December 2013).
- Huesmann, D.; Birke, A.; Klinker, K.; Türk, S.; Räder, H.J.; Barz, M. Revisiting secondary structures in NCA polymerization: Influences on the analysis of protected polylysines. Macromolecules 2014, 2014. [Google Scholar] [CrossRef]
- Deming, T.J. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci. 2007, 32, 858–875. [Google Scholar] [CrossRef]
- Galic, V.L.; Wright, J.D.; Lewin, S.N.; Herzog, T.J. Paclitaxel poliglumex for ovarian cancer. Expert Opin. Investig. Drugs 2011, 20, 813–821. [Google Scholar] [CrossRef]
- Li, C.; Wallace, S. Polymer-drug conjugates: Recent development in clinical oncology. Adv. Drug Deliv. Rev 2008, 60, 886–898. [Google Scholar] [CrossRef]
- Buescher, J.M.; Margaritis, A. Microbial biosynthesis of polyglutamic acid biopolymer and applications in the biopharmaceutical, biomedical and food industries. Crit. Rev. Biotechnol. 2007, 27, 1–19. [Google Scholar] [CrossRef]
- Springett, G.M.; Takimoto, C.; McNamara, M.; Doroshow, J.H.; Syed, S.; Eastham, E.; Spriggs, D.; Pezzulli, S.; Michelson, G.; Dupont, J. Phase I study of CT-2106 (polyglutamate camptothecin) in patients with advanced malignancies. J. Clin. Oncol. 2004, 22, 3127. [Google Scholar]
- CT-2106 (Polyglutamate Camptothecin). Available online: http://www.wikinvest.com/stock/Cell_Therapeutics_(CTIC)/Ct-2106_Polyglutamate_Camptothecin (accessed on 27 December 2013).
- Eldar-Boock, A.; Miller, K.; Sanchis, J.; Lupu, R.; Vicent, M.J.; Satchi-Fainaro, R. Integrin-assisted drug delivery of nano-scaled polymer therapeutics bearing paclitaxel. Biomaterials 2011, 32, 3862–3874. [Google Scholar] [CrossRef]
- Fante, C.; Eldar-Boock, A.; Satchi-Fainaro, R.; Osborn, H.M.I.; Greco, F. Synthesis and biological evaluation of a polyglutamic acid–dopamine conjugate: A new antiangiogenic agent. J. Med. Chem. 2011, 54, 5255–5259. [Google Scholar] [CrossRef]
- Akagi, T.; Baba, M.; Akashi, M. Biodegradable nanoparticles as vaccine adjuvants and delivery systems: Regulation of immune responses by nanoparticle-based vaccine. Adv. Polym. Sci. 2012, 247, 31–64. [Google Scholar] [CrossRef]
- Heller, P.; Huesmann, D.; Scherer, M.; Barz, M. From Polymers to Nanomedicines: New Materials for Future Vaccines. In Molecular Vaccine: From Prophylaxis to Therapy; Springer International Publication: Berlin, Germany, 2013. [Google Scholar]
- Ghosh, S.C.; Auzenne, E.; Khodadadian, M.; Farquhar, D.; Klostergaard, J. N,N-Dimethylsphingosine conjugates of poly-l-glutamic acid: Synthesis, characterization, and initial biological evaluation. Bioorg. Med. Chem. Lett. 2009, 19, 1012–1017. [Google Scholar] [CrossRef]
- Vicent, M.J. Polymer-drug conjugates as modulators of cellular apoptosis. AAPS J. 2007, 9, E200–E207. [Google Scholar] [CrossRef]
- Malet, G.; Martin, A.G.; Orzaez, M.; Vicent, M.J.; Masip, I.; Sanclimens, G.; Ferrer-Montiel, A.; Mingarro, I.; Messeguer, A.; Fearnhead, H.O.; et al. Small molecule inhibitors of Apaf-1-related caspase- 3/-9 activation that control mitochondrial-dependent apoptosis. Cell. Death Differ. 2006, 13, 1523–1532. [Google Scholar] [CrossRef]
- Vicent, M.J.; Perez-Paya, E. Poly-l-glutamic acid (PGA) aided inhibitors of apoptotic protease activating factor 1 (Apaf-1): An antiapoptotic polymeric nanomedicine. J. Med. Chem. 2006, 49, 3763–3765. [Google Scholar] [CrossRef]
- Santamaria, B.; Benito-Martin, A.; Ucero, A.C.; Aroeira, L.S.; Reyero, A.; Vicent, M.J.; Orzaez, M.; Celdran, A.; Esteban, J.; Selgas, R.; et al. A nanoconjugate Apaf-1 inhibitor protects mesothelial cells from cytokine-induced injury. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Greco, F.; Vicent, M.J.; Penning, N.A.; Nicholson, R.I.; Duncan, R. HPMA copolymer-aminoglutethimide conjugates inhibit aromatase in MCF-7 cell lines. J. Drug Target. 2005, 13, 459–470. [Google Scholar] [CrossRef]
- Deladriere, C.; Masia, E.; Greco, F.; Lucas, R.; Vicent, M.J. Polymer-Drug Conjugates as Platforms for Combination Therapy in the Treatment of Hormone-Dependent Breast Cancer. In Proceedings of the 37th Annual Meeting and Exposition of the Controlled Release Society, Portland, OR, USA, 10–14 July 2010.
- Greco, F.; Vicent, M.J. Combination therapy: Opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv. Drug Deliv. Rev. 2009, 61, 1203–1213. [Google Scholar] [CrossRef]
- Wadhwa, S.; Mumper, R.J. Polypeptide conjugates of d-penicillamine and idarubicin for anticancer therapy. J. Control. Release 2012, 158, 215–223. [Google Scholar] [CrossRef]
- Banta, S.; Wheeldon, I.R.; Blenner, M. Protein engineering in the development of functional hydrogels. Ann. Rev. Biomed. Eng. 2010, 12, 167–186. [Google Scholar] [CrossRef]
- Bae, Y.; Buresh, R.A.; Williamson, T.P.; Chen, T.H.; Furgeson, D.Y. Intelligent biosynthetic nanobiomaterials for hyperthermic combination chemotherapy and thermal drug targeting of HSP90 inhibitor geldanamycin. J. Control. Release 2007, 122, 16–23. [Google Scholar] [CrossRef]
- McDaniel, J.R.; Callahan, D.J.; Chilkoti, A. Drug delivery to solid tumors by elastin-like polypeptides. Adv. Drug Deliv. Rev. 2010, 62, 1456–1467. [Google Scholar] [CrossRef]
- Nettles, D.L.; Chilkoti, A.; Setton, L.A. Applications of elastin-like polypeptides in tissue engineering. Adv. Drug Deliv. Rev. 2010, 62, 1479–1485. [Google Scholar] [CrossRef]
- Bidwell, G.L.; Raucher, D. Application of thermally responsive polypeptides directed against c-Myc transcriptional function for cancer therapy. Mol. Cancer Ther. 2005, 4, 1076–1085. [Google Scholar] [CrossRef]
- Dreher, M.R.; Raucher, D.; Balu, N.; Colvin, O.M.; Ludeman, S.M.; Chilkoti, A. Evaluation of an elastin-like polypeptide-doxorubicin conjugate for cancer therapy. J. Control. Release 2003, 91, 31–43. [Google Scholar] [CrossRef]
- MacKay, J.A.; Chen, M.; McDaniel, J.R.; Liu, W.; Simnick, A.J.; Chilkoti, A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Natl. Mater. 2009, 8, 993–999. [Google Scholar]
- Furgeson, D.Y.; Dreher, M.R.; Chilkoti, A. Structural optimization of a “smart” doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. J. Control. Release 2006, 110, 362–369. [Google Scholar] [CrossRef]
- Massodi, I.; Bidwell, G.L.; Raucher, D. Evaluation of cell penetrating peptides fused to elastin-like polypeptide for drug delivery. J. Control. Release 2005, 108, 396–408. [Google Scholar] [CrossRef]
- Varkony, H.; Weinstein, V.; Klinger, E.; Sterling, J.; Cooperman, H.; Komlosh, T.; Ladkani, D.; Schwartz, R. The glatiramoid class of immunomodulator drugs. Expert Opin. Pharmacother. 2009, 10, 657–668. [Google Scholar] [CrossRef]
- Johnson, K.P.; Brooks, B.R.; Ford, C.C.; Goodman, A.; Guarnaccia, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Pruitt, A.; Rose, J.W.; et al. Sustained clinical benefits of glatiramer acetate in relapsing multiple sclerosis patients observed for 6 years. Copolymer 1 multiple sclerosis study group. Mult. Scler. 2000, 6, 255–266. [Google Scholar]
- De Stefano, N.; Filippi, M.; Confavreux, C.; Vermersch, P.; Simu, A.M.E. The results of two multicenter, open-label studies assessing efficacy, tolerability and safety of protiramer, a high molecular weight synthetic copolymeric mixture, in patients with relapsing–remitting multiple sclerosis. Mult. Scler. 2009, 15, 238–243. [Google Scholar]
- Saiki, I.; Murata, J.; Iida, J.; Sakurai, T.; Nishi, N.; Matsuno, K.; Azuma, I. Antimetastatic effects of synthetic polypeptides containing repeated structures of the cell adhesive Arg-Gly-Asp (RGD) and Tyr-Ile-Gly-Ser-Arg (YIGSR) sequences. Br. J. Cancer 1989, 60, 722–728. [Google Scholar]
- Saiki, I.; Murata, J.; Matsuno, K.; Ogawa, R.; Nishi, N.; Tokura, S.; Azuma, I. Anti-metastatic and anti-invasive effects of polymeric Arg-Gly-Asp (RGD) peptide, poly(RGD), and its analogues. Jpn. J. Cancer Res. Gann 1990, 81, 660–667. [Google Scholar] [CrossRef]
- Shaunak, S.; Thornton, M.; John, S.; Teo, I.; Peers, E.; Mason, P.; Krausz, T.; Davies, D.S. Reduction of the viral load of HIV-1 after the intraperitoneal administration of dextrin 2-sulphate in patients with AIDS. AIDS 1998, 12, 399–409. [Google Scholar] [CrossRef]
- Trivedi, R.; Kompella, U.B. Nanomicellar formulations for sustained drug delivery: Strategies and underlying principles. Nanomedicine 2010, 5, 485–505. [Google Scholar] [CrossRef]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2012, 64, 37–48. [Google Scholar] [CrossRef]
- Hirano, T.; Klesse, W.; Ringsdorf, H. Polymeric derivatives of activated cyclophosphamide as drug delivery systems in antitumor chemotherapy. Pharmacologically active polymers, 20. Die Makromol. Chem. 1979, 180, 1125–1131. [Google Scholar] [CrossRef]
- Osada, K.; Christie, R.J.; Kataoka, K. Polymeric micelles from poly(ethylene glycol)-poly(amino acid) block copolymer for drug and gene delivery. J. R. Soc. Interface R. Soc. 2009, 6, S325–S339. [Google Scholar] [CrossRef]
- Bae, Y.; Kataoka, K. Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv. Drug Deliv. Rev. 2009, 61, 768–784. [Google Scholar] [CrossRef]
- Osada, K.; Kataoka, K. Drug and Gene Delivery Based on Supramolecular Assembly of PEG-Polypeptide Hybrid Block Copolymers. In Peptide Hybrid Polymers; Klok, H.-A., Schlaad, H., Eds.; Springer: Berlin Heidelberg, Germany, 2006; pp. 113–153. [Google Scholar]
- NC-6004 Nanoplatin™. Available online: http://www.nanocarrier.co.jp/en/research/pipeline/02.html (accessed on 27 December 2013).
- Inoue, T.; Yamashita, Y.; Nishihara, M.; Sugiyama, S.; Sonoda, Y.; Kumabe, T.; Yokoyama, M.; Tominaga, T. Therapeutic efficacy of a polymeric micellar doxorubicin infused by convection-enhanced delivery against intracranial 9L brain tumor models. Neuro Oncol. 2009, 11, 151–157. [Google Scholar]
- Bae, Y.; Diezi, A.; Zhao, A.; Kwon, G.S. Mixed polymeric micelles for combination cancer chemotherapy through the concurrent delivery of multiple chemotherapeutic agents. J. Control. Release 2007, 122, 324–330. [Google Scholar] [CrossRef]
- Epirubicin Micelle (pH-Sensitive Micelle). Available online: http://www.nanocarrier.co.jp/en/research/pipeline/04.html (accessed on 27 December 2013).
- Bae, Y.H.; Yin, H. Stability issues of polymeric micelles. J. Control. Release 2008, 131, 2–4. [Google Scholar] [CrossRef]
- Miller, T.; Rachel, R.; Besheer, A.; Uezguen, S.; Weigandt, M.; Goepferich, A. Comparative investigations on in vitro serum stability of polymeric micelle formulations. Pharm. Res. 2012, 29, 448–459. [Google Scholar] [CrossRef]
- Torchilin, V.P. Micellar nanocarriers: Pharmaceutical perspectives. Pharm. Res. 2007, 24, 1–16. [Google Scholar]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Meng, F.; Cheng, R.; Deng, C.; Zhong, Z. Intracellular drug release nanosystems. Mater. Today 2012, 15, 436–442. [Google Scholar] [CrossRef]
- Kakizawa, Y.; Harada, A.; Kataoka, K. Environment-sensitive stabilization of core−shell structured polyion complex micelle by reversible cross-linking of the core through disulfide bond. J. Am. Chem. Soc. 1999, 121, 11247–11248. [Google Scholar] [CrossRef]
- Christie, R.J.; Nishiyama, N.; Kataoka, K. Delivering the code: Polyplex carriers for deoxyribonucleic acid and ribonucleic acid interference therapies. Endocrinology 2009, 151, 466–473. [Google Scholar] [CrossRef]
- Yu, H.; Wagner, E. Bioresponsive polymers for nonviral gene delivery. Curr. Opin. Mol. Ther. 2009, 11, 165–178. [Google Scholar]
- Wagner, E.; Kloeckner, J. Gene delivery using polymer therapeutics. Polym. Therap. I Adv. Polym. Sci. 2006, 192, 135–173. [Google Scholar] [CrossRef]
- CALAA-01. Available online: http://www.calandopharma.com/ (accessed on 27 December 2013).
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Formation of polyion complex micelles in an aqueous milieu from a pair of oppositely-charged block copolymers with poly(ethylene glycol) segments. Macromolecules 1995, 28, 5294–5299. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Novel polyion complex micelles entrapping enzyme molecules in the core: preparation of narrowly-distributed micelles from lysozyme and poly(ethylene glycol)−poly(aspartic acid) block copolymer in aqueous medium. Macromolecules 1998, 31, 288–294. [Google Scholar] [CrossRef]
- Nishiyama, N.; Yokoyama, M.; Aoyagi, T.; Okano, T.; Sakurai, Y.; Kataoka, K. Preparation and characterization of self-assembled polymer−metal complex micelle from cis-dichlorodiammineplatinum(II) and poly(ethylene glycol)−poly(α,β-aspartic acid) block copolymer in an aqueous medium. Langmuir 1998, 15, 377–383. [Google Scholar]
- Nishiyama, N.; Okazaki, S.; Cabral, H.; Miyamoto, M.; Kato, Y.; Sugiyama, Y.; Nishio, K.; Matsumura, Y.; Kataoka, K. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003, 63, 8977–8983. [Google Scholar]
- Uchino, H.; Matsumura, Y.; Negishi, T.; Koizumi, F.; Hayashi, T.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br.J.Cancer 2005, 93, 678–687. [Google Scholar] [CrossRef]
- Dhal, P.K.; Polomoscanik, S.C.; Vassiliadis, J.; Wawersik, S.; Ledbetter, S.; Arbeeny, C.; Schiavi, S.; Miller, R.J. Biocompatible cationic block copolymers as vectors for kidney specific siRNA delivery. In Proceedings of the 9th International Symposium Polymer Therapy: From Lab to Clinic, Valencia, Spain, 28–30 May 2012.
- Xia, J.; Chen, L.; Chen, J.; Tian, H.; Li, F.; Zhu, X.; Li, G.; Chen, X. Hydrophobic polyphenylalanine-grafted hyperbranched polyethylenimine and its in vitro gene transfection. Macromol. Biosci. 2011, 11, 211–218. [Google Scholar] [CrossRef]
- Shibata, A.; Murata, S.; Ueno, S.; Liu, S.; Futaki, S.; Baba, Y. Synthetic copoly(Lys/Phe) and poly(Lys) translocate through lipid bilayer membranes. BBA-Biomembranes 2003, 1616, 147–155. [Google Scholar] [CrossRef]
- Tung, C.; Mueller, S.; Weissleder, R. Novel branching membrane translocational peptide as gene delivery vector. Bioorg. Med. Chem. 2002, 10, 3609–3614. [Google Scholar] [CrossRef]
- Bayele, H.K.; Sakthivel, T.; O'Donell, M.; Pasi, K.J.; Wilderspin, A.F.; Lee, C.A.; Toth, I.; Florence, A.T. Versatile peptide dendrimers for nucleic acid delivery. J. Pharm. Sci. 2005, 94, 446–457. [Google Scholar] [CrossRef]
- Coles, D.J.; Yang, S.; Esposito, A.; Mitchell, D.; Minchin, R.F.; Toth, I. The synthesis and characterisation of a novel dendritic system for gene delivery. Tetrahedron 2007, 63, 12207–12214. [Google Scholar] [CrossRef]
- Yang, S.; Coles, D.J.; Esposito, A.; Mitchell, D.J.; Toth, I.; Minchin, R.F. Cellular uptake of self-assembled cationic peptide–DNA complexes: Multifunctional role of the enhancer chloroquine. J. Control. Release 2009, 135, 159–165. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, L.; Stass, S.A.; Mixson, A.J. Branched co-polymers of histidine and lysine are efficient carriers of plasmids. Nucleic Acid Res. 2001, 29, 1334–1340. [Google Scholar] [CrossRef]
- Leng, Q.; Mixson, A.J. Modified branched peptides with a histidine-rich tail enhance in vitro gene transfection. Nucleic Acid Res. 2005, 33. [Google Scholar] [CrossRef]
- Leng, Q.; Scaria, P.; Zhu, J.; Ambulos, N.; Campbell, P.; Mixson, A.J. Highly branched HK peptides are effective carriers of siRNA. J. Gene Med. 2005, 7, 977–986. [Google Scholar] [CrossRef]
- Nicolle, G.M.; Tóth, E.; Schmitt-Willich, H.; Radüchel, B.; Merbach, A.E. The impact of rigidity and water exchange on the relaxivity of a dendritic MRI contrast agent. Chem. Eur. J. 2002, 8, 1040–1048. [Google Scholar] [CrossRef]
- Jaspers, K.; Versluis, B.; Leiner, T.; Dijkstra, P.; Oostendorp, M.; van Golde, J.M.; Post, M.J.; Backes, W.H. MR angiography of collateral arteries in a hind limb ischemia model: Comparison between blood pool agent gadomer and small contrast agent Gd-DTPA. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Polcyn, P.; Jurczak, M.; Rajnisz, A.; Solecka, J.; Urbanczyk-Lipkowska, Z. Design of antimicrobially active small amphiphilic peptide dendrimers. Molecules 2009, 14, 3881–3905. [Google Scholar] [CrossRef]
- Jackson, E.F.; Esparza-Coss, E.; Wen, X.; Ng, C.S.; Daniel, S.L.; Price, R.E.; Rivera, B.; Charnsangavej, C.; Gelovani, J.G.; Li, C. Magnetic resonance imaging of therapy-induced necrosis using gadolinium-chelated polyglutamic acids. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 830–838. [Google Scholar] [CrossRef]
- Melancon, M.P.; Lu, W.; Huang, Q.; Thapa, P.; Zhou, D.; Ng, C.; Li, C. Targeted imaging of tumor-associated M2 macrophages using a macromolecular contrast agent PG-Gd-NIR813. Biomaterials 2010, 31, 6567–6573. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.; Yang, J. Antimicrobial dendrimeric peptides. Eur. J. Biochem. 2002, 269, 923–932. [Google Scholar] [CrossRef]
- Liu, Z.; Brady, A.; Young, A.; Rasimick, B.; Chen, K.; Zhou, C.; Kallenbach, N.R. Length effects in antimicrobial peptides of the (RW)n series. Antimicrob. Agents Chemother. 2007, 51, 597–603. [Google Scholar] [CrossRef]
- Zhu, J.; Luther, P.W.; Leng, Q.; Mixson, A.J. Synthetic histidine-rich peptides inhibit Candida species and other fungi in vitro: Role of endocytosis and treatment implications. Antimicrob. Agent Chemother. 2006, 50, 2797–2805. [Google Scholar] [CrossRef]
- Delort, E.; Darbre, T.; Reymond, J. A strong positive dendritic effect in a peptide dendrimer-catalyzed ester hydrolysis reaction. J. Am. Chem. Soc. 2004, 126, 15642–15643. [Google Scholar] [CrossRef]
- Delort, E.; Nguyen-Trung, N.; Darbre, T.; Reymond, J. Synthesis and activity of histidine-containing catalytic peptide dendrimers. J. Org. Chem. 2006, 71, 4468–4480. [Google Scholar] [CrossRef]
- Maillard, N.; Clouet, A.; Darbre, T.; Reymond, J.L. Combinatorial libraries of peptide dendrimers: Design, synthesis, on-bead high-throughput screening, bead decoding and characterization. Natl. Protoc. 2009, 4, 132–142. [Google Scholar] [CrossRef]
- Sommer, P.; Uhlich, N.A.; Reymond, J.; Darbre, T. A peptide dendrimer model for vitamin B12 transport proteins. ChemBioChem 2008, 9, 689–693. [Google Scholar] [CrossRef]
- Tam, J.P. Synthetic peptide vaccine design: Synthesis and properties of a high-density multiple antigenic peptide system. Proc. Natl. Acad. Sci. USA 1988, 85, 5409–5413. [Google Scholar] [CrossRef]
- Sun, W.C.; Davis, P.B. Reducible DNA nanoparticles enhance in vitro gene transfer via an extracellular mechanism. J. Control. Release 2010, 146, 118–127. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Vu, H.N.; Pack, D.W. A top-down approach for construction of hybrid polymer-virus gene delivery vectors. J. Control. Release 2010, 144, 39–45. [Google Scholar] [CrossRef]
- Lee, M.; Veiseh, O.; Bhattarai, N.; Sun, C.; Hansen, S.J.; Ditzler, S.; Knoblaugh, S.; Lee, D.; Ellenbogen, R.; Zhang, M.; Olson, J.M. Rapid pharmacokinetic and biodistribution studies using cholorotoxin-conjugated iron oxide nanoparticles: A novel non-radioactive method. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, A.H.; Murray, L.S.; Hilditch, TE.; Murray, T.; Burtles, S.; Fraier, D.; Frigerio, E.; Cassidy, J. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin. Cancer Res. 1999, 5, 83–94. [Google Scholar]
- Baker, M. Whole-animal imaging: The whole picture. Nature 2010, 463, 977–980. [Google Scholar] [CrossRef]
- Herth, M.M.; Barz, M.; Jahn, M.; Zentel, R.; Rosch, F. 72/74As-labeling of HPMA based polymers for long-term in vivo PET imaging. Bioorg. Med. Chem. Lett. 2010, 20, 5454–5458. [Google Scholar] [CrossRef]
- Herth, M.M.; Barz, M.; Moderegger, D.; Allmeroth, M.; Jahn, M.; Thews, O.; Zentel, R.; Rösch, F. Radioactive labeling of defined HPMA-based polymeric structures using [18F]FETos for in vivo imaging by positron emission tomography. Biomacromolecules 2009, 10, 1697–1703. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Dendrimer-based contrast agents for molecular imaging. Curr. Top. Med. Chem. 2008, 8, 1180–1186. [Google Scholar] [CrossRef]
- Richardson, S.C.; Wallom, K.L.; Ferguson, E.L.; Deacon, S.P.; Davies, M.W.; Powell, A.J.; Riper, R.C.; Duncan, R. The use of fluorescence microscopy to define polymer localisation to the late endocytic compartments in cells that are targets for drug delivery. J. Control. Release 2008, 127, 1–11. [Google Scholar] [CrossRef]
- Chen, X.; Conti, P.S.; Moats, R.A. In vivo near-infrared fluorescence imaging of integrin alphavbeta3 in brain tumor xenografts. Cancer Res. 2004, 64, 8009–8014. [Google Scholar] [CrossRef]
- Bhaskar, S.; Tian, F.; Stoeger, T.; Kreyling, W.; de la Fuente, J.M.; Grazu, V.; Borm, P.; Estrada, G.; Ntziachristos, V.; Razansky, D. Multifunctional nanocarriers for diagnostics, drug delivery and targeted treatment across blood-brain barrier: Perspectives on tracking and neuroimaging. Fibre Toxicol. 2010, 2010. [Google Scholar] [CrossRef]
- Gindy, M.E.; Prud'homme, R.K. Multifunctional nanoparticles for imaging, delivery and targeting in cancer therapy. Expert Opin. Drug Deliv. 2009, 6, 865–878. [Google Scholar] [CrossRef]
- Melancon, M.P.; Stafford, R.J.; Li, C. Challenges to effective cancer nanotheranostics. J. Control. Release 2012, 164, 177–182. [Google Scholar] [CrossRef]
- Wen, X.; Jackson, E.F.; Price, R.E.; Kim, E.E.; Wu, Q.; Wallace, S.; Charnsangavej, C.; Gelovani, J.G.; Li, C. Synthesis and characterization of poly(l-glutamic acid) gadolinium chelate: A new biodegradable MRI contrast agent. Bioconjug. Chem. 2004, 15, 1408–1415. [Google Scholar] [CrossRef]
- Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Frank, J.; Nijsen, W.; Hennink, W.E. Polymeric micelles in anticancer therapy: Targeting, imaging and triggered release. Pharm. Res. 2010, 27, 2569–2589. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. [Google Scholar] [CrossRef]
- Fujita, M.; Lee, B.S.; Khazenzon, N.M.; Penichet, M.L.; Wawrowsky, K.A.; Patil, R.; Ding, H.; Holler, E.; Black, K.L.; Ljubimova, J.Y. Brain tumor tandem targeting using a combination of monoclonal antibodies attached to biopoly(beta-l-malic acid). J. Control. Release 2007, 122, 356–363. [Google Scholar] [CrossRef]
- Ljubimova, J.Y.; Fujita, M.; Ljubimov, A.V.; Torchilin, V.P.; Black, K.L.; Holler, E. Poly(malic acid) nanoconjugates containing various antibodies and oligonucleotides for multitargeting drug delivery. Nanomedicine 2008, 3, 247–265. [Google Scholar] [CrossRef]
- Orzaez, M.; Mondragon, L.; Marzo, I.; Sanclimens, G.; Messeguer, A.; Perez-Paya, E.; Vicent, M.J. Conjugation of a novel Apaf-1 inhibitor to peptide-based cell-membrane transporters: Effective methods to improve inhibition of mitochondria-mediated apoptosis. Peptides 2007, 28, 958–968. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ming, X.; Nakagawa, O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug. Chem. 2012, 23, 147–157. [Google Scholar] [CrossRef]
- Shu, J.Y.; Panganiban, B.; Xu, T. Peptide-polymer conjugates: From fundamental science to application. Ann. Rev. Phys. Chem. 2013, 64, 631–657. [Google Scholar] [CrossRef]
- Stern, L.; Perry, R.; Ofek, P.; Many, A.; Shabat, D.; Satchi-Fainaro, R. A novel antitumor prodrug platform designed to be cleaved by the endoprotease legumain. Bioconjug. Chem. 2009, 20, 500–510. [Google Scholar] [CrossRef]
- Vartak, D.G.; Gemeinhart, R.A. Matrix metalloproteases: Underutilized targets for drug delivery. J. Drug Target. 2007, 15, 1–20. [Google Scholar] [CrossRef]
- Blencowe, C.A.; Russell, A.T.; Greco, F.; Hayes, W.; Thornthwaite, D.W. Self-immolative linkers in polymeric delivery systems. Polym. Chem. 2011, 2, 773–790. [Google Scholar] [CrossRef]
- Apostolovic, B.; Deacon, S.P.; Duncan, R.; Klok, H.A. Hybrid polymer therapeutics incorporating bioresponsive, coiled coil peptide linkers. Biomacromolecules 2010, 11, 1187–1195. [Google Scholar] [CrossRef]
- Klok, H.; Hernández, J.R.; Becker, S.; Müllen, K. Star-shaped fluorescent polypeptides. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 1572–1583. [Google Scholar] [CrossRef]
- Inoue, K.; Sakai, H.; Ochi, S.; Itaya, T.; Tanigaki, T. Preparation and conformation of hexaarmed star poly(beta-benzyl-l-aspartates) synthesized utilizing hexakis(4-aminophenoxy) cyclotriphosphazene. J. Am. Chem. Soc. 1994, 116, 10783–10784. [Google Scholar] [CrossRef]
- Inoue, K.; Horibe, S.; Fukae, M.; Muraki, T.; Ihara, E.; Kayama, H. Synthesis and conformation of star-shaped poly(γ-benzyl-l-glutamate)s on a cyclotriphosphazene core. Macromol. Biosci. 2003, 3, 26–33. [Google Scholar] [CrossRef]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N.; Messman, J.; Mays, J. Synthesis of 3- and 4- arm star-block copolypeptides using multifunctional amino initiators and high vacuum techniques. Macromol. Symp. 2006, 240, 12–17. [Google Scholar] [CrossRef]
- Karatzas, A.; Bilalis, P.; Iatrou, H.; Pitsikalis, M.; Hadjichristidis, N. Synthesis of well-defined functional macromolecular chimeras based on poly(ethylene oxide) or poly(N-vinyl pyrrolidone). React.Funct.Polym. 2009, 69, 435–440. [Google Scholar] [CrossRef]
- Sulistio, A.; Gurr, P.A.; Blencowe, A.; Qiao, G.G. Peptide-based star polymers: The rising star in functional polymers. Aust. J. Chem. 2012, 65, 978–984. [Google Scholar] [CrossRef]
- Sulistio, A.; Lowenthal, J.; Blencowe, A.; Bongiovanni, M.N.; Ong, L.; Gras, L.; Zhang, X.; Qiao, G.G. Folic acid conjugated amino acid-based star polymers for active targeting of cancer cells. Biomacromolecules 2011, 12, 3469–3477. [Google Scholar] [CrossRef]
- Sulistio, A.; Blencowe, A.; Widjaya, A.; Zhang, X.; Qiao, G. Development of functional amino acid-based star polymers. Polym. Chem. 2012, 3, 224–234. [Google Scholar] [CrossRef]
- Xing, T.; Lai, B.; Ye, X.; Yan, L. Disulfide core cross-linked PEGylated polypeptide nanogel prepared by a one-step ring opening copolymerization of N-carboxyanhydrides for drug delivery. Macromol. Biosci. 2011, 11, 962–969. [Google Scholar] [CrossRef]
- Sheiko, S.S.; Sumerlin, B.S.; Matyjaszewski, K. Cylindrical molecular brushes: Synthesis, characterization, and properties. Progr. Polym. Sci. 2008, 33, 759–785. [Google Scholar] [CrossRef]
- Sahl, M.; Muth, S.; Branscheid, R.; Fischer, K.; Schmidt, M. Helix-coil transition in cylindrical brush polymers with poly-l-lysine side chains. Macromolecules 2012, 45, 5167–5175. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Mays, J. Macromolecular architectures by living and controlled/living polymerizations. Progr. Polym. Sci. 2006, 31, 1068–1132. [Google Scholar] [CrossRef]
- Peleshanko, S.; Tsukruk, V.V. The architectures and surface behavior of highly branched molecules. Progr. Polym. Sci. 2008, 33, 523–580. [Google Scholar] [CrossRef]
- Weller, D.; McDaniel, J.R.; Fischer, K.; Chilkoti, A.; Schmidt, M. Cylindrical polymer brushes with elastin-like polypeptide side chains. Macromolecules 2013, 46, 4966–4971. [Google Scholar] [CrossRef]
- Denkewalter, R.G.; Kolc, J.; Lukasavage, W.J. Macromolecule Highly Branched Homogeneous Compound Based on Lysine Units. US4289872 A, 15 September 1981. [Google Scholar]
- Aharoni, S.M.; Crosby, C.R.; Walsh, E.K. Size and solution properties of globular tert-butyloxycarbonyl-poly(α,iε-l-lysine). Macromolecules 1982, 15, 1093–1098. [Google Scholar] [CrossRef]
- Denkewalter, R.G.; Kolc, J.F.; Lukasavage, W.J. Macromolecular Highly Branched Α,Ω-Diamino Carboxylic Acids. US 4410688 A, 18 October 1983. [Google Scholar]
- Twyman, L.J.; Beezer, A.E.; Mitchell, J.C. The synthesis of chiral dendritic molecules based on the repeat unit l-glutamic acid. Tetrahedron Lett. 1994, 35, 4423–4424. [Google Scholar] [CrossRef]
- Crespo, L.; Sanclimens, G.; Pons, M.; Giralt, E.; Royo, M.; Albericio, F. Peptide and amide bond-containing dendrimers. Chem. Rev. 2005, 105, 1663–1681. [Google Scholar] [CrossRef]
- Twyman, L.J.; Beezer, A.E.; Esfand, R.; Mathews, B.T.; Mitchell, J.C. The synthesis of chiral dendrimeric molecules based on amino acid repeat units. J. Chem. Res. Synopses 1998, 1998, 758–759. [Google Scholar]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A new class of polymers: Starburst-dendritic macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef]
- Buhleier, E.; Wehner, W.; VÖGtle, F. “Cascade”- and “Nonskid-chain-like” syntheses of molecular cavity topologies. Synthesis-stuttgart 1978, 1978, 155–158. [Google Scholar] [CrossRef]
- Hawker, C.J.; Frechet, J.M.J. Preparation of polymers with controlled molecular architecture. A new convergent approach to dendritic macromolecules. J. Am. Chem. Soc. 1990, 112, 7638–7647. [Google Scholar] [CrossRef]
- Hawker, C.J.M.; Frechet, J. A new convergent approach to monodisperse dendritic macromolecules. J. Chem. Soc. Chem. Commun. 1990, 1990, 1010–1013. [Google Scholar] [CrossRef]
- Jensen, K.J.; Barany, G. Carbopeptides: Carbohydrates as potential templates for de novo design of protein models. J. Peptide Res. 2000, 56, 3–11. [Google Scholar] [CrossRef]
- Wang, L.; Ni, J.J.; Singh, S. Carbohydrate-centered maleimide cluster as a new type of templates for multivalent peptide assembling: Synthesis of multivalent HIV-1 gp41 peptides. Bioorg. Med. Chem. 2003, 11, 159–166. [Google Scholar] [CrossRef]
- Dirksen, A.; Meijer, E.W.; Adriaens, W.; Hackeng, T.M. Strategy for the synthesis of multivalent peptide-based nonsymmetric dendrimers by native chemical ligation. Chem. Commun. 2006, 2006, 1667–1669. [Google Scholar]
- Tam, J.P.; Lu, Y.A. Vaccine engineering: enhancement of immunogenicity of synthetic peptide vaccines related to hepatitis in chemically defined models consisting of T- and B-cell epitopes. Proc. Natl. Acad. Sci. USA 1989, 86, 9084–9088. [Google Scholar] [CrossRef]
- Tam, J.P. Recent advances in multiple antigen peptides. J. Immunol. Meth. 1996, 196, 17–32. [Google Scholar] [CrossRef]
- Liu, Z.; Young, A.W.; Hu, P.; Rice, A.J.; Zhou, C.; Zhang, Y.; Kallenbach, N.R. Tuning the membrane selectivity of antimicrobial peptides by using multivalent design. ChemBioChem 2007, 8, 2063–2065. [Google Scholar] [CrossRef]
- Hou, S.; Zhou, C.; Liu, Z.; Young, A.W.; Shi, Z.; Ren, D.; Kallenbach, N.R. Antimicrobial dendrimer active against Escherichia coli biofilms. Bioorg. Med. Chem. Lett. 2009, 19, 5478–5481. [Google Scholar] [CrossRef]
- Janiszewska, J.; Swieton, J.; Lipkowski, A.; Urbanczyk-Lipkowska, Z. Low molecular mass peptide dendrimers that express antimicrobial properties. Bioorg. Med. Chem. Lett. 2003, 13, 3711–3713. [Google Scholar] [CrossRef]
- Falciani, C.; Lozzi, L.; Pini, A.; Bracci, L. Bioactive peptides from libraries. Chem. Biol. 2005, 12, 417–426. [Google Scholar] [CrossRef]
- Pini, A.; Giuliani, A.; Falciani, C.; Runci, Y.; Ricci, C.; Lelli, B.; Malossi, M.; Neri, P.; Rossolini, G.M.; Bracci, L. Antimicrobial activity of novel dendrimeric peptides obtained by phage display selection and rational modification. Antimicrob. Agent Chemother. 2005, 49, 2665–2672. [Google Scholar] [CrossRef]
- Klajnert, B.; Janiszewska, J.; Urbanczyk-Lipkowska, Z.; Bryszewska, M.; Shcharbin, D.; Labieniec, M. Biological properties of low molecular mass peptide dendrimers. Int. J. Pharm. 2006, 309, 208–217. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Duro-Castano, A.; Conejos-Sánchez, I.; Vicent, M.J. Peptide-Based Polymer Therapeutics. Polymers 2014, 6, 515-551. https://doi.org/10.3390/polym6020515
Duro-Castano A, Conejos-Sánchez I, Vicent MJ. Peptide-Based Polymer Therapeutics. Polymers. 2014; 6(2):515-551. https://doi.org/10.3390/polym6020515
Chicago/Turabian StyleDuro-Castano, Aroa, Inmaculada Conejos-Sánchez, and María J. Vicent. 2014. "Peptide-Based Polymer Therapeutics" Polymers 6, no. 2: 515-551. https://doi.org/10.3390/polym6020515
APA StyleDuro-Castano, A., Conejos-Sánchez, I., & Vicent, M. J. (2014). Peptide-Based Polymer Therapeutics. Polymers, 6(2), 515-551. https://doi.org/10.3390/polym6020515