Supramolecular Functionalities Influence the Thermal Properties, Interactions and Conductivity Behavior of Poly(ethylene glycol)/LiAsF6 Blends

Abstract
:
1. Introduction

2. Experimental Section
2.1. Materials
2.2. LiAsF6/U-PEG and LiAsF6/U-PEG-U Polymer Electrolytes
2.3. Characterization
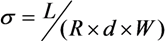
3. Results and Discussion
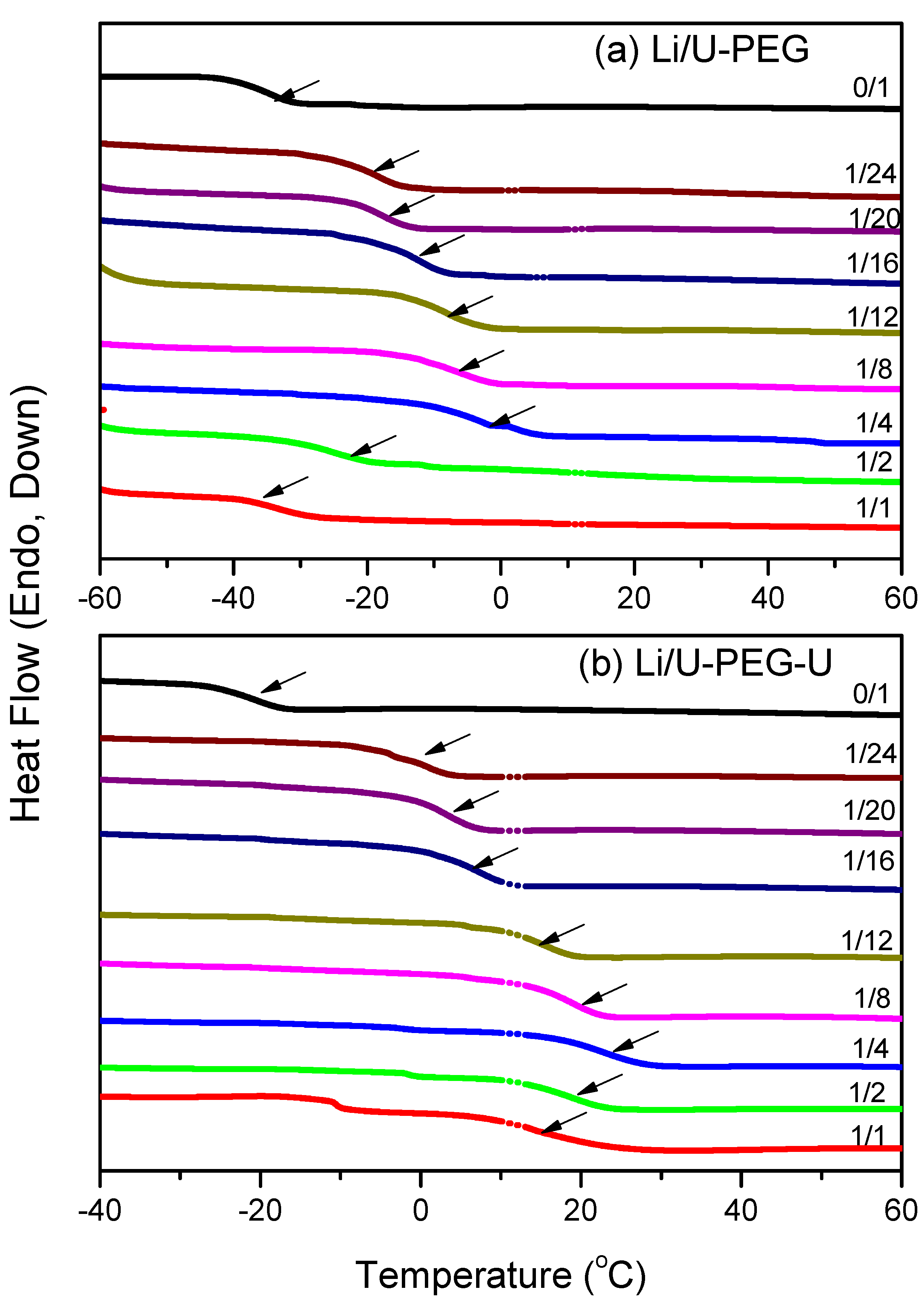
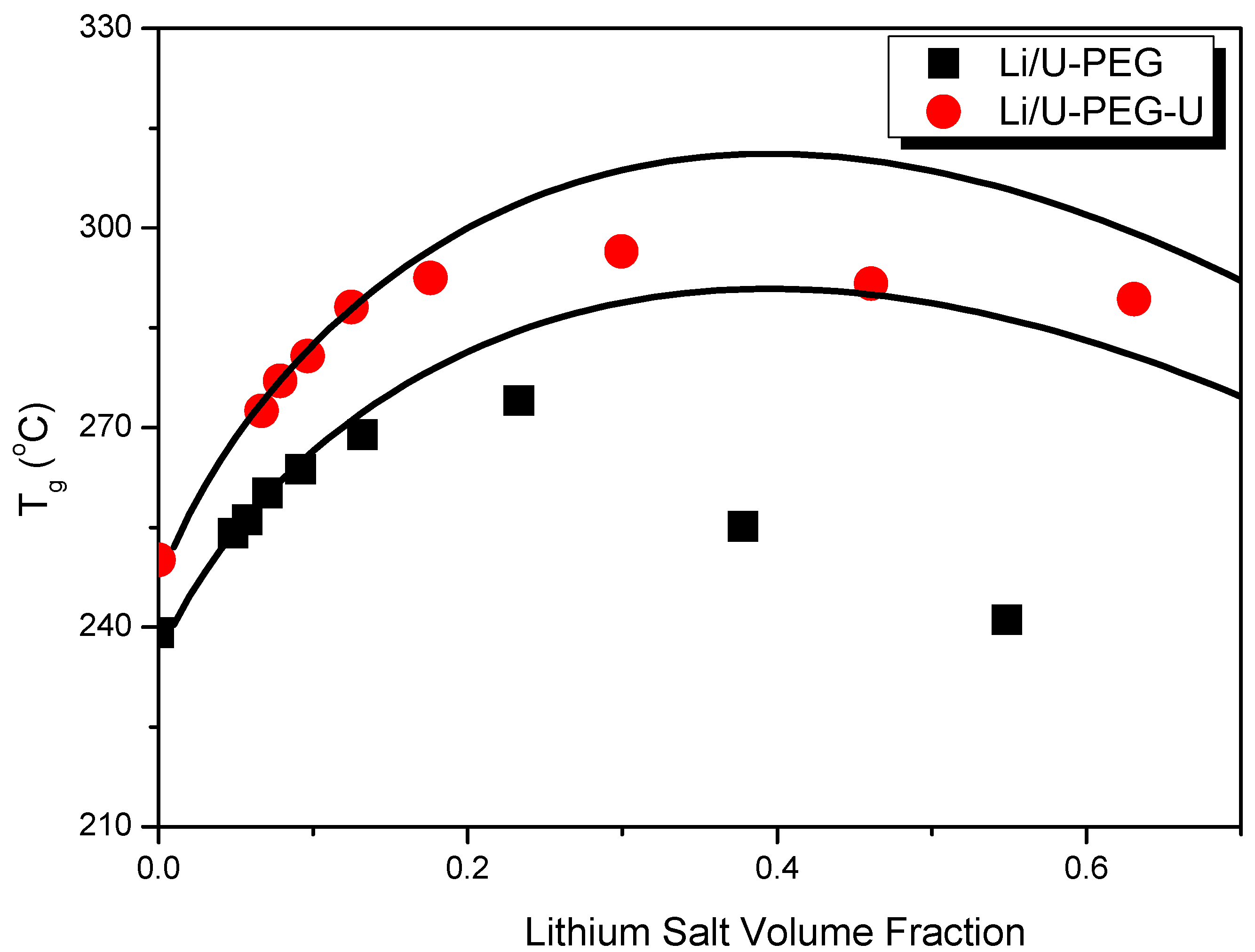
3.1. Thermal Analyses
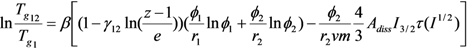
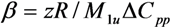
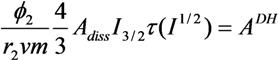
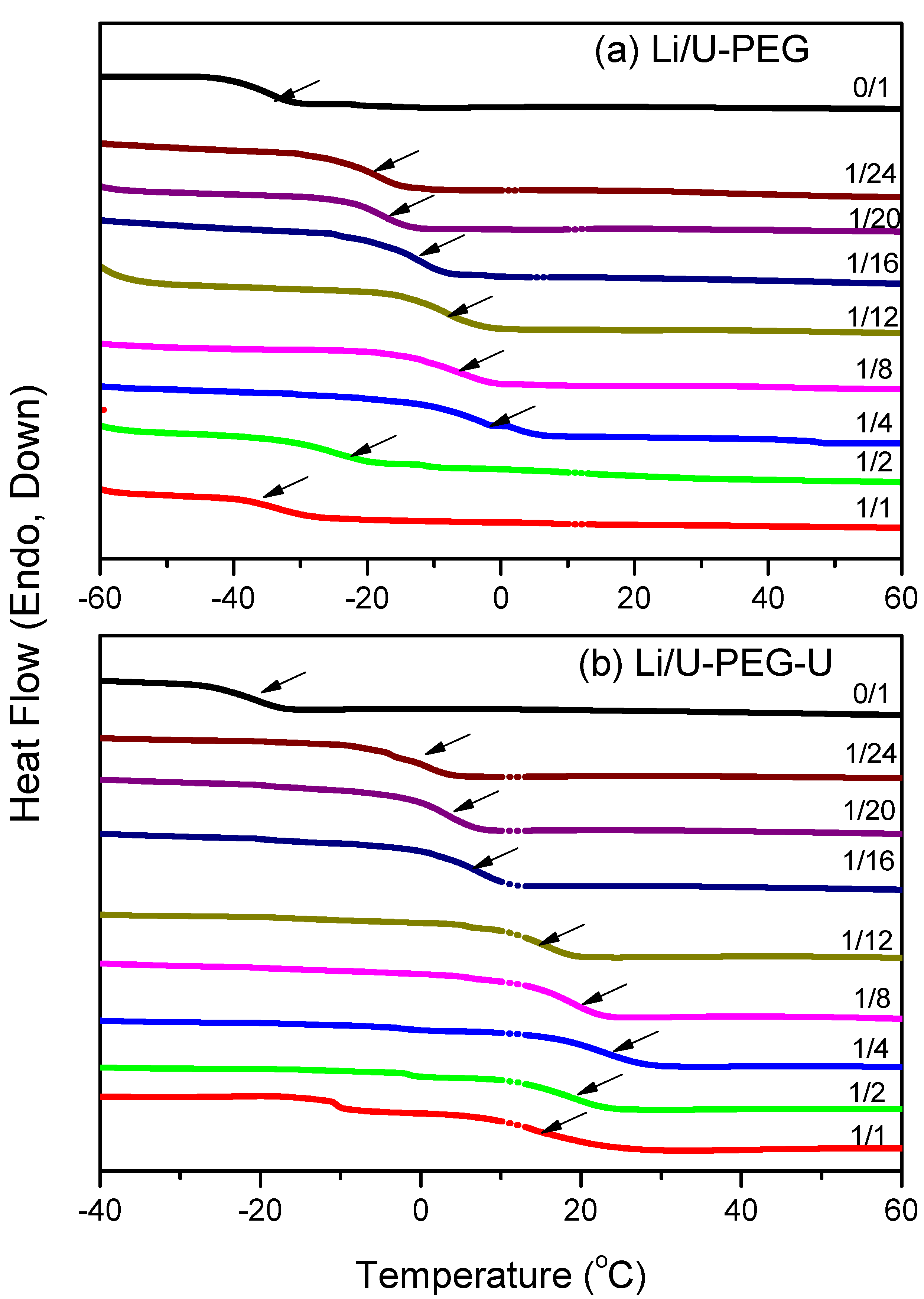

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
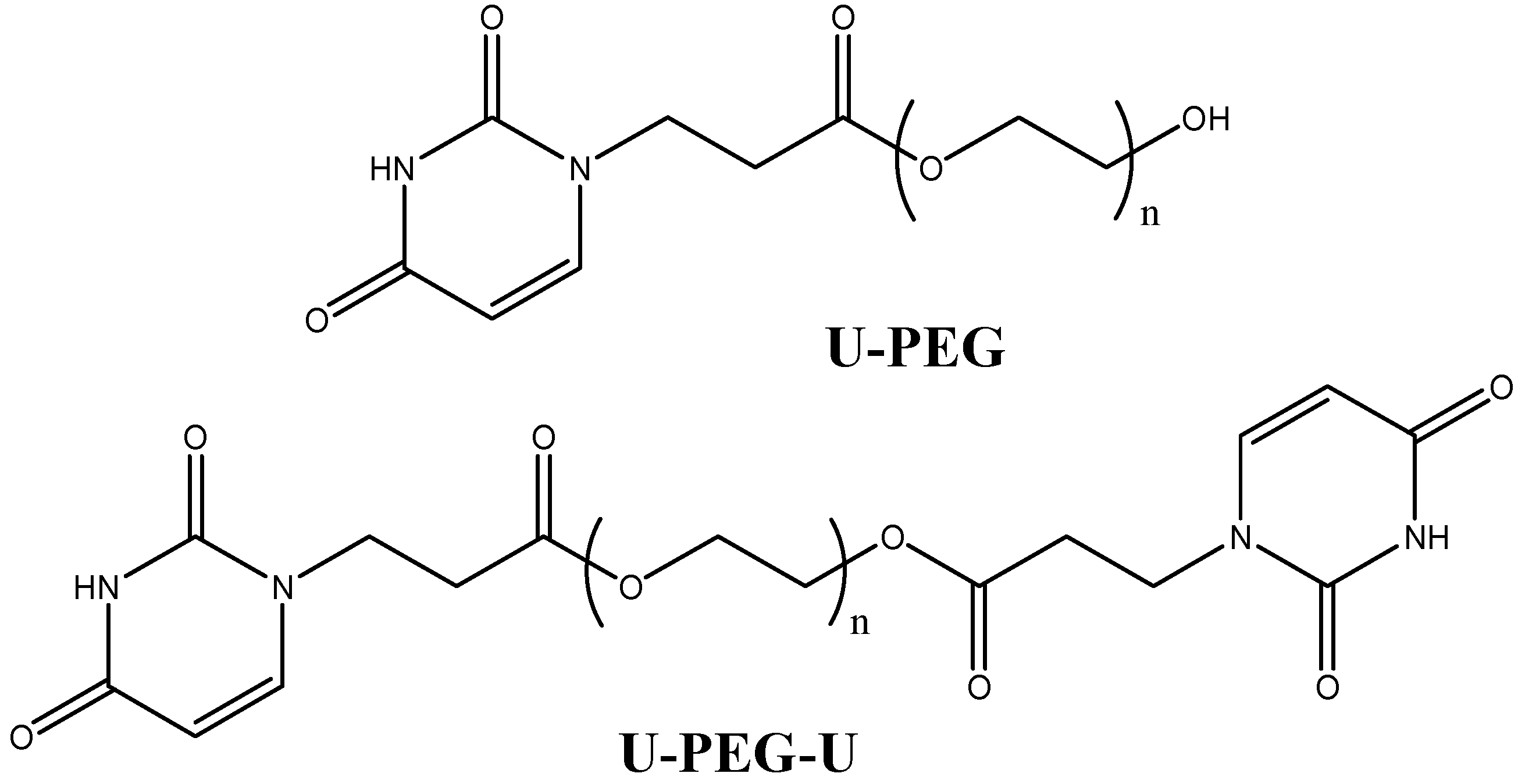{kind=link}
{kind=link}
| Polymers | M (g/mol) | Tg (°C) | Δ Cpp [J/(kg K)] a | Density (g/cm3) |
|---|---|---|---|---|
| U-PEG | 465 | −34 | 1,261 | 1.368 a |
| U-PEG-U | 647 | −23 | 1,261 | 1.374 a |
| LiAsF6 | 196 | – | – | 2.252 |
| Blend System | z | β | r12 | ADH |
| LiAsF6/U-PEG | 6 | 0.899 | 2.6 | 0.02 |
| LiAsF6/U-PEG-U | 6 | 0.899 | 2.7 | 0.02 |
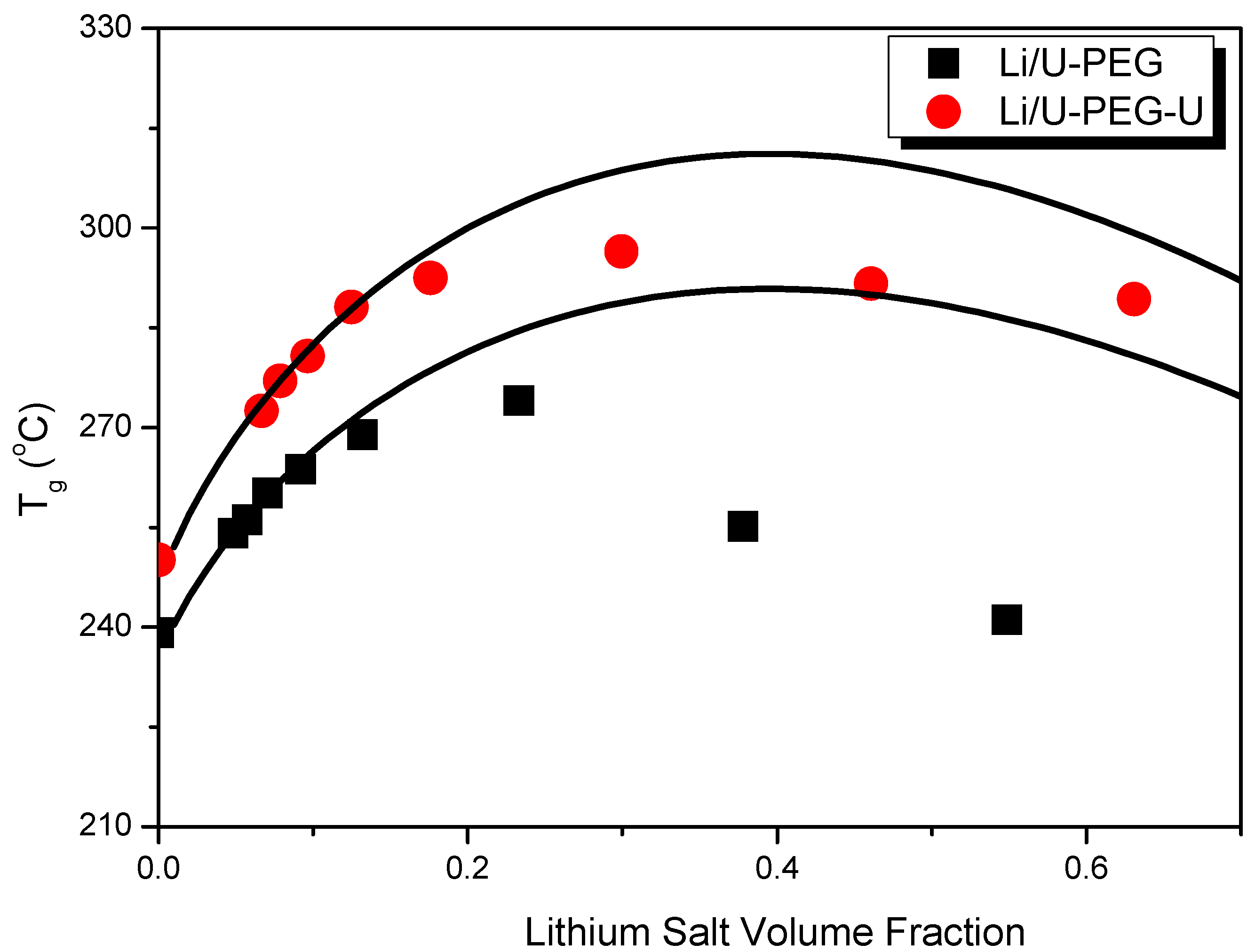
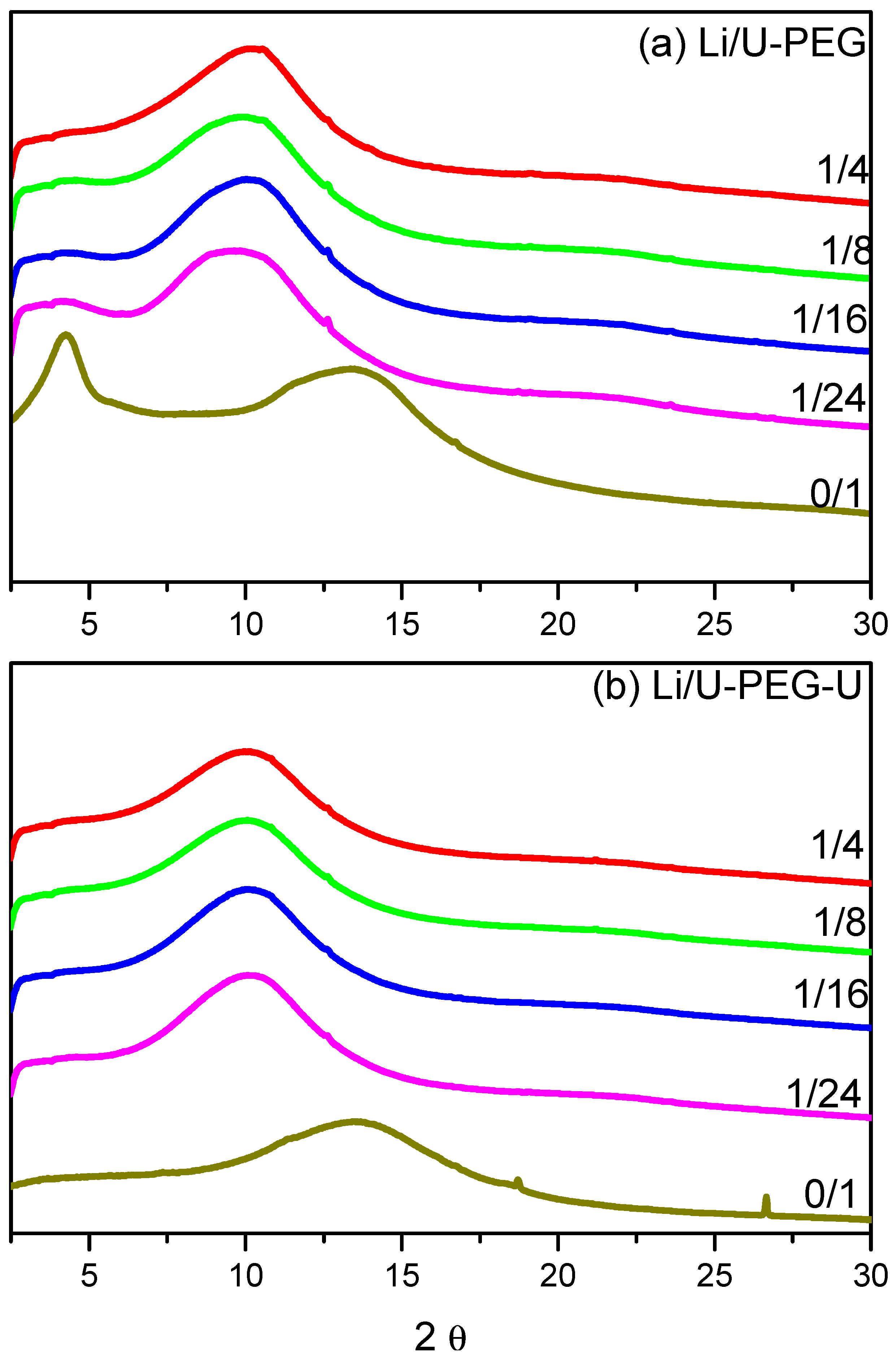
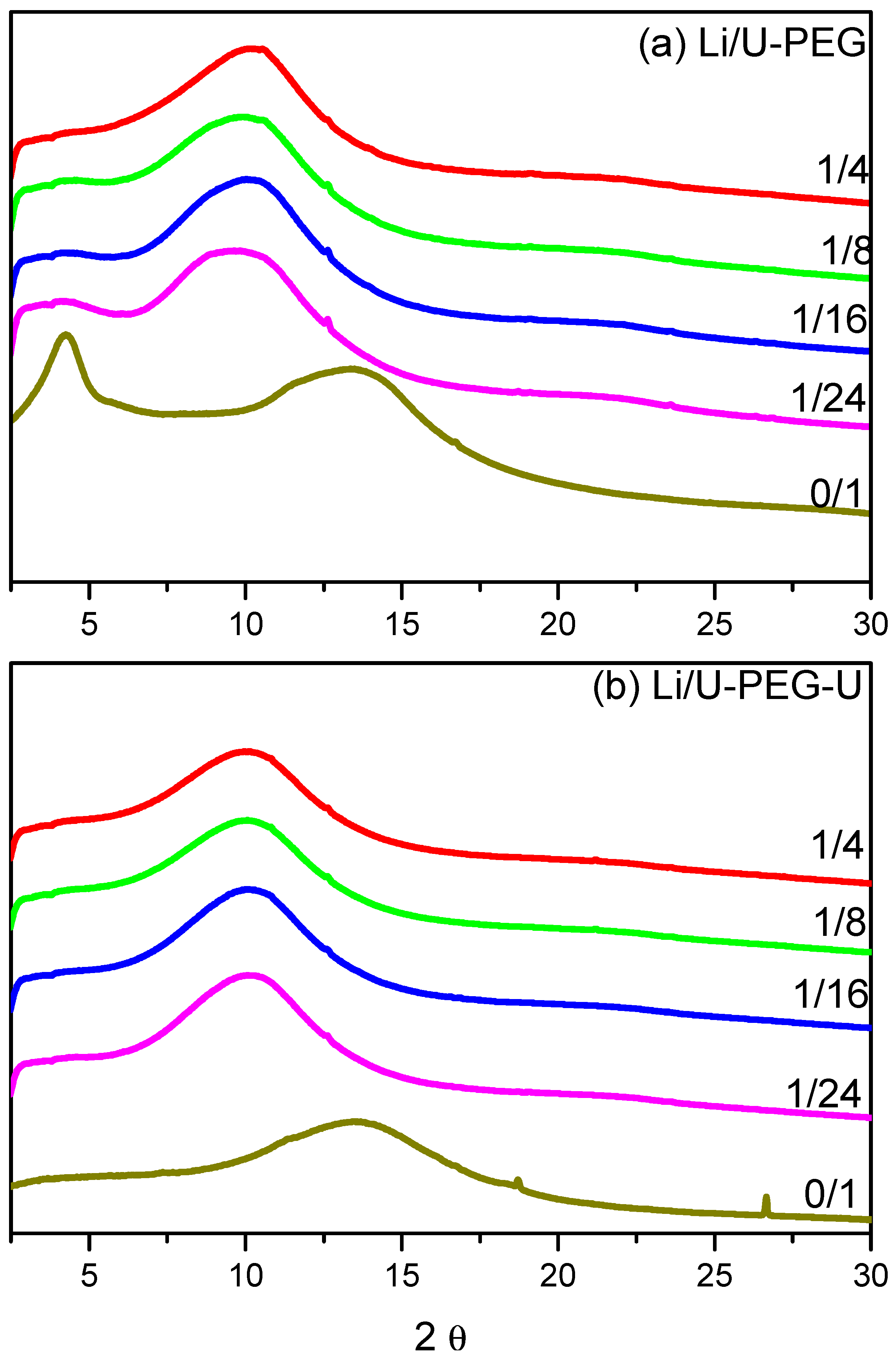
3.2. WAXD Analyses
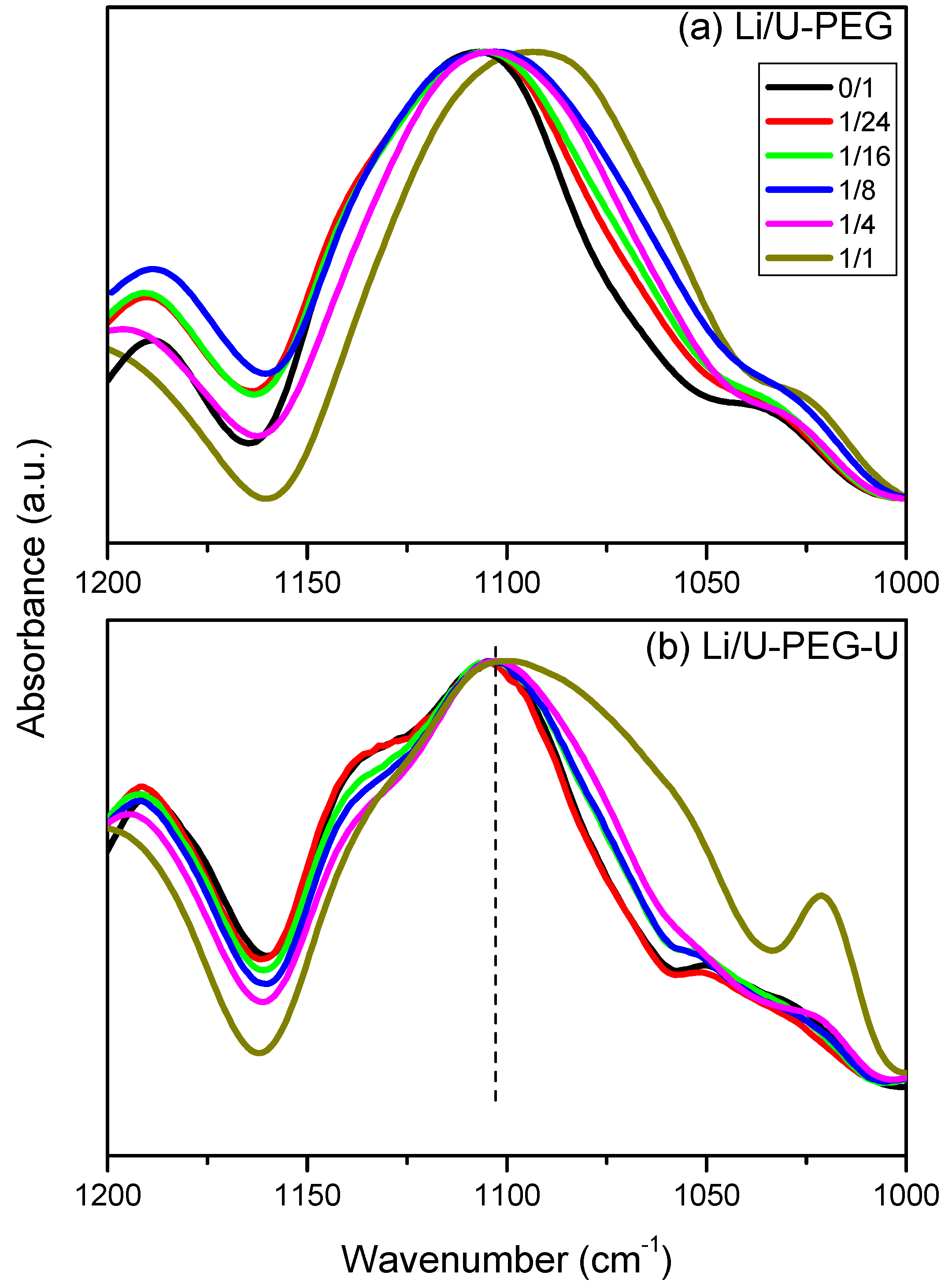
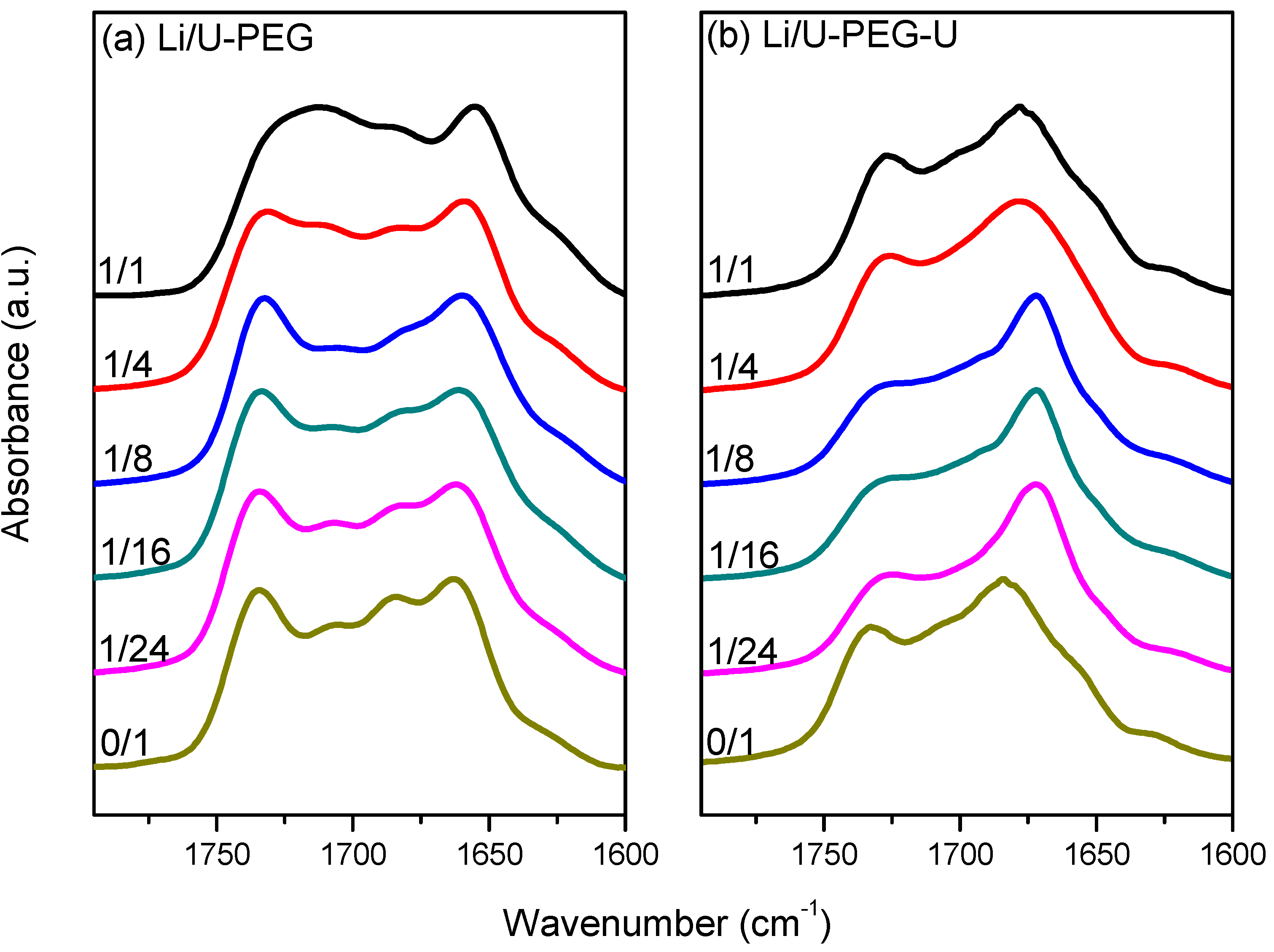
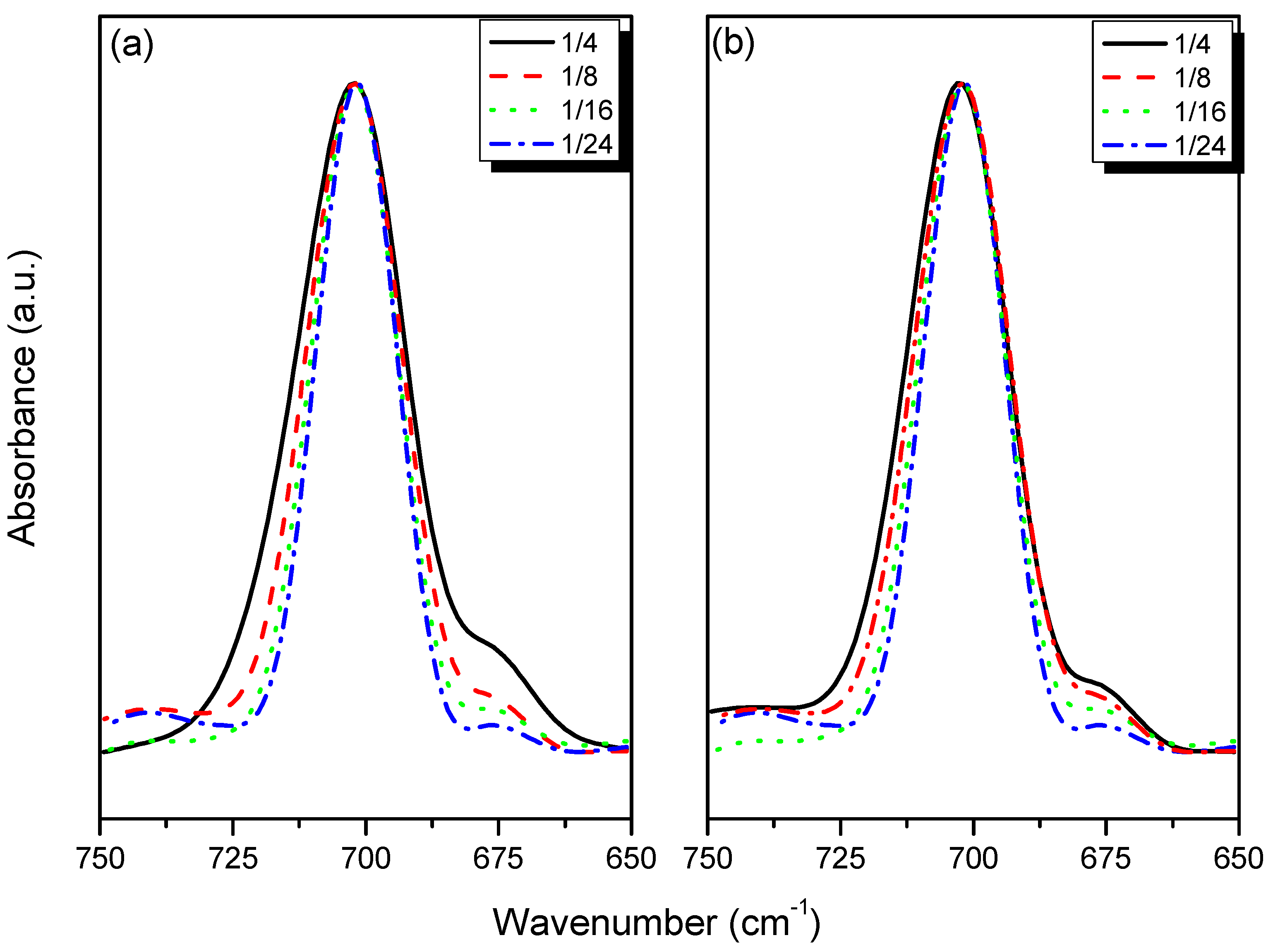
3.3. FTIR Spectroscopic Analyses
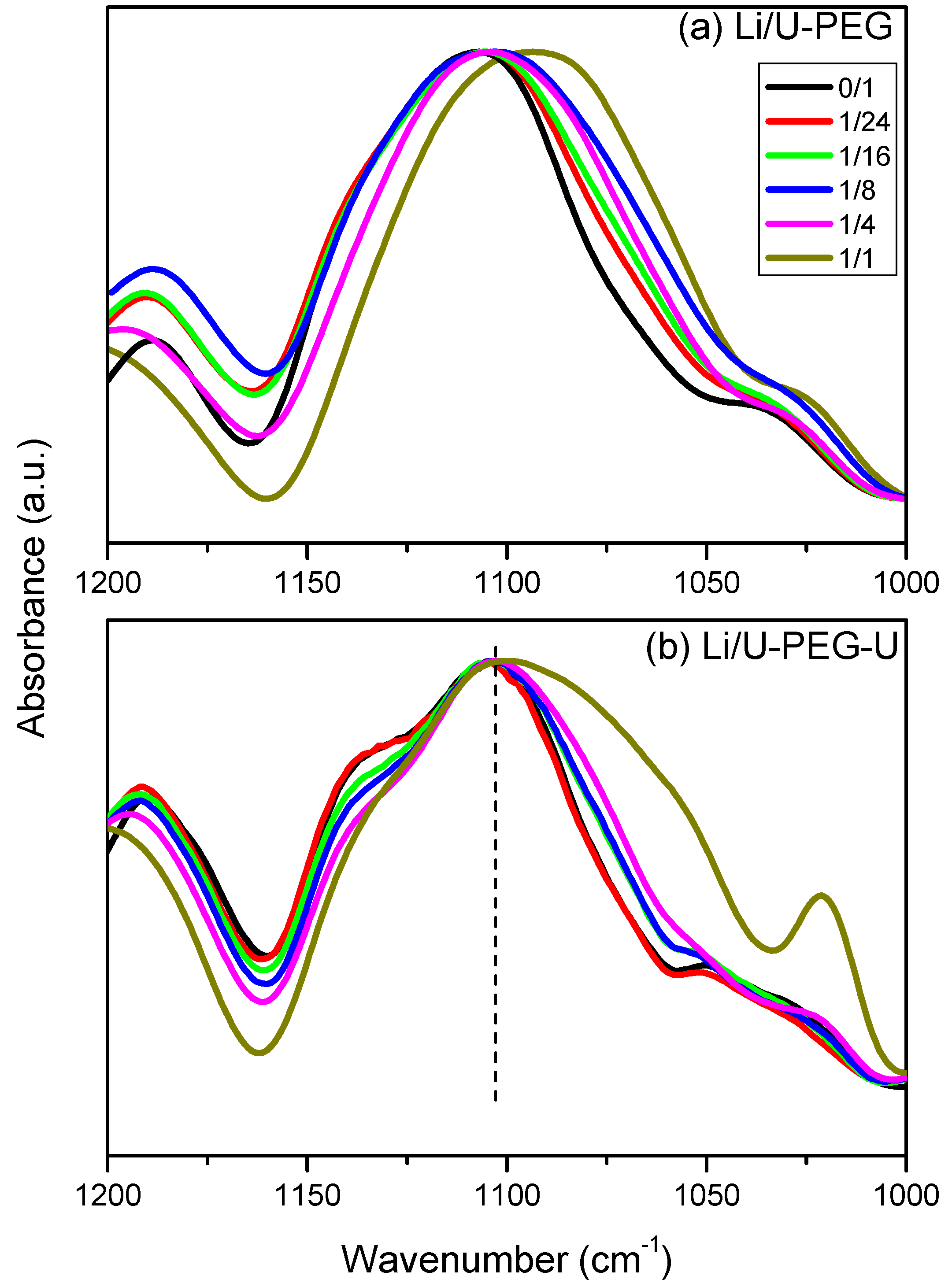
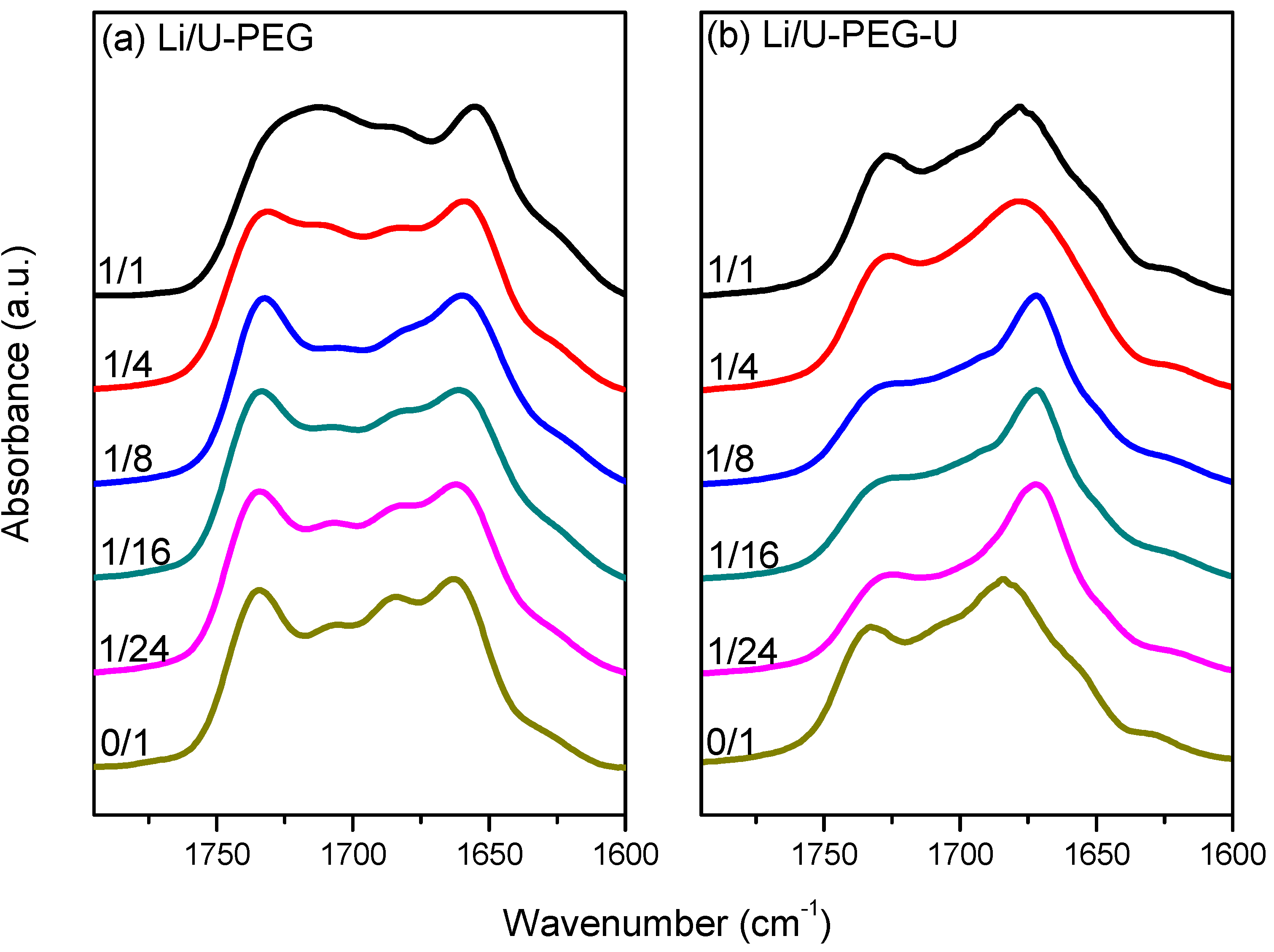

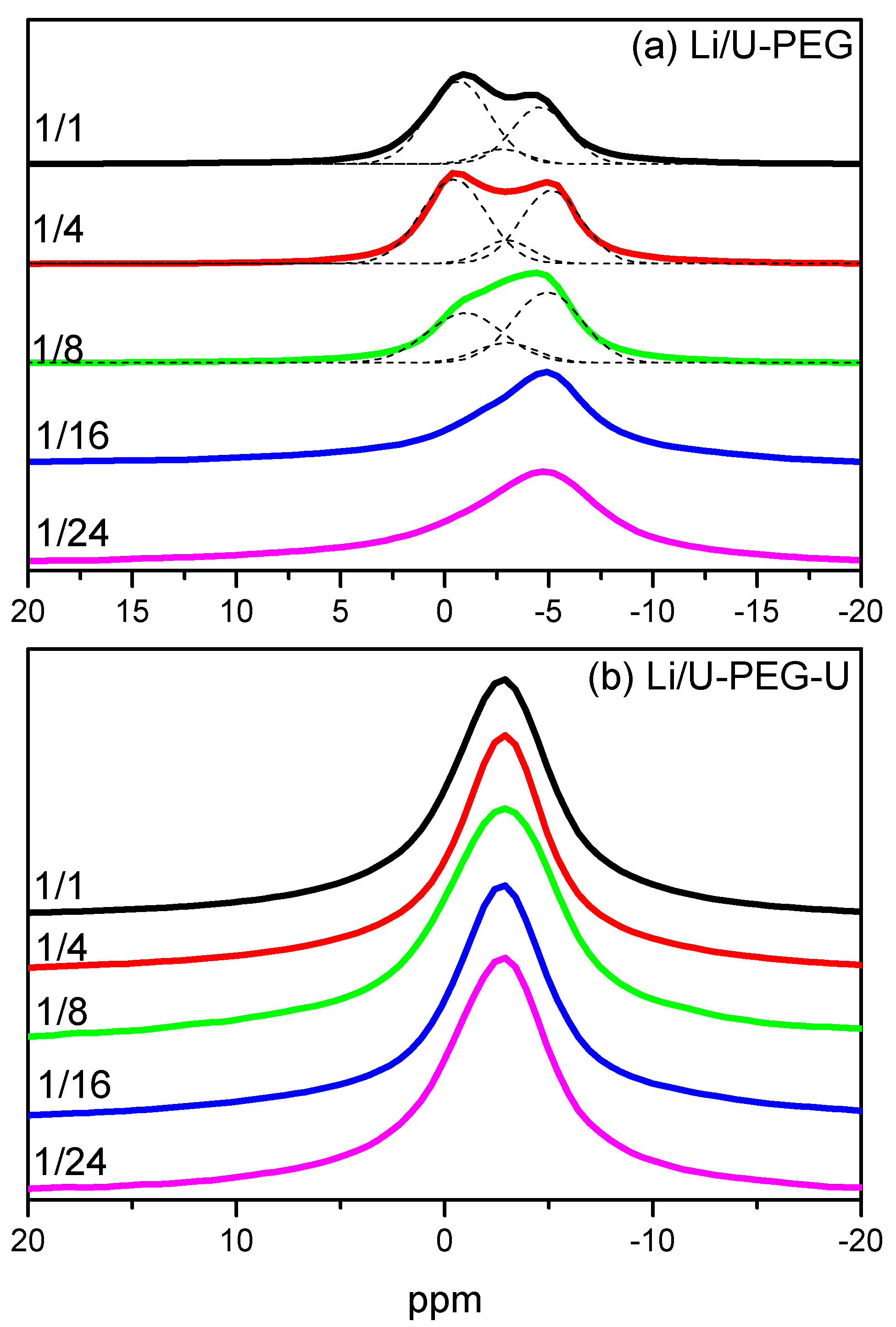
3.4. 7Li MAS NMR Spectroscopy
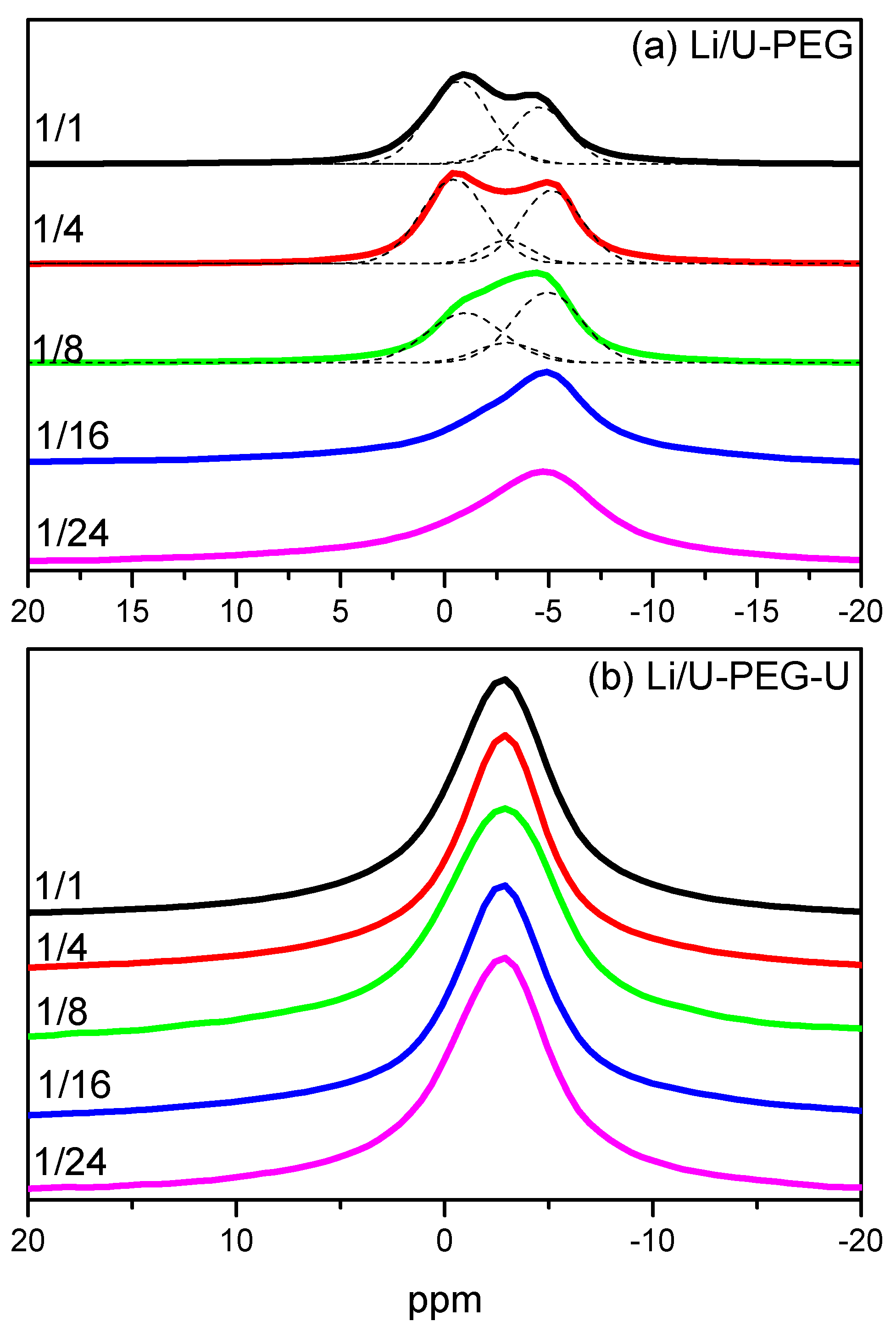
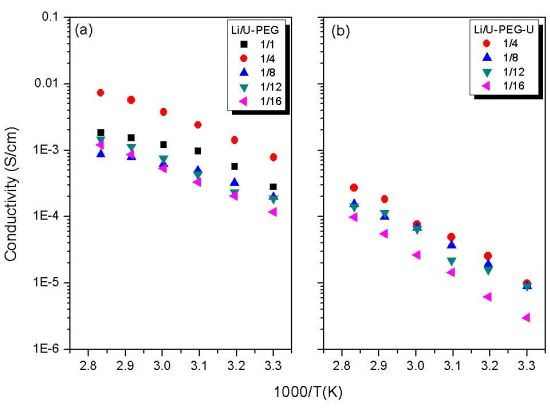
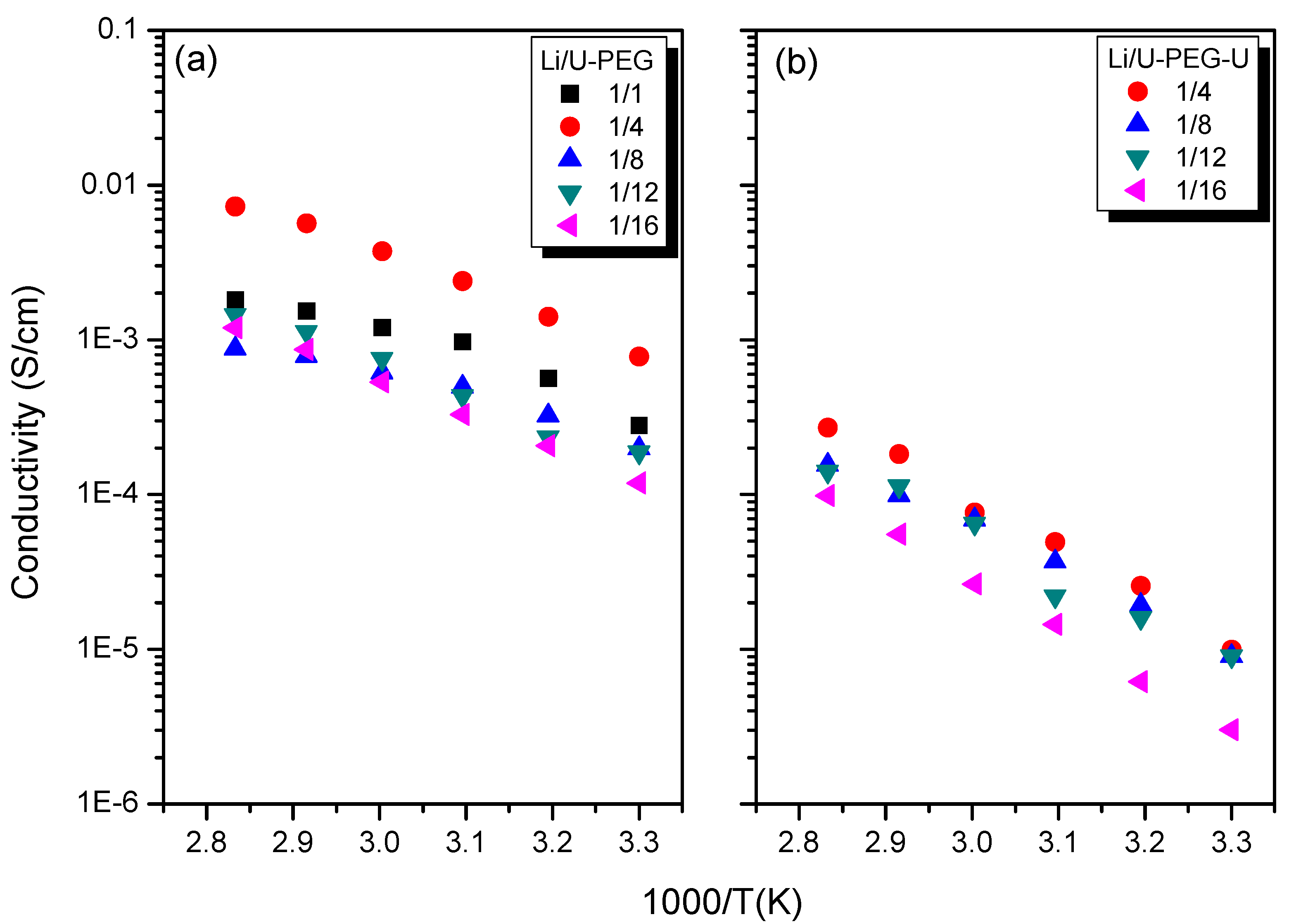
3.5. Ionic Conductivity
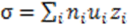
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Ye, Y.S.; Huang, Y.J.; Cheng, C.C.; Chang, F.C. A new supramolecular sulfonated polyimide for use in proton exchange membranes for fuel cells. Chem. Commun. 2010, 46, 7554–7556. [Google Scholar] [CrossRef]
- Ye, Y.S.; Yen, Y.C.; Cheng, C.C.; Chen, W.Y.; Tsai, L.T.; Chang, F.C. Sulfonated poly(ether ether ketone) membranes crosslinked with sulfonic acid containing benzoxazine monomer as proton exchange membranes. Polymer 2009, 50, 3196–3203. [Google Scholar] [CrossRef]
- Saikia, D.; Wu, H.Y.; Lin, C.P.; Pan, Y.C.; Fang, J.; Tsai, L.D.; Fey, G.T.K.; Kao, H.M. New highly conductive organic-inorganic hybrid electrolytes based on star-branched silica based architectures. Polymer 2012, 53, 6008–6020. [Google Scholar] [CrossRef]
- Murata, K.; Izuchi, S.; Yoshihisa, Y. An overview of the research and development of solid polymer electrolyte batteries. Electrochim. Acta 2000, 45, 1501–1508. [Google Scholar] [CrossRef]
- Croce, F.; Appetecchi, G.B.; Persi, L. Nanocomposite polymer electrolytes for lithium batteries. Nature 1998, 394, 456–458. [Google Scholar] [CrossRef]
- Zhang, H.; Kullarni, S.; Wnder, S.L. Polyethylene glycol functionalized polyoctahedral silsesquioxanes as electrolytes for lithium batteries, fuel cells, and energy conversion. J. Electrochem. Soc. 2006, 153, A239–A248. [Google Scholar] [CrossRef]
- Lin, C.K.; Wu, I.D. Investigating the effect of interaction behavior on the ionic conductivity of Polyester/LiClO4 blend systems. Polymer 2011, 52, 4106–4113. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Yen, Y.J.; Kuo, S.W.; Chen, H.W.; Chang, F.C. Complicated phase behavior and ionic conductivities of PVP-co-PMMA based polymer electrolytes. Polymer 2007, 48, 1329–1342. [Google Scholar] [CrossRef]
- Pivovar, B.S. An overview of electro–osmosis in fuel cell polymer electrolytes. Polymer 2006, 47, 4194–4202. [Google Scholar] [CrossRef]
- Xue, C.C.; Meador, M.A.B.; Zhu, Z.; Ge, J.J.; Cheng, S.Z.D.; Putthanarat, S. Morphology of PI–PEO block copolymers for lithium batteries. Polymer 2006, 47, 6149–6155. [Google Scholar] [CrossRef]
- Xi, J.; Qiu, X.; Zheng, S.; Tang, X. Nanocomposite polymer electrolyte comprising PEO/LiClO4 and solid super acid: Effect of sulphated–zirconia on the crystallization kinetics of PEO. Polymer 2005, 46, 5702–5706. [Google Scholar] [CrossRef]
- Rocco, A.M.; Carias, A.D.; Pereira, R.P. Polymer electrolytes based on a ternary miscible blend of poly(ethylene oxide), poly(bisphenol A-co-epichlorohydrin) and poly(vinyl ethyl ether). Polymer 2010, 51, 5151–5164. [Google Scholar] [CrossRef]
- Zardalids, G.; Ioannou, E.; Pispas, S.; Floudas, G. Relating structure, viscoelasticity, and local mobility to conductivity in PEO/LiTf electrolytes. Macromolecules 2013, 46, 2705–2714. [Google Scholar] [CrossRef]
- Berthier, C.; Gorecki, W.; Minier, M.; Armand, M.B.; Chabagno, J.M.; Rigaud, P. Microscopic investigation of ionic conductivity in alkali metal salts-poly(ethylene oxide) adducts. Solid State Ion. 1983, 11, 91–95. [Google Scholar] [CrossRef]
- Chen, H.W.; Chiu, C.Y.; Wu, H.D.; Shen, I.W.; Chang, F.C. Solid-state electrolyte nanocomposites based on poly(ethylene oxide), poly(oxypropylene) diamine, mineral clay and lithium perchlorate. Polymer 2002, 43, 5011–5016. [Google Scholar] [CrossRef]
- Xia, D.W.; Smid, J. Solid polymer electrolyte complexes of polymethacrylates carrying pendant oligo-oxyethylene(glyme) chains. J. Polym. Sci. Polym. Lett. Ed. 1984, 22, 617–621. [Google Scholar]
- Li, J.; Khan, I.M. Highly conductive solid polymer electrolytes prepared by blending high molecular weight poly(ethylene oxide), poly(2- or 4-vinylpyridine), and lithium perchlorate. Macromolecules 1993, 26, 4544–4550. [Google Scholar] [CrossRef]
- Wieczorek, W.; Such, K.; Zalewska, A.; Raducha, D.; Florjanczyk, Z.; Stevens, J.R. Composite polyether electrolytes with Lewis acid type additives. J. Phys. Chem. B 1998, 102, 352–360. [Google Scholar] [CrossRef]
- Chen, H.W.; Xu, H.; Huang, C.F.; Chang, F.C. Novel polymer electrolyte composed of poly(ethylene oxide), lithium triflate, and benzimidazole. J. Appl. Polym. Sci. 2004, 91, 719–725. [Google Scholar] [CrossRef]
- Chen, H.W.; Jiang, C.H.; Wu, H.D.; Chang, F.C. Hydrogen bonding effect on the poly(ethylene oxide), phenolic resin, and lithium perchlorate-based solid-state electrolyte. J. Appl. Polym. Sci. 2004, 91, 1207–1216. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Chen, H.W.; Kuo, S.W.; Huang, C.F.; Chang, F.C. Investigating the effect of miscibility on the ionic conductivity of LiClO4/PEO/PCL ternary blends. Macromolecules 2004, 37, 8424–8430. [Google Scholar] [CrossRef]
- Yen, Y.C.; Cheng, C.C.; Kuo, S.W.; Chang, F.C. A new poly(amide urethane) solid state electrolyte containing supramolecular structure. Macromolecules 2010, 43, 2634–2637. [Google Scholar] [CrossRef]
- Sijbesma, R.P.; Meijer, E.W. Self-assembly of well-defined structures by hydrogen bonding. Curr. Opin. Colloid Interface Sci. 1999, 4, 24–32. [Google Scholar] [CrossRef]
- Elemans, J.A.; Rowan, A.E.; Nolte, R.J.M. Mastering molecular matter: Supramolecular architectures by hierarchical self-assembly. J. Mater. Chem. 2003, 13, 2661–2670. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Grzybowski, B. Self-assembly at all scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef]
- Wang, Y.J.; Pan, Y.; Wang, L.; Pang, M.J.; Chen, L. Characterization of (PEO)LiClO4-Li1.3Al0.3Ti1.7(PO4)3 composite polymer electrolytes with different molecular weights of PEO. J. Appl. Polym. Sci. 2006, 102, 4269–4275. [Google Scholar]
- Xu, W.; Belieres, J.P.; Angell, C.A. Ionic conductivity and electrochemical stability of poly[oligo(ethylene glycol)oxalate]-lithium salt complexes. Chem. Mater. 2001, 13, 575–580. [Google Scholar] [CrossRef]
- Wang, J.H.; Altukhov, O.; Cheng, C.C.; Chang, F.C.; Kuo, S.W. Supramolecular structures of uracil-functionalized PEG with multi-diamidopyridine POSS through complementary hydrogen bonding interactions. Soft Matter 2013, 9, 5196–5206. [Google Scholar] [CrossRef]
- Eisenberg, A.; Rinaudo, M. Polyelectrolytes and ionomers. Polym. Bull. 1990, 24, 671. [Google Scholar] [CrossRef]
- Kim, J.H.; Min, B.R.; Kim, C.K.; Won, J.; Kang, Y.S. New insights into the coordination mode of silver ions dissolved in poly(2-ethyl-2-oxazoline) and its relation to facilitated olefin transport. Macromolecules 2002, 35, 5250–5255. [Google Scholar] [CrossRef]
- Kim, J.H.; Min, B.R.; Won, J.; Kang, Y.S. Analysis of the glass transition behavior of polymer-salt complexes: An extended configurational entropy model. J. Phys. Chem. B 2003, 107, 5901–5905. [Google Scholar] [CrossRef]
- Koh, J.H.; Park, J.T.; Koh, J.K.; Kim, J.H. Prediction of the glass transition temperature of semicrystalline polymer/salt complexes. J. Polym. Sci. B Polym. Phys. 2009, 47, 793–798. [Google Scholar] [CrossRef]
- Kuo, S.W.; Wu, C.H.; Chang, F.C. Thermal properties, interactions, morphologies, and conductivity behavior in blends of poly(vinylpyridine)s and zinc perchlorate. Macromolecules 2004, 37, 192–200. [Google Scholar] [CrossRef]
- Carlsson, P.; Mattsson, B.; Swenson, J.; Borjesson, L.; Torell, L.M.; McGreevy, R.L.; Howell, W.S. Intermediate range structural correlations in polymer electrolyte PPO-LiClO4 from neutron diffraction experiments and reverse Monte Carlo simulations. Electrochim. Acta 1998, 43, 1545–1550. [Google Scholar] [CrossRef]
- Deng, Z.; Irish, D.E. A Raman spectral study of solvation and ion association in the systems LiAsF6/CH3CO2CH3 and LiAsF6/HCO2CH3. Can. J. Chem. 1991, 69, 1766–1773. [Google Scholar] [CrossRef]
- Meyer, F.; Raquez, J.M.; Verge, P.; de Arenaze, I.M.; Coto, B.; Voort, P.V.D.; Meaurio, E.; Dervaux, B.; Sarasua, J.R.; Prez, F.D.; et al. Poly(ethylene oxide)-b-poly(l-lactide) diblock copolymer/carbon nanotube-based nanocomposites: LiCl as supramolecular structure-directing agent. Biomacromolecules 2011, 12, 4086–4094. [Google Scholar] [CrossRef]
- Szekrenyes, Z.; Kamara, K.; Tarczay, G.; Pallas, A.L.; Marangoni, T.; Prato, M.; Bonifazi, D.; Bjork, J.; Hanke, F.; Persson, M. Melting of hydrogen bonds in uracil derivatives probed by infrared spectroscopy and ab initio molecular dynamics. J. Phys. Chem. B 2012, 116, 4626–4633. [Google Scholar] [CrossRef]
- Fu, R.; Ma, Z.; Zheng, J.P.; Au, G.; Plichta, E.J.; Ye, C. High-resolution 7Li solid-state NMR study of LixV2O5 cathode electrodes for Li-rechargeable batteries. J. Phys. Chem. B 2003, 107, 9730–9735. [Google Scholar]
- Wang, H.L.; Kao, H.M.; Wen, T.C. Direct 7Li-NMR spectral evidence for different Li+ local environments in a polyether poly(urethane urea) electrolyte. Macromolecules 2000, 33, 6910–6912. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, Y.; Greenbaum, S.G.; Bajue, S.A.; Golodnitsky, D.; Ardel, G.; Strauss, E.; Peled, E. Electrical, thermal and NMR investigation of composite solid electrolytes based on PEO, LiI and high surface area inorganic oxides. Electrochim. Acta 1998, 43, 1557–1561. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, J.-H.; Cheng, C.-C.; Altukhov, O.; Chang, F.-C.; Kuo, S.-W. Supramolecular Functionalities Influence the Thermal Properties, Interactions and Conductivity Behavior of Poly(ethylene glycol)/LiAsF6 Blends. Polymers 2013, 5, 937-953. https://doi.org/10.3390/polym5030937
Wang J-H, Cheng C-C, Altukhov O, Chang F-C, Kuo S-W. Supramolecular Functionalities Influence the Thermal Properties, Interactions and Conductivity Behavior of Poly(ethylene glycol)/LiAsF6 Blends. Polymers. 2013; 5(3):937-953. https://doi.org/10.3390/polym5030937
Chicago/Turabian StyleWang, Jui-Hsu, Chih-Chia Cheng, Oleksii Altukhov, Feng-Chih Chang, and Shiao-Wei Kuo. 2013. "Supramolecular Functionalities Influence the Thermal Properties, Interactions and Conductivity Behavior of Poly(ethylene glycol)/LiAsF6 Blends" Polymers 5, no. 3: 937-953. https://doi.org/10.3390/polym5030937