1. Introduction
Organic semiconductors are based on conjugated organic small molecules or polymers, which can be processed from solution into electronics devices, allowing a rapid inexpensive roll-to-roll fabrication [
1]. Especially, polymers have shown great advantages, due to their good film forming properties and mechanical flexibility applicable to bendable electronics [
2]. Recently, field-effect transistor (FET) devices based on high-performance polymers have achieved charge-carrier mobilities higher than 1 cm
2/Vs, already surpassing the benchmark performance of amorphous silicon [
3,
4]. In most of the cases, these organic semiconductors lead to a unipolar transport of the hole or electron as charge carriers in the transistor. Recently, devices with a balanced migration of both types of carriers, so-called ambipolar FETs, have been reported. This kind of transistor behavior is especially required in complementary-like logic circuits [
5,
6].
Ambipolarity with balanced hole and electron mobilities in one single active layer is an intrinsic property of organic semiconductors and a matter of the adequate material design and transistor geometry. Various strategies exist to realize ambipolar devices. One is based on a heterojunction structure of a binary blend of two materials with corresponding unipolar electron- and hole-transporting behavior [
7]. In another approach, an adaption of the work function of the electrodes or a functionalization of the dielectric/semiconductor interface decrease the charge trapping [
8,
9]. Interestingly, only a few reports have described the effect of the microstructure on the ambipolar device characteristics [
10,
11]. It was observed that certain morphologies seem to favor ambipolarity, and that the behavior from ambipolar to unipolar is switchable/controllable by changing the microstructure. A further aspect is the molecular energy levels of the organic semiconductor, which are optimized for ambipolar FETs by lowering the bandgap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). This property can be realized by low-bandgap donor-acceptor copolymers, being also highly attractive for solar cells, due to their broad light absorption profiles, and have revealed high ambipolar mobilities over 1 cm²/Vs for both charge-carrier species [
12]. In this context, benzothiadiazole-cyclopentadithiophene (
CDT-BTZ) copolymers are one promising type of polymer leading to ultrahigh hole mobilities up to 5.5 cm²/Vs (in single-fiber FETs) when substituted with linear alkyl side chains and possessing higher molecular weight fractions [
13,
14]. It has to be noted that despite the relatively low bandgap of these derivatives of around 1.8 eV, only a unipolar hole transport was observed in standard device architectures (Au electrodes and hexamethyldisilazane (HMDS)-treated SiO
2 surface).
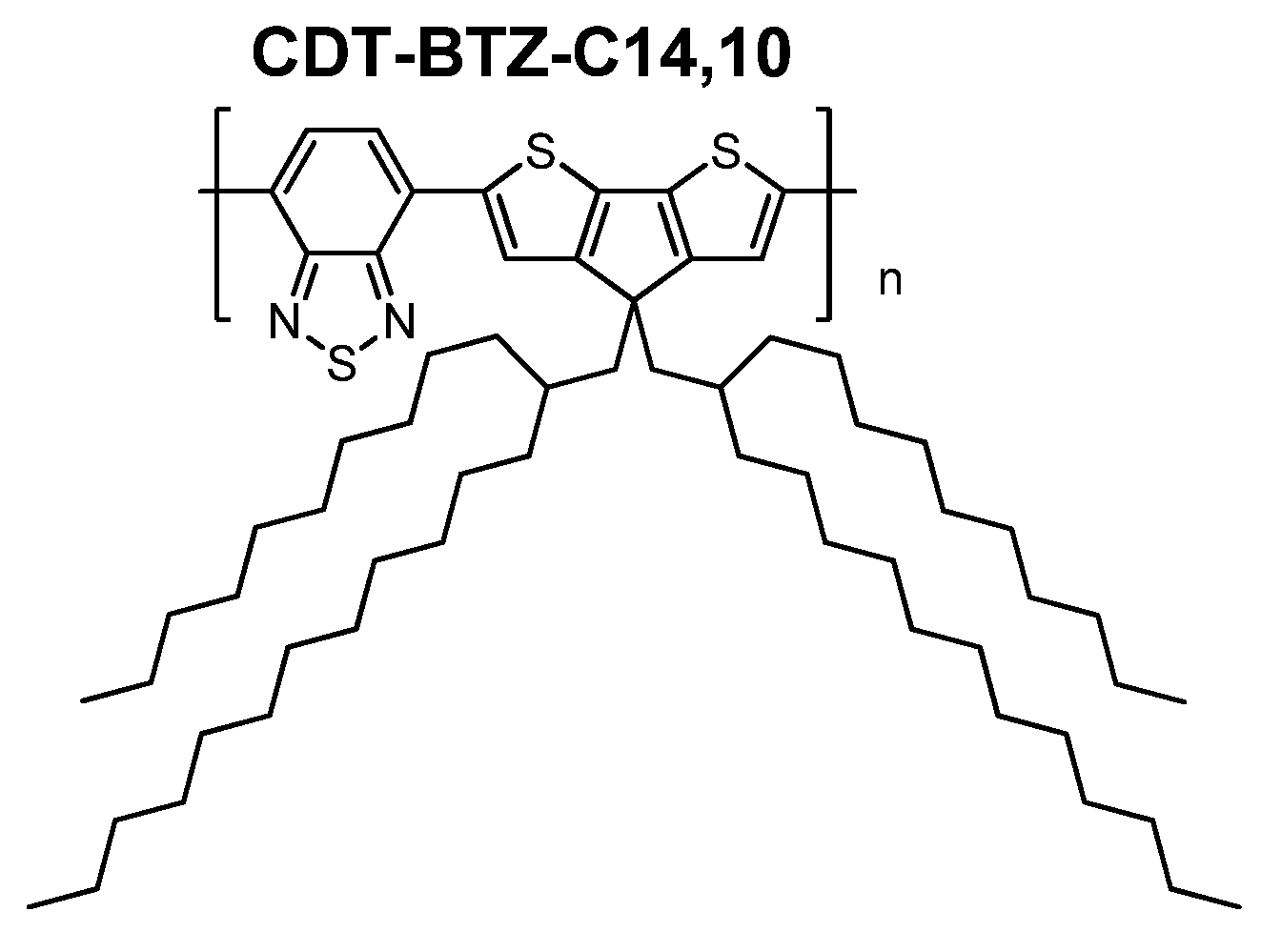
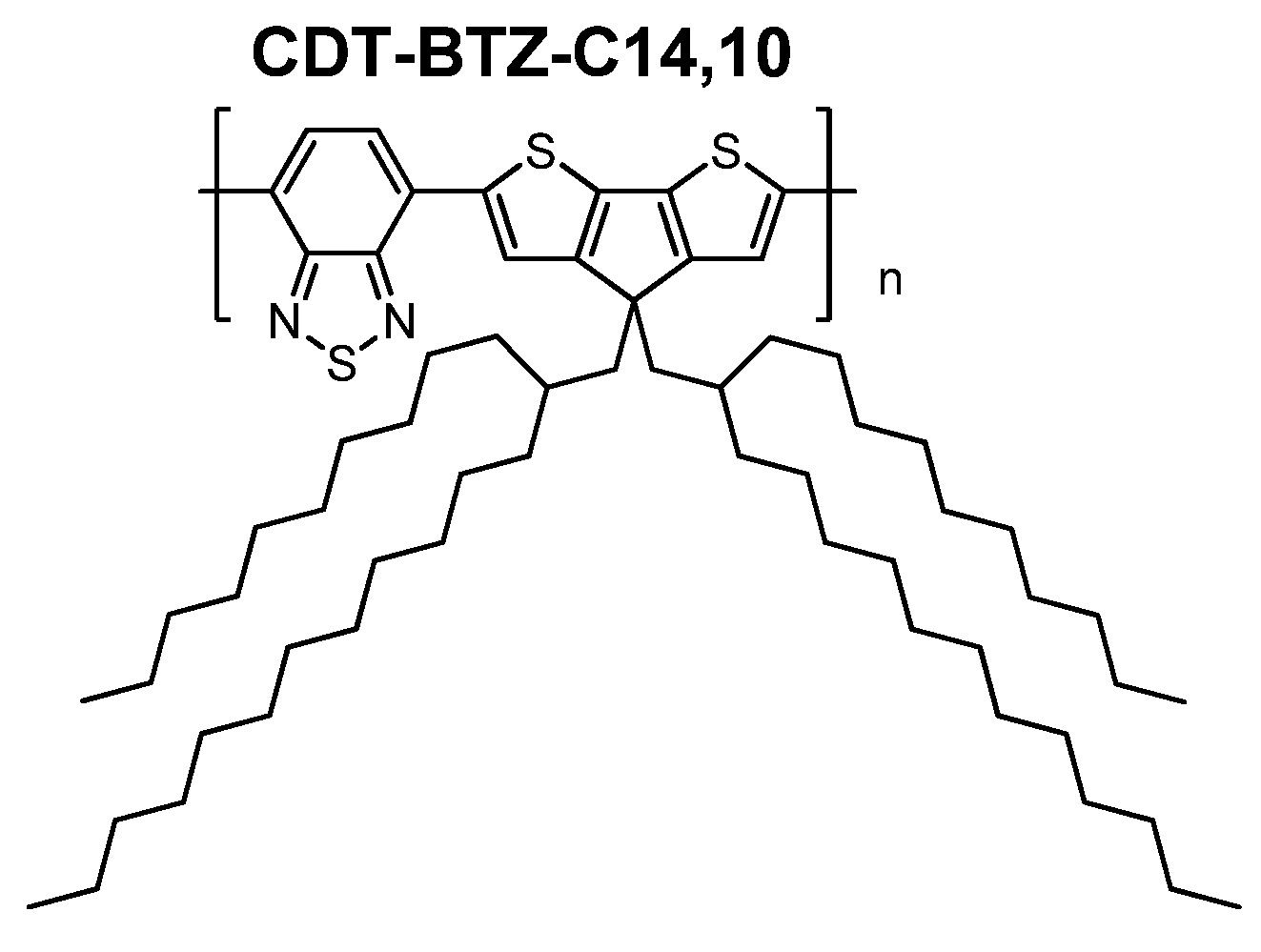
In this work, we have investigated the influence of branched, bulky side chains (
C14,10) on the polymer packing of the
CDT-BTZ polymer (
Figure 1), thin film microstructure and charge carrier transport in a FET. As expected, pronounced differences in the packing and microstructure were found in comparison to the polymers with linear chains. Surprisingly,
CDT-BTZ-C14,10 shows ambipolar device characteristics under the same transistor geometry, as previously reported, despite unchanged energy levels in comparison to polymers with hexadecyl (
C16) and 3,7-dimethyloctyl (
C8,2). In this work, it is shown for the first time that the ambipolarity is related to a lateral shift of the conjugated backbones towards each other induced by the steric demand of the
C14,10 chains. These results indicate that the electronic properties of a conjugated polymer might be dependent on the relative arrangement of the backbones, which can be well-controlled by the steric influence of the substituents.
Figure 1.
Structure of CDT-BTZ with branched side chains.
Figure 1.
Structure of CDT-BTZ with branched side chains.
2. Experimental
The cyclic voltammetry experiments were carried out with an EG&G Princeton Applied Research potentiostat, model 273. The working electrode consisted of glassy carbon electrode (GCE) (3.0 mm dia.). A platinum wire was used as the counter electrode, and an Ag wire was used as the reference electrode internally calibrated with ferrocene/ferrocenium (Fc/Fc+) in the measurement. The thin film was prepared on the surface of working electrode by drop-casting. After that, the CV measurements were executed in a solution of tetrabutylammonium hexafluorophosphate (Bu4NPF6, 0.1 M) in acetonitrile with a scan rate of 100 mV·s−1 at room temperature under argon.
The HOMO and LUMO levels were determined from the analysis of redox potential behavior observed in cyclic voltammogram curves in comparison with that of ferrocene, using the following expressions:
Transmission: two-dimensional wide-angle X-ray scattering measurements were performed using copper anode X-ray tube, operated at 35 kV/30 mA, as the X-ray source, Osmic confocal Max-Flux optics and a two pin-hole collimation system. The patterns were recorded on a two-dimensional (2D) area detector (Bruker HI-STAR) for macroscopically oriented fibers [
15] with a diameter of 0.7 mm, which were prepared by extrusion. The fiber samples were mounted vertically towards the 2D detector.
Field-effect transistors: All FETs were fabricated employing the bottom-gate, bottom-contact architecture. The 200 nm-thick SiO2 dielectric covering the highly doped Si acting as the gate electrode was functionalized with hexamethyldisilazane (HMDS) to minimize interfacial trapping sites. The channel lengths and widths are 20 and 1400 μm, respectively. All the electrical measurements (using Keithley 4200 SCS) are performed in a glove box under nitrogen atmosphere. The devices were fabricated by drop-casting the polymer from a 2 mg/mL solution with subsequent annealing at 200 °C for 30 minutes.
The morphology of the drop-cast films was examined using atomic force microscopy (AFM) (Veeco Digitals Instruments) in the tapping mode at room temperature.
Solid-State NMR: Solid-state magic-angle spinning (MAS) NMR experiments were carried out on a Bruker 850 Avance III spectrometer using a commercial 2.5 mm double resonance probe from Bruker. The 2D
1H–
1H double quantum-single quantum (DQ-SQ) correlation spectrum was recorded at a MAS frequency of 20.0 kHz, employing the back-to-back (BaBa) sequence [
16] with a 50.0 μs period to excite/reconvert the DQ signals with 16 accumulations for a total of 128 t
1 increments. The 2D
13C{
1H} frequency-switched Lee-Goldburg heteronuclear correlation (FSLG-HETCOR) experiment [
17] used a MAS frequency of 15.0 kHz, a recycle delay of 2 s, a cross polarization (CP) contact time of 3.0 ms and 1,152 scans for a total of 36 t
1 increments. Each t
1 increment had a span of five basic FSLG [
18] blocks (81.6 μs). High-power
1H SPINAL-64 decoupling was used during acquisition [
19]. The CP transfer step utilized Lee-Goldburg CP (LG-CP) conditions [
20,
21] to suppress
1H spin diffusion during CP. The Hartmann-Hahn matching condition was pre-optimized on L-alanine. A scaling factor of 0.574 was determined for the
1H-
1H FSLG homonuclear decoupling sequence by recording a 2D
13C{
1H} FLSG-HETCOR spectrum for adamantane using identical experimental conditions and used to rescale the indirect
1H dimension for the investigated sample. All spectra are referenced with respect to tetramethyl silane (TMS) using solid adamantane (
13C, 29.46 ppm;
1H, 1.85 ppm) as a secondary reference [
22,
23].
DFT and NICS Calculations: DFT calculations at the B97-D/6-311g(d,p) [
24] level have been performed to model the molecular structure and geometry (bond lengths, angles and torsions), as well as to estimate the isotropic (liquid-state)
1H and
13C-NMR chemical shifts of the
CDT-BTZ copolymer. The optimized structures were used to build the copolymer model stack for solid-state NMR calculations under PBC (periodic boundary conditions). For the analysis of NMR current screening effects, caused by the close proximity of the neighboring π-conjugated polymer chains, we have performed nucleus-independent chemical shift (NICS) calculations of stacked model structures. To understand the magnetic shielding effect dependence upon sliding along the backbone direction, we have composed six models based on a unit cell with parallel and anti-parallel acceptors (BTZs) arrangements. Furthermore, the model unit cells consisted of stacks of five polymer layers with slides along the backbone direction of 3.0 Å, 1.5 Å and 0.0 Å, respectively. These layers were isolated from their periodic replicas in order to purely quantify the magnetic shielding within the isolated stack. The model stacks were further simplified by truncating the alkyl-chains to methyl groups, since these do not affect the aromatic ring currents of the backbones and, thus, can be neglected for the sake of efficient use of computational resources. In every NICS calculation, performed under full PBC, we have removed the third layer to evaluate the NICS shifts at this specific position,
i.e., we take into account the magnetic shielding effects from two layers above and two layers below. The resulting NICS maps represent the planes taken at the third layer position. All NICS map calculations were performed with the CPMD package [
25] within the DFT framework using the gradient-corrected exchange and correlation functionals (BLYP) [
26,
27], Goedecker-Teter-Hutter pseudopotentials [
24] together with a plane wave cutoff of 70 Ry.
3. Results and Discussion
The
CDT-BTZ polymer with branched, sterically demanding 2-decyltetradecyl (
C14,10) substituents was synthesized accordingly to the previously described route (
Figure 1) [
13]. A molecular weight of Mn = 13,000 (using polystyrene/trichlorobenzene gel permeation chromatography standard) and a polydispersity of a polydispersity index (PDI) = 1.29 are determined. The levels for the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO)
versus vacuum were determined from the value of onset oxidation potential and onset reduction potential during cyclic voltammetry (CV) scans on thin films showing reversibility in both reduction and oxidation behavior. For comparison to
CDT-BTZ-C14,10, the HOMO and LUMO levels of the recently reported
CDT-BTZs with linear hexadecyl (
C16) and 3,7-dimethyloctyl (
C8,2) side chains were investigated. The electrochemical energy levels and energy gaps are summarized in
Table 1. All polymers reveal identical electrochemical energy gaps in the range between 1.88 eV and 1.91 eV, being in the same range (taking into account the experimental error), as previously reported [
28]. The HOMO and LUMO are also located in similar levels with −4.91 eV for the lowest HOMO for
CDT-BTZ-C8,2 and the highest LUMO of −3.27 eV for
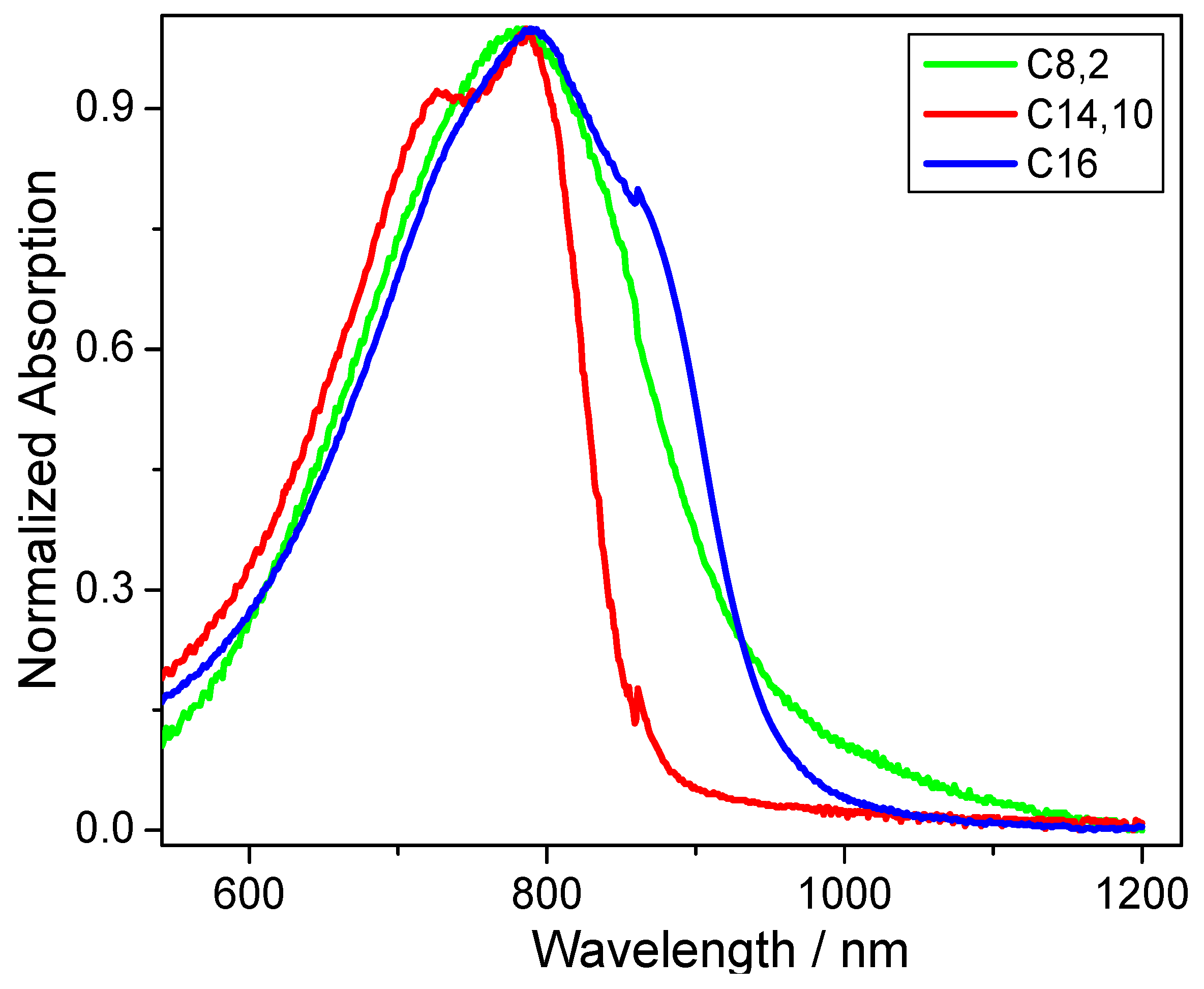
CDT-BTZ-C14,10. Additionally, the optical band gap was derived from optical absorption data acquired for thin films (
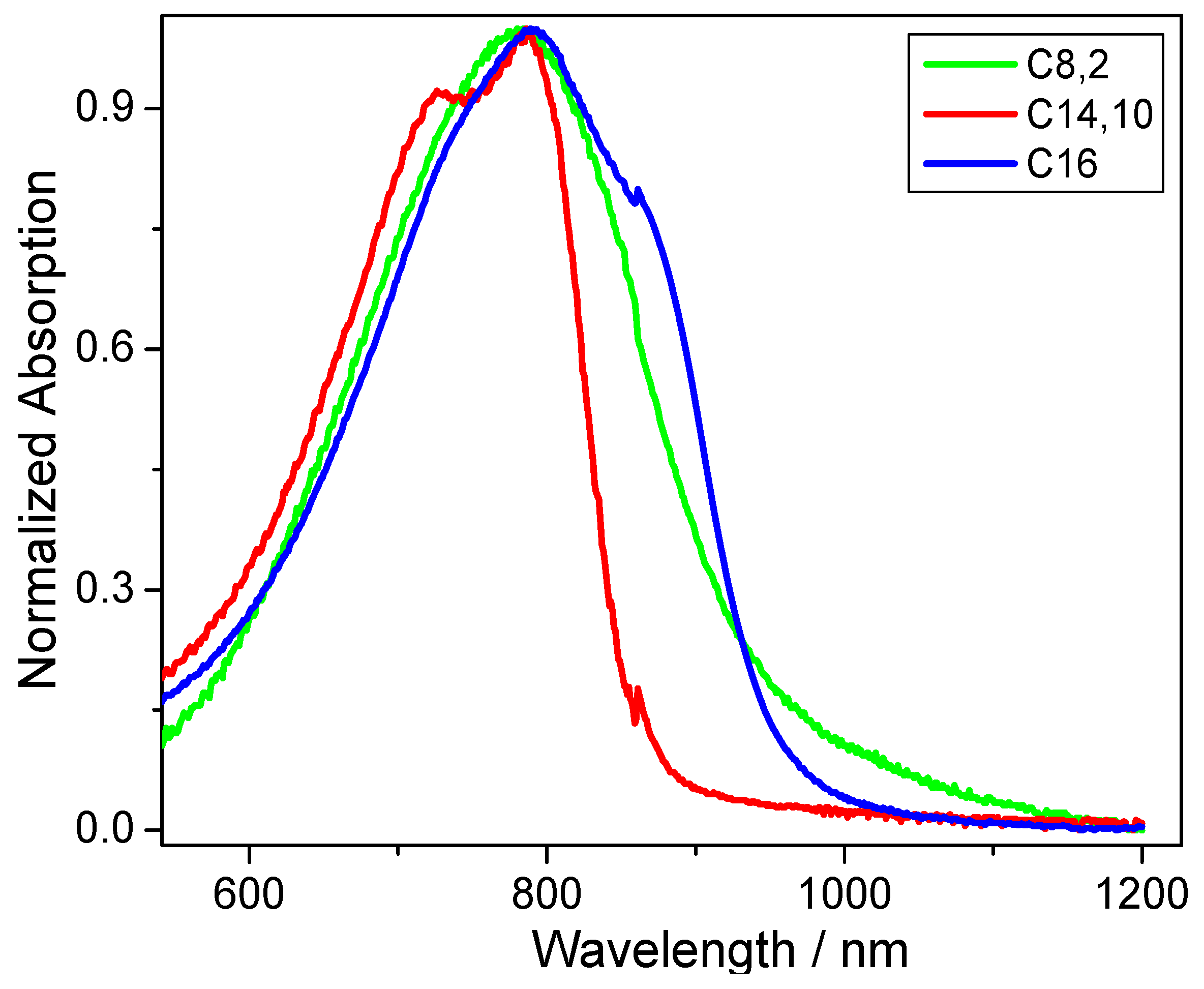
Figure 2). While no large differences are obvious for the solid-state absorption maximum peak of around 785 nm (±3 nm),
CDT-BTZ-C14,10 (at 727 nm) and
CDT-BTZ-C16 (at 861 nm) reveal small shoulders. The optical band gap of 1.94 eV for
CDT-BTZ-C14,10 is identical to the value determined by CV; however, the values for the other two polymers are significantly smaller with 1.3 eV for
CDT-BTZ-C16 and 1.32 eV for
CDT-BTZ-C8,2 (
Table 1). It has to be emphasized that the strong tailing in the solid-state absorption of the latter polymers makes the determination of the onset difficult. The difference between optical and electrochemical band gap is in agreement with other reports [
28].
Table 1.
Electrochemical derived highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels and energy gaps of CDT–BTZ polymers with different side chains.
Table 1.
Electrochemical derived highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels and energy gaps of CDT–BTZ polymers with different side chains.
| Polymer | HOMOCV eV | LUMOCV eV | ![Polymers 05 00833 i003]() eV eV | ![Polymers 05 00833 i004]() eV eV |
|---|
| CDT-BTZ-C14,10 | −5.15 | −3.27 | 1.88 | 1.94 |
| CDT-BTZ-C8,2 | −4.91 | −3.02 | 1.89 | 1.32 |
| CDT-BTZ-C16 | −5.06 | −3.15 | 1.91 | 1.30 |
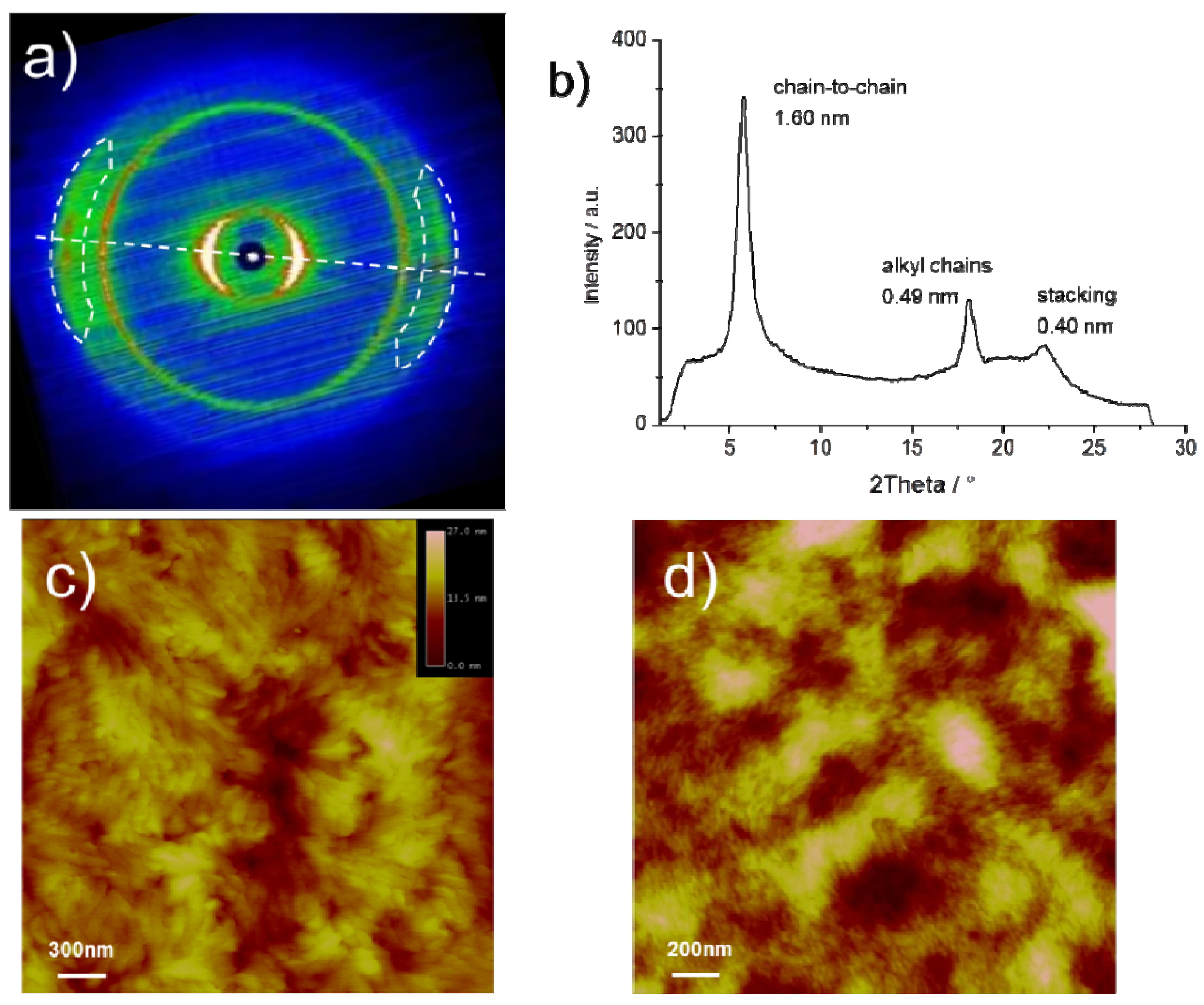
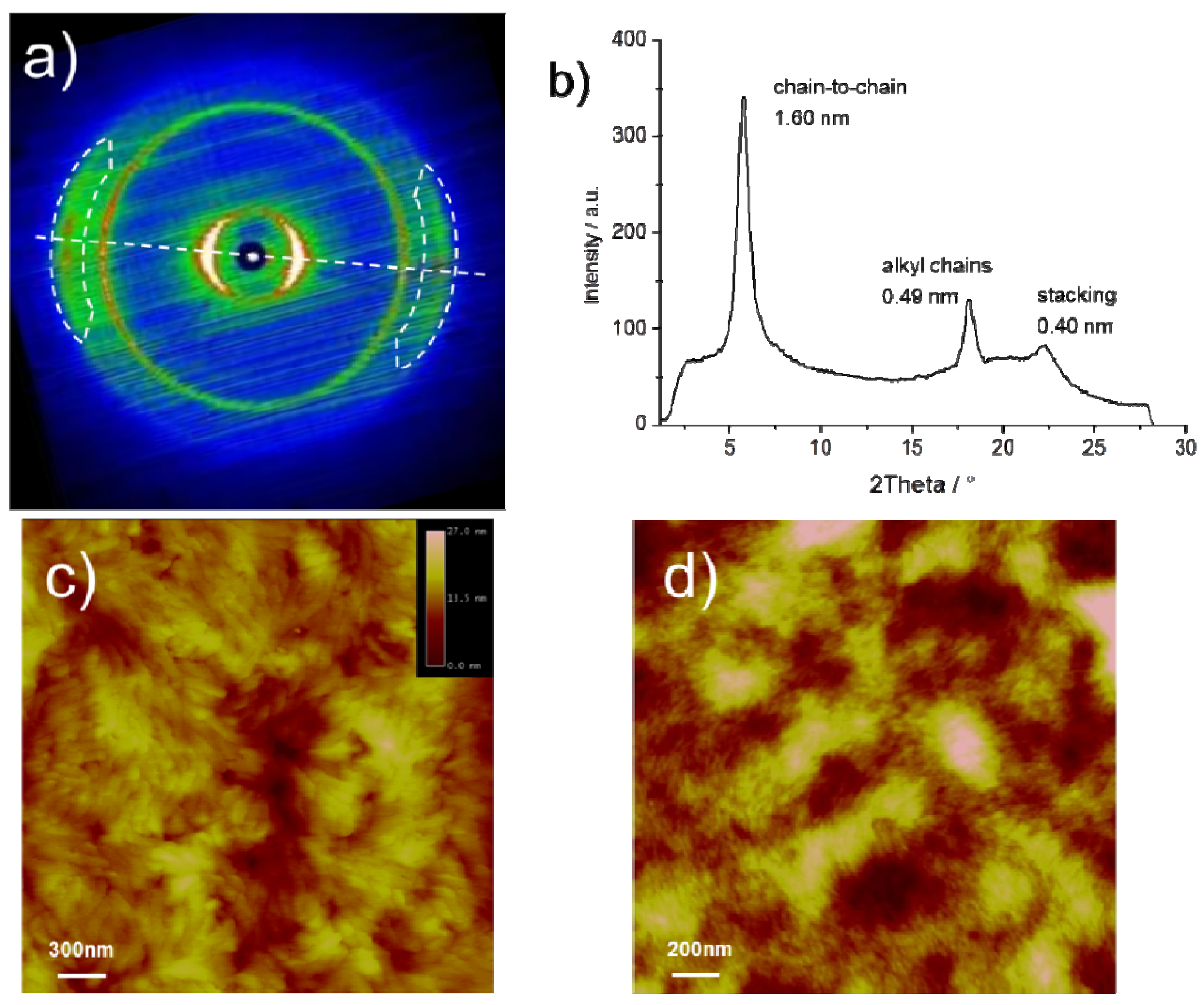
The bulk organization of
CDT-BTZ-C14,10 was determined by two-dimensional wide-angle X-ray scattering (2DWAXS) on mechanically extruded samples with the polymers macroscopically oriented. The 2DWAXS data for
CDT-BTZ-C16 and
CDT-BTZ-C8,2 have been previously published [
13]. Briefly, the polymers are arranged in lamellar structures and possess a π-stacking distance of around 0.37 nm. In contrast to this, the intermolecular distance of 0.40 nm for
CDT-BTZ-C14,10 is significantly increased, as derived from the characteristic wide-angle reflections located in the equatorial plane of the corresponding s at the proximity of the conjugated backbone, which reduce the non-covalent interactions between backbones. Interestingly, these scattering intensities are much sharper (smaller full width at half maximum and, thus, longer coherence length along the stacking direction) for
CDT-BTZ-C14,10, although the alkyl substituents are considerably extended in comparison to
CDT-BTZ-C16 and
CDT-BTZ-C8,2. The relative isotropic reflection corresponding to a spacing of 0.49 nm is related to the alkyl side chains. Typically, the alkyl chains are disordered in the periphery of the lamellas and contribute only to an amorphous halo in the 2D pattern. In the case of
CDT-BTZ-C14,10, the distinct reflection indicates a higher order of crystallized substituents, despite their bulkiness. As expected, the chain-to-chain distance between lamellae depends on the length of the side chains and is derived from the equatorial small-angle reflections to be only 1.60 nm for
CDT-BTZ-C14,10 in comparison to 2.70 nm for
CDT-BTZ-C16. Additional 2DSAXS measurements for
CDT-BTZ-C14,10 did not expose further reflections related to larger d-spacings. The dramatically reduced chain-to-chain distance for this polymer can only be attributed to the crystallized, tightly packed side chains in the backbone periphery.
Figure 2.
UV-Vis absorption spectra for thin films of CDT-BTZ polymers with different side chains.
Figure 2.
UV-Vis absorption spectra for thin films of CDT-BTZ polymers with different side chains.
The influence of the side chain crystallization on the microstructure of solution processed thin layers was studied by atomic force microscopy (AFM). The images display significant differences between
CDT-BTZ-C14,10 and
CDT-BTZ-C8,2 or
CDT-BTZ-C16, the two latter polymers assembling into randomly oriented nanofibers (
Figure 3d) [
13]. This stands in contrast to
CDT-BTZ-C14,10 being organized in larger, more distinct domains with higher crystallinity (
Figure 3c). The surface roughness is higher in comparison to the polymer with linear side chains. The increased crystallinity of the thin film is in agreement with the bulk organization.
Figure 3.
(a) Two-dimensional wide-angle X-ray scattering (2DWAXS) pattern (dashed line indicates the equatorial integration; reflections attributed to polymer stacking are marked by dashed circles) of CDT-BTZ-C14,10; (b) equatorial integration with characteristic d-spacings; (c) tapping mode AFM height image of thin film; (d) tapping mode AFM height image of CDT-BTZ-C16.
Figure 3.
(a) Two-dimensional wide-angle X-ray scattering (2DWAXS) pattern (dashed line indicates the equatorial integration; reflections attributed to polymer stacking are marked by dashed circles) of CDT-BTZ-C14,10; (b) equatorial integration with characteristic d-spacings; (c) tapping mode AFM height image of thin film; (d) tapping mode AFM height image of CDT-BTZ-C16.
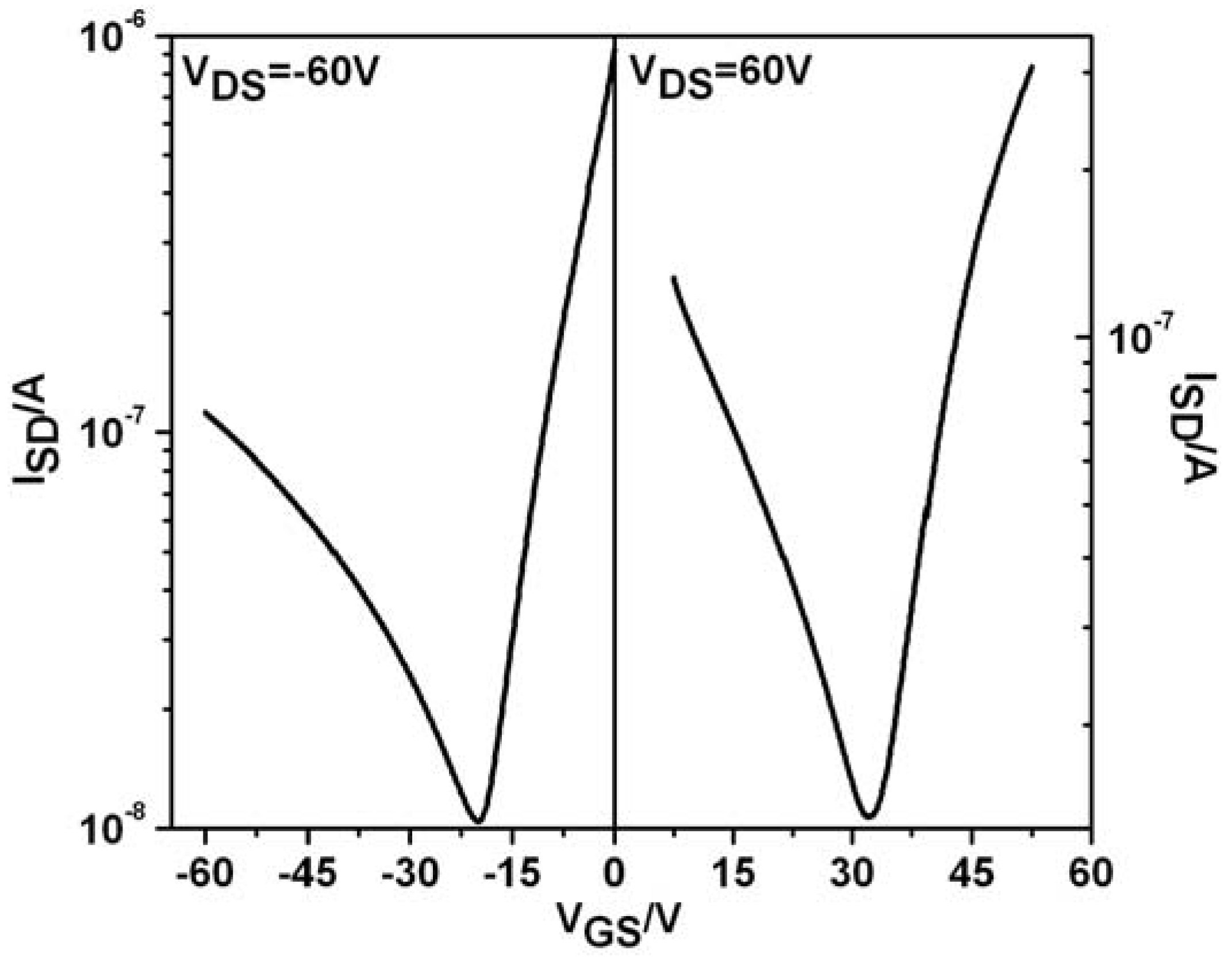
To probe the charge-carrier transport of
CDT-BTZ-C14,10, thin film field-effect transistors were investigated. Bottom-gate, bottom-contact HMDS-treated devices were fabricated by drop-casting the polymer from a 2 mg/mL solution with subsequent annealing at 200 °C for 30 minutes. In stark contrast to the
CDT-BTZ polymers with linear side chains, the mobility of
CDT-BTZ-C14,10 decreases drastically with several orders of magnitude. This drop in device performance is directly related to the increase in π-stacking distance observed by 2DWAXS. Surprisingly, the
CDT-BTZ polymer changes its operation behavior from unipolar (only holes) for the
C16 and
C8,2 substituted ones to ambipolar (electron and holes) for the derivative with
C14,10 chains. Such a side chain-dependent switch in field-effect response for the same conjugated backbone has not been reported, so far. From the transfer curves of both
p-type and for
n-type operation modes for the positive and negative gate voltages mobilities of μ
e = 6 × 10
−4 cm²/Vs and μ
h = 6 × 10
−5 cm²/Vs are determined for
CDT-BTZ-C14,10 (
Figure 4). The symmetric transfer characteristics in the saturation regime confirm a quite balanced transport of hole and electron carriers. The crossover point from electron- to hole-dominated current is approximately V
GS = −20 V at V
DS = −60 V. Below this gate voltage, the transistor shows a typical
p-type behavior in the accumulation mode. On the other hand, at V
DS = 60 V, an electron domination and
n-type properties of the device are apparent from V
g > 30 V.
Figure 4.
Transfer field-effect transistor (FET) curves for CDT-BTZ-C14,10.
Figure 4.
Transfer field-effect transistor (FET) curves for CDT-BTZ-C14,10.
Since the electrochemical energy levels do not differ between the three polymers and the devices were prepared under the same conditions, other reasons for the ambipolar transport for
CDT-BTZ-C14,10 must be taken into account. For instance, changes in the microstructure can lead from ambipolar to unipolar (or
vice versa) device operation without the modification of the energy levels [
10]. In the case of
CDT-BTZ, the polymer with long and branched side chains (
C14,10) possesses a significantly larger π-stacking distance, influencing the electron migration through the active layer. It has also to be noted that the substituents might induce a lateral shift of the backbones with respect to each other, leading to a variation in the π-orbital overlap between the donor and acceptor units. In this case, X-ray scattering does not provide detailed information about the donor-acceptor arrangement, because of insufficient polymer ordering.
To characterize the local molecular arrangements of donor (CDT) and acceptor (BTZ) groups in
CDT-BTZ-C14,10, solid-state NMR experiments in combination with DFT and nucleus-independent chemical shift (NICS) calculations have been performed. Solid-state NMR is a unique method that offers access to molecular packing details for polymers and other organic-based materials [
29,
30,
31,
32,
33] and to details about the donor-acceptor packing in
CDT-BTZ polymers [
34,
35]. For
CDT-BTZ-C16, we were able to show from a combination of X-ray diffraction and solid-state NMR that the
CDT-BTZ polymer chains self-assemble into π-stacks, where the single polymer chain includes alternating CDT units, pointing in opposite directions (packing in a staggered fashion) and BTZ moieties stacked in an antiparallel configuration along the π-stacking direction [
13]. Thus, where X-ray diffraction techniques only provide information about the long-range ordering of semi-crystalline polymers, solid-state NMR gives access to complementary information on the molecular scale [
36]. In π-conjugated polymers, the sensitivity of solid-state NMR towards molecular properties relies strongly on the chemical environments, where, specifically, the influence of aromatic and anti-aromatic ring currents of neighboring π-conjugated polymer chains lead to significant high-field and low-field shifts of δ
iso for
1H, respectively [
37]. To quantify the impact of such ring currents on the
1H chemical shift caused by π-stacking of
CDT-BTZ polymer chains, we have used NICS calculations [
38,
39].
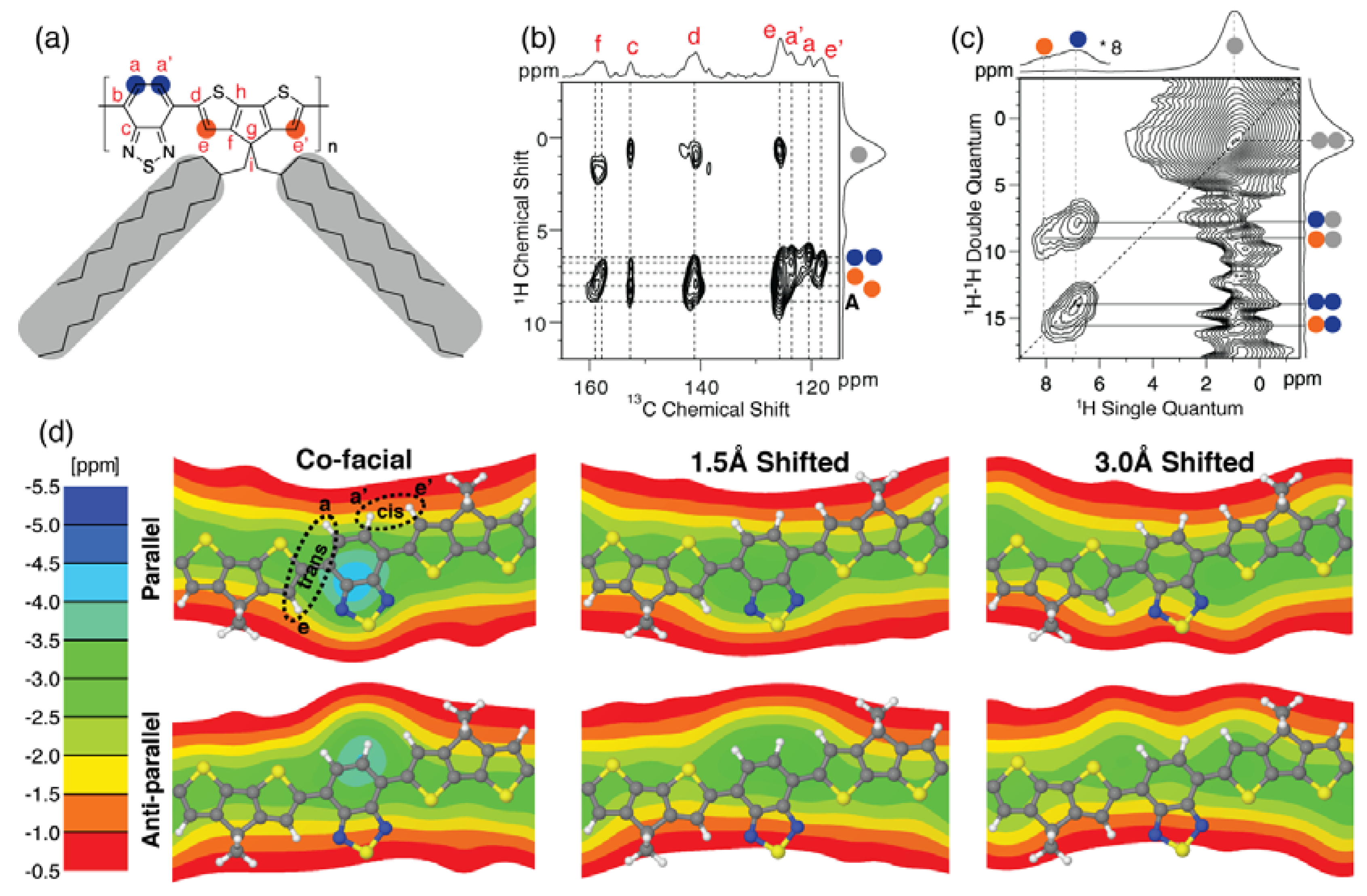
Figure 5a,b,c show the resulting 2D
13C{
1H} FSLG-HETCOR and 2D
1H–
1H DQ-SQ correlation spectra recorded for
CDT-BTZ-C14,10. As illustrated in
Figure 5d, the
CDT-BTZ-C14,10 polymer can include both
cis and
trans conformations of CDT and BTZ groups within a single polymer chain. These conformations can be distinguished via the
1H–
13C correlation signals in
Figure 5b. All four
1H resonances associated with the CDT and BTZ groups fall within the range from 6.4 to 8.0 ppm, resulting in the narrow
1H–
1H correlation range centered at the diagonal in
Figure 5c. The
1H resonances in liquid-state NMR are in the range 8.0–8.2 ppm. Nevertheless, the
1H spectral features in
Figure 5c for
CDT-BTZ-C14,10 show similarities to our recent results reported for
CDT-BTZ-C16, where the
1H signals from the
1H–
1H BTZ and CDT correlations centered at the diagonal are more elongated and go down to 5.5 ppm [
13,
34,
35]. For
CDT-BTZ-C14,10, the
1H–
1H correlations at the diagonal only goes to 6.4 ppm. Thus, the main difference between the two
CDT-BTZ-C14,10 and
CDT-BTZ-C16 polymers is the reduced high-field shift for the polymer backbone protons of
CDT-BTZ-C14. As revealed from the 2DWAXS experiments above, the π-stacking distance of
CDT-BTZ-C14,10 is 0.3 Å larger than for
CDT-BTZ-C16. This implies that the BTZ and CDT protons in
CDT-BTZ-C14,10 will experience less magnetic shielding from the stacked BTZ-CDT main chains as compared to
CDT-BTZ-C16.
To relate the high-field shift of the
1H resonances to the local polymer chain packing in
CDT-BTZ-C14,10, we have performed NICS map calculations for a number of model systems with a π-stacking distance of 4.0 Å, as shown in
Figure 5d. These models include co-facial stacking of parallel and anti-parallel BTZ acceptors in addition to a translation (or shift) of every
CDT-BTZ-C14,10 polymer chain by 1.5 Å and 3.0 Å. The NICS maps shown in
Figure 5d, in general, confirm that the main origin of the lower high-field shift for
CDT-BTZ-C14,10 is a result of an increase in π-stacking distance. All three packing models with parallel-stacked BTZ acceptors (
Figure 5d, upper row) show that the
cis and
trans conformations of BTZ and CDT groups only result in a narrow range of high-field shifts of 1.0–2.0 ppm. For the anti-parallel stacking of BTZ acceptors (
Figure 5d, lower row), the spread is larger, giving high-field shifts in the range 1.0–4.0 ppm. These differences in high-field shifts between parallel and antiparallel stacking of BTZ acceptors show that the main polymer chain packing of
CDT-BTZ-C14,10 includes anti-parallel BTZ acceptor groups as also determined for
CDT-BTZ-C16 [
13]. In the co-facial scenario with anti-parallel stacked BTZ acceptors in
Figure 5d, the
1H chemical shifts of the acceptor group are shifted to high-field by 3–4 ppm. This shift is too large, since the experimental high-field shift for
CDT-BTZ-C14,10 is only 0.2–1.8 ppm. For the translational models in
Figure 5d, the high-field shift is lower and of similar magnitude as the experimental
1H high-field shifts, illustrating that the packing of
CDT-BTZ-C14,10 polymer chains includes a translational shift in the range between 1.0 and 3.0 Å.
To correlate the translational shift between π-stacked
CDT-BTZ-C14,10 polymer chains to the charge-carrier transport properties, we compare the current results of the calculated electronic coupling in terms of the charge-transfer integral between stacked polymer chains in
CDT-BTZ-C16 [
35]. To recall, these calculations showed a cosine-like behavior of the HOMO-HOMO (hole-) and LUMO-LUMO (electron-) transfer integrals as a function of the translational shift between
CDT-BTZ polymer chains with local minima at −1.0 and −1.8 Å, respectively. Both transfer integrals have a local maximum at 0.0 Å, corresponding to co-facially stacked acceptors, in addition to a local maximum at −2.0 Å for the HOMO-HOMO overlap (see Ref. 35 Figure 7). Further, the amplitude and period of the HOMO-HOMO charge-transfer integral is larger and shorter, respectively, compared to the LUMO-LUMO charge-transfer integral. The different shapes of the HOMOs and LUMOs and their corresponding wave-function overlap can explain these differences in amplitude and period: the HOMOs are strongly localized on the BTZ acceptor groups, whereas the LUMOs are spread along the polymer backbone [
35]. Although the charge-transfer integral calculations were performed at a shorter π-stacking distance of 3.6 Å for
CDT-BTZ-C16 [
35], we expect the trends to be comparable for
CDT-BTZ-C14,10 at a stacking distance of 4.0 Å, however, with a reduced charge-transfer integral. Thus, the origin of the ambipolarity for
CDT-BTZ-C14,10 is a result of increased donor-acceptor interactions caused by sliding of the CDT-BTZ polymer chains away from co-facially stacked BTZ acceptors. This leads to a significantly reduced hole mobility as compared to
CDT-BTZ-C16, but with an increased electron mobility.
Figure 5.
(
a) Chemical structure and assignment scheme used for
CDT-BTZ-C14,10; (
b) 2D
13C{
1H} frequency-switched Lee-Goldburg heteronuclear correlation (FSLG-HETCOR) obtained using a cross polarization (CP) contact time of 3.0 ms; (
c) 2D
1H-
1H DQ-SQ correlation spectrum recorded using the BaBa pulse sequence with one rotor period of dipolar recoupling (50.0 μs) [
16]; (
d) nucleus-independent chemical shift (NICS) maps calculated for co-facial stacking of parallel and anti-parallel
BTZ acceptors in combination with shift/translation of every polymer chain (as given) in
CDT-BTZ-C14,10 at a π-stacking distance of 4.0 Å. The NICS color bar quantifies the effect of chain packing on the NMR chemical shift induced by the aromatic ring currents of neighboring polymer chains. The
trans (a–c) and
cis (a′–c′) conformations are illustrated in (
d) for the co-facial, parallel acceptors. The amorphous fraction is denoted with an A in (
b). All solid-state NMR spectra were recorded at 20.0 T.
Figure 5.
(
a) Chemical structure and assignment scheme used for
CDT-BTZ-C14,10; (
b) 2D
13C{
1H} frequency-switched Lee-Goldburg heteronuclear correlation (FSLG-HETCOR) obtained using a cross polarization (CP) contact time of 3.0 ms; (
c) 2D
1H-
1H DQ-SQ correlation spectrum recorded using the BaBa pulse sequence with one rotor period of dipolar recoupling (50.0 μs) [
16]; (
d) nucleus-independent chemical shift (NICS) maps calculated for co-facial stacking of parallel and anti-parallel
BTZ acceptors in combination with shift/translation of every polymer chain (as given) in
CDT-BTZ-C14,10 at a π-stacking distance of 4.0 Å. The NICS color bar quantifies the effect of chain packing on the NMR chemical shift induced by the aromatic ring currents of neighboring polymer chains. The
trans (a–c) and
cis (a′–c′) conformations are illustrated in (
d) for the co-facial, parallel acceptors. The amorphous fraction is denoted with an A in (
b). All solid-state NMR spectra were recorded at 20.0 T.




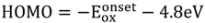


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 eV
eV eV
eV