Polymer Directed Protein Assemblies

Abstract
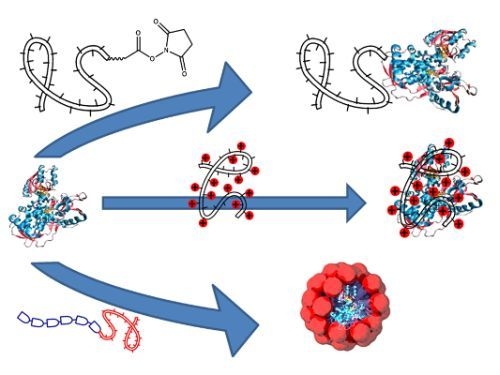
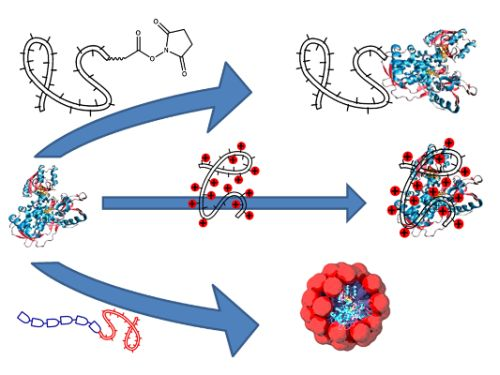
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
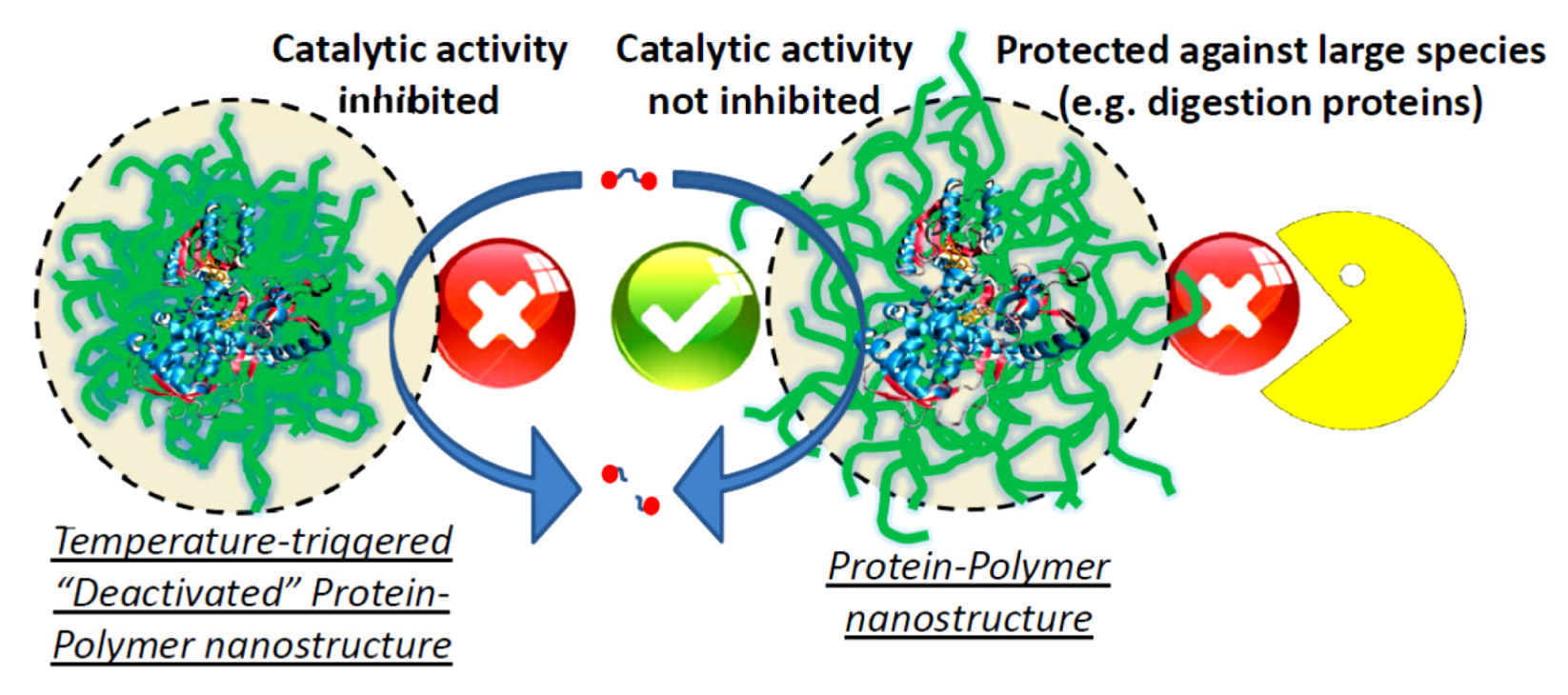{kind=link}
1. Introduction
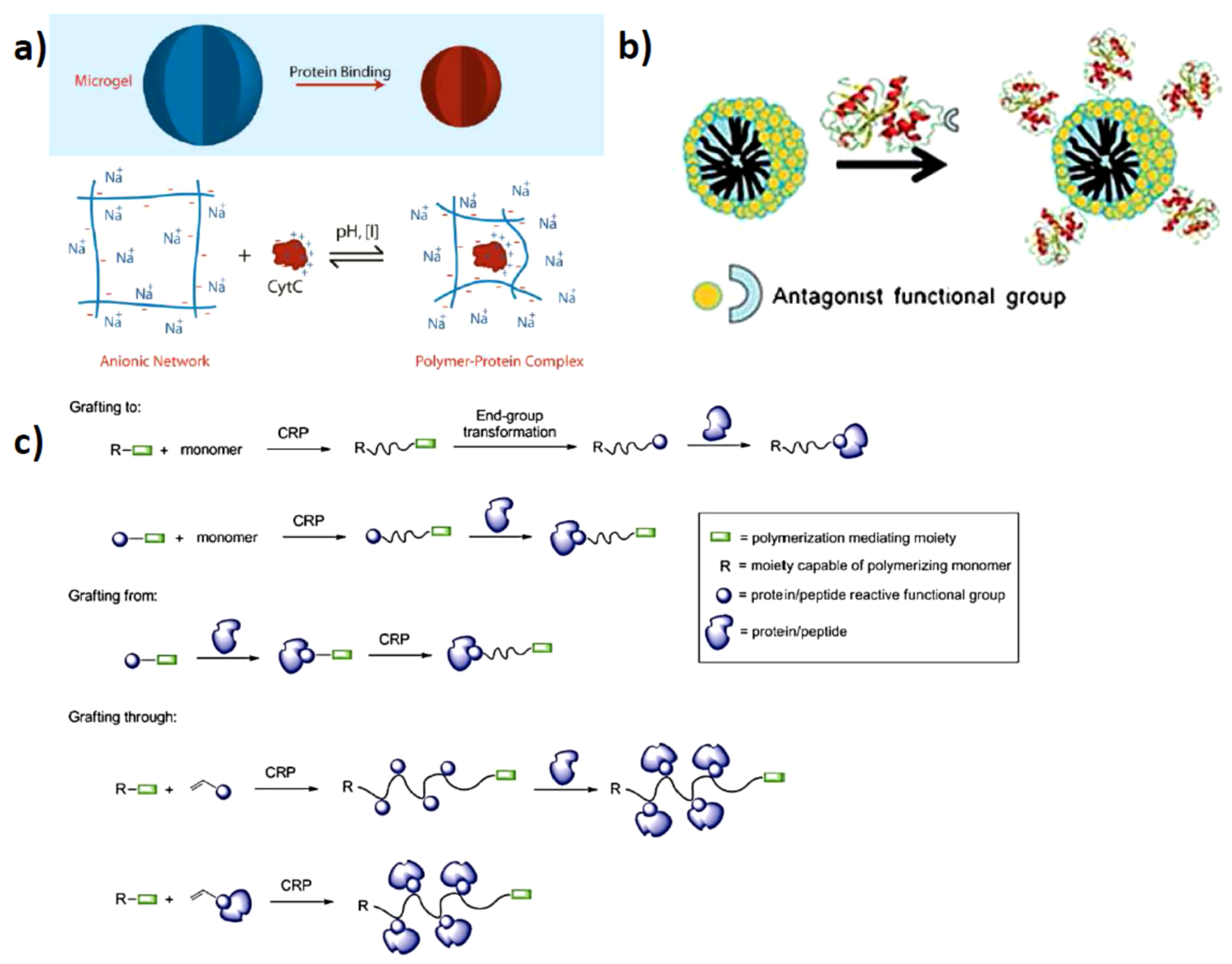
2. Electrostatically Directed Protein-Polymer Assembly
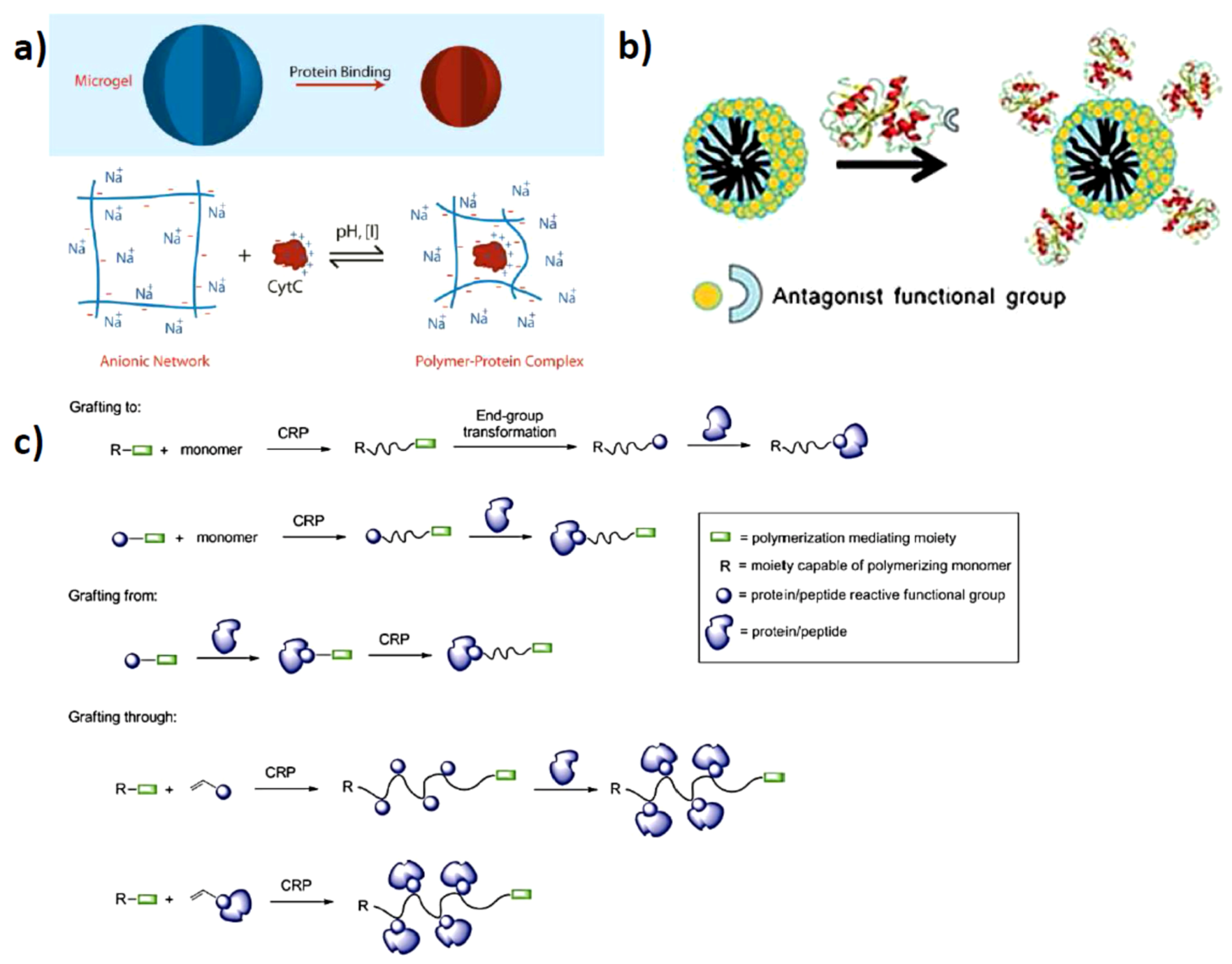
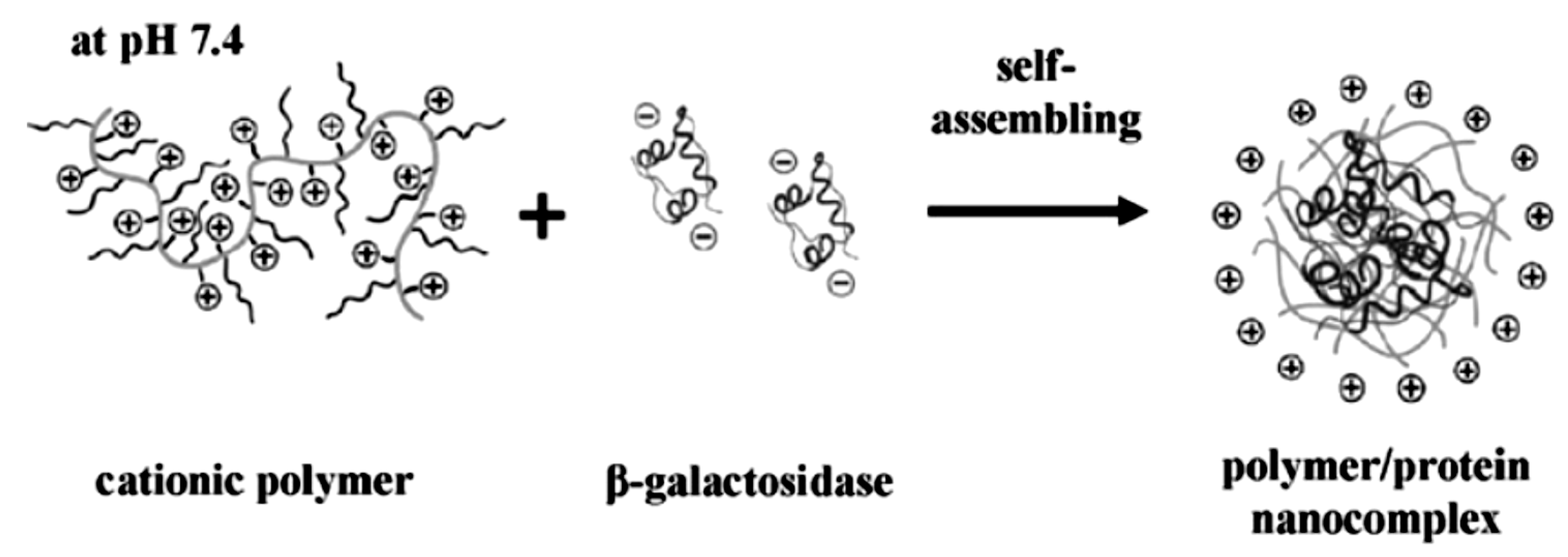
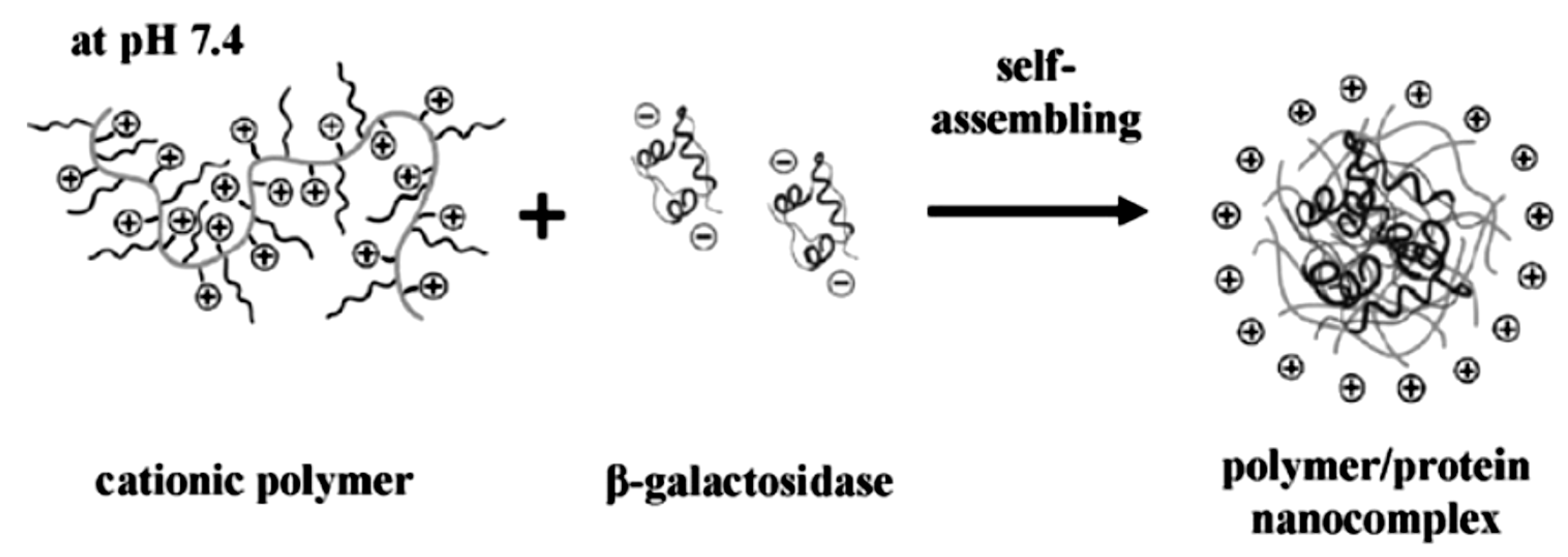
2.1. Solution Assembled Structures via Electrostatics
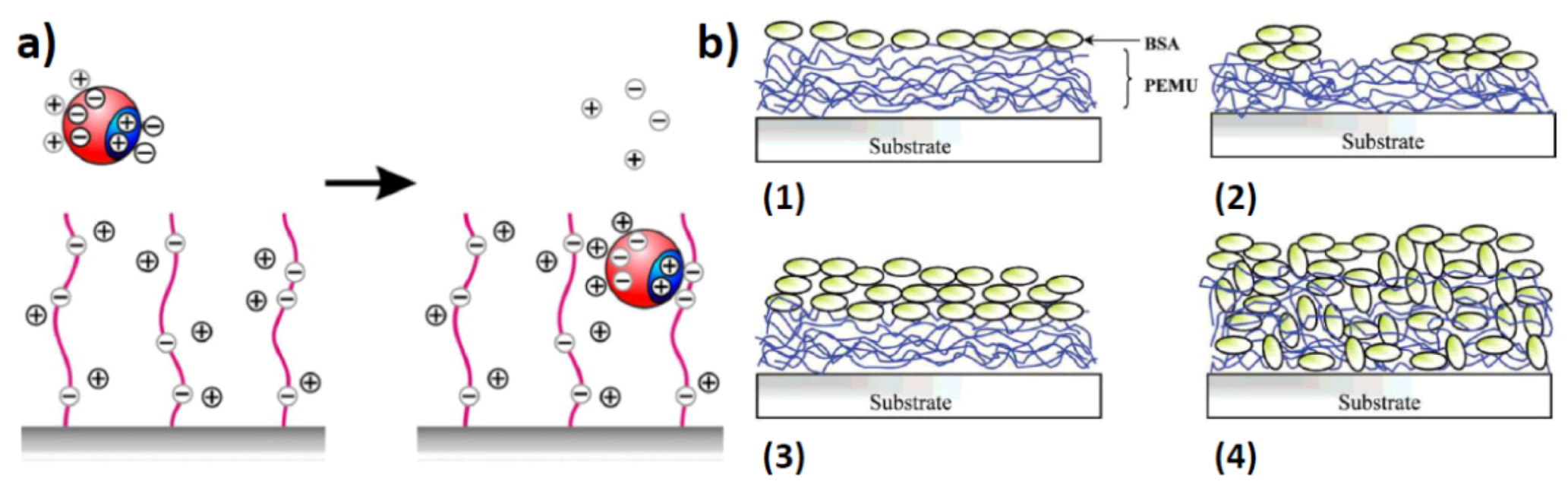
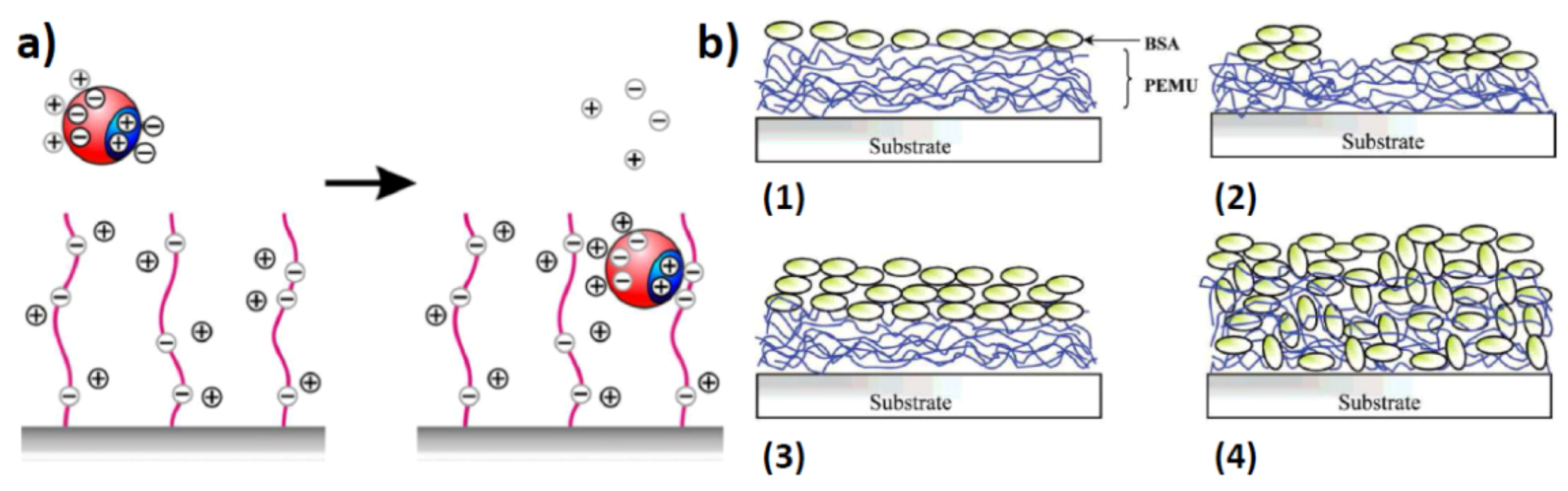
2.2. Surface Assembled Electrostatic Structures
2.3. Artificial Assembly of Virus Particles
3. Directed Assembly of Polymer-Protein Conjugates
3.1. Protein-Polymer Block-Copolymers
3.1.1. Self-Assembly in Phase-Separating Thin Films

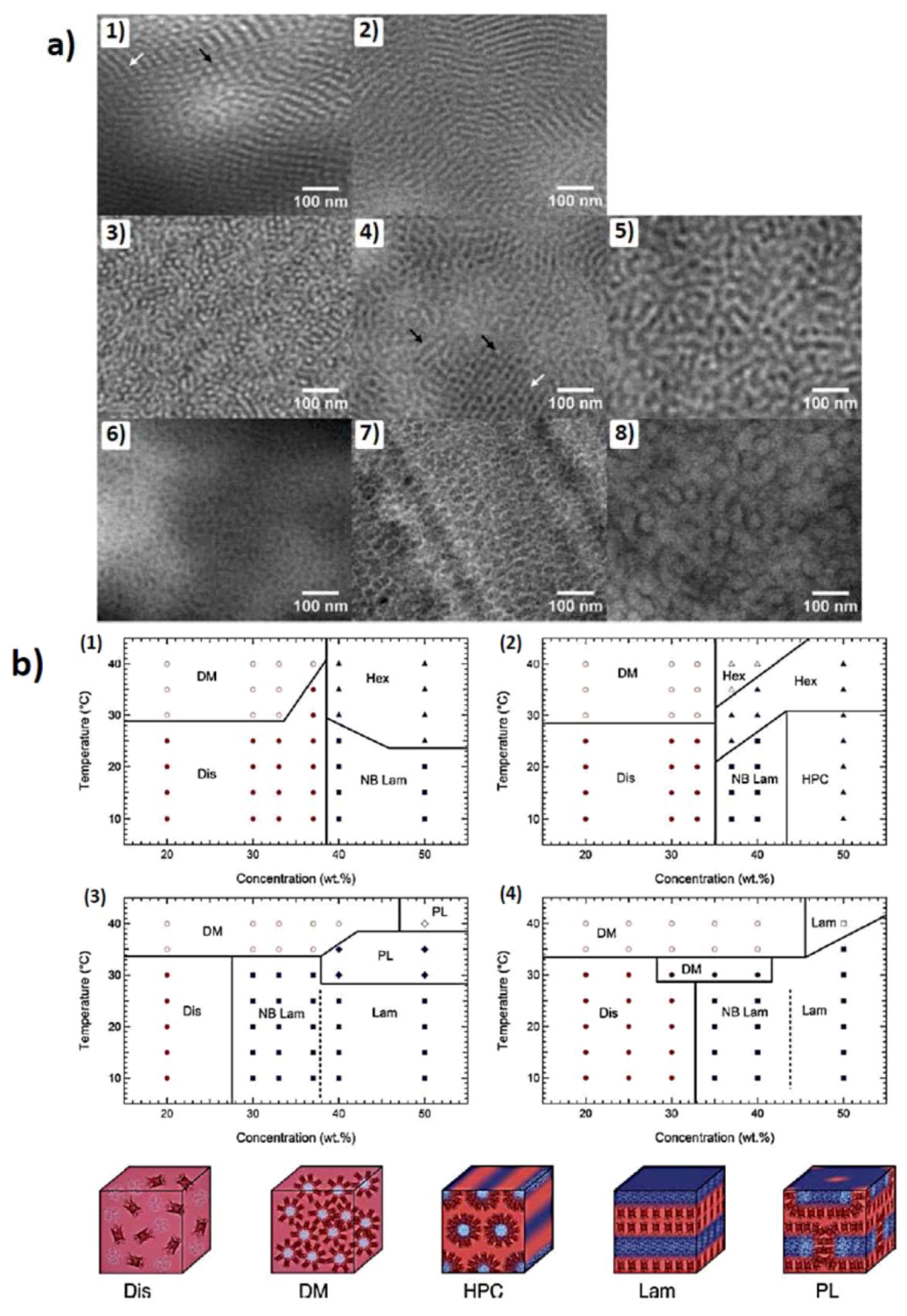
3.1.2. Self-Assembly in Solution
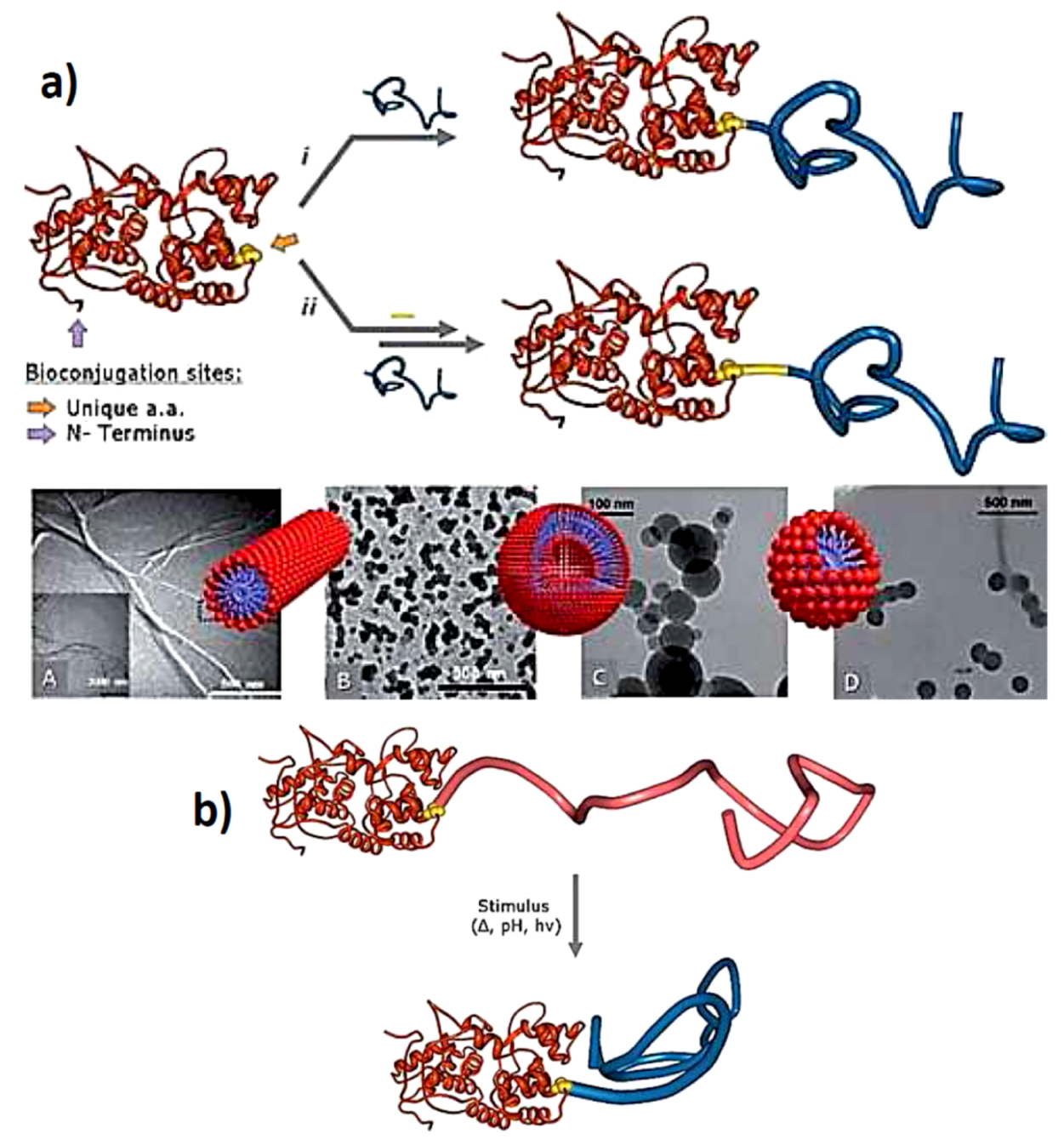
3.2. Protein Centralized Multiple Polymer Grafts
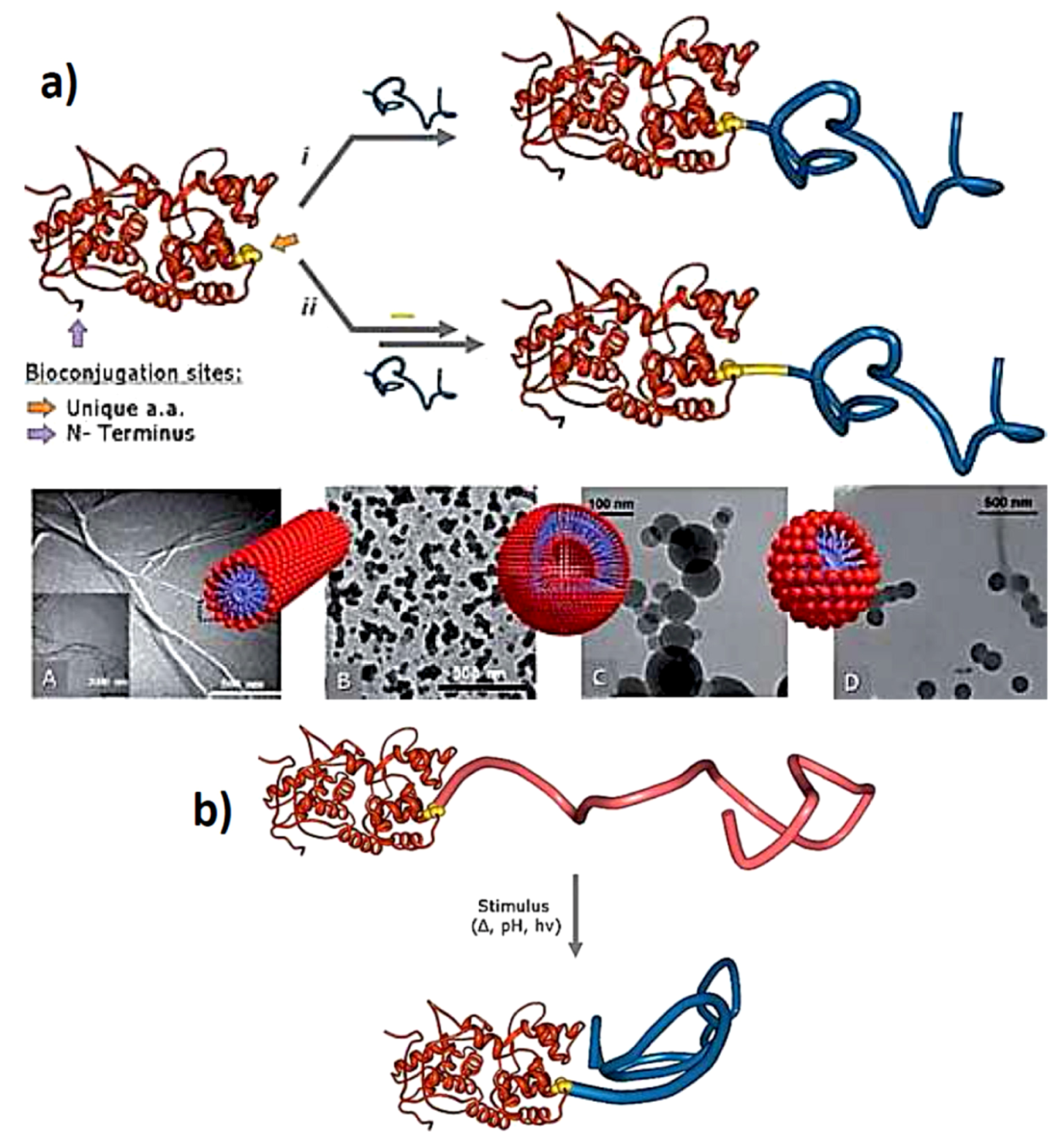
3.3. Virus-Polymer Conjugates
4. Potential Applications of Polymer-Driven Protein Assembly
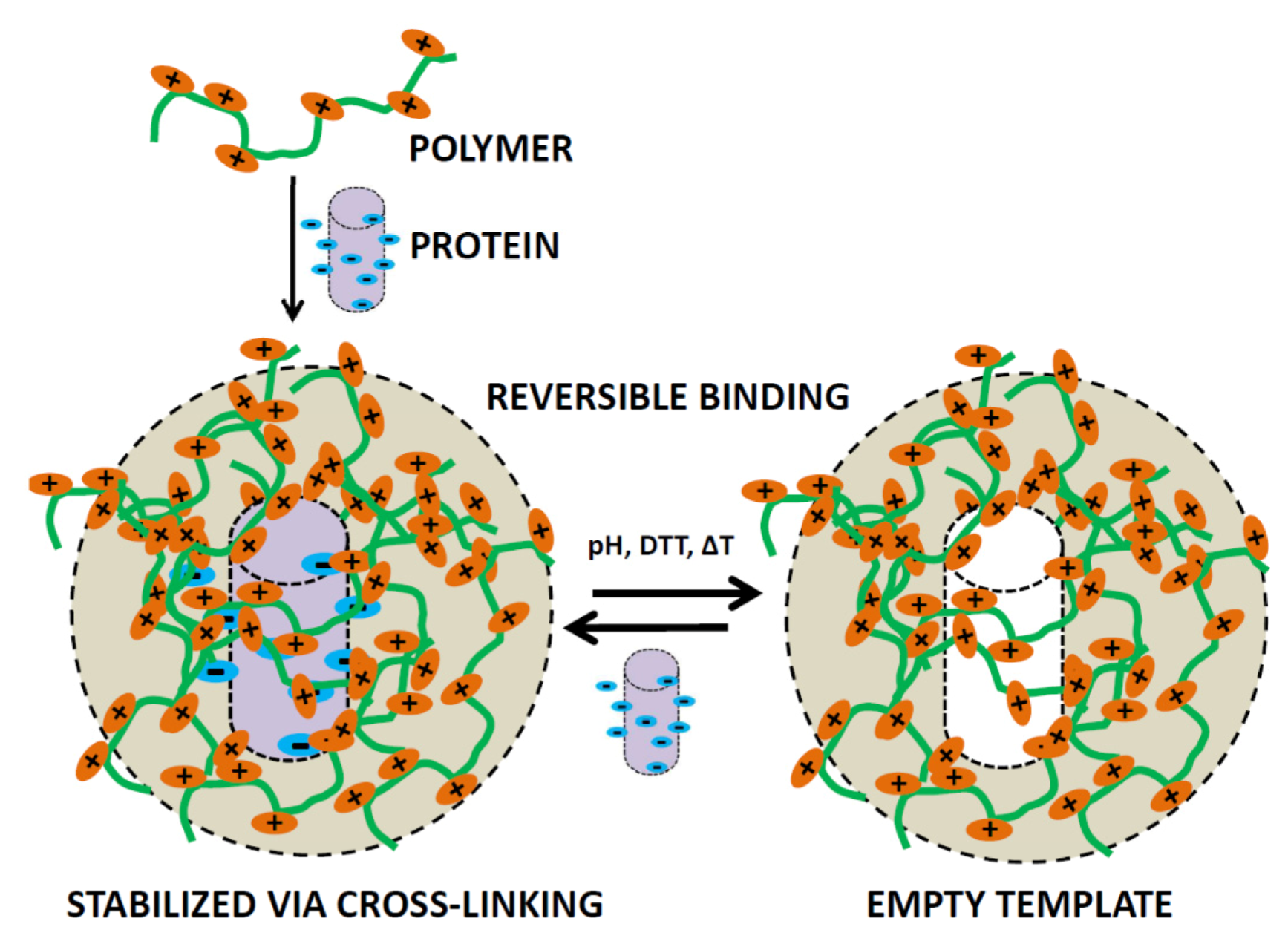
4.1. Protein-Imprinting into Polymer Surfaces

4.2. Amyloid Formation and Prevention
4.3. Therapeutic and Smart Hybrid Bioactive Systems

4.4. Bio-Interfaces
5. Conclusions
Conflict of Interest
References
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Perutz, M.F.; Windle, A.H. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 2001, 412, 143–144. [Google Scholar] [CrossRef]
- Pokorski, J.K.; Steinmetz, N.F. The art of engineering viral nanoparticles. Mol. Pharmaceut. 2011, 8, 29–43. [Google Scholar] [CrossRef]
- van Rijn, P.; Böker, A. Bionanoparticles and hybrid materials: Tailored structural properties, self-assembly, materials and developments in the field. J. Mater. Chem. 2011, 21, 16735–16747. [Google Scholar] [CrossRef]
- Klug, A. The tobacco mosaic virus particle: Structure and assembly. Philos. Trans. R. Soc. B 1999, 354, 531–535. [Google Scholar] [CrossRef]
- Broyer, R.M.; Grover, G.N.; Maynard, H.D. Emerging synthetic approaches for protein-polymer conjugations. Chem. Commun. 2011, 47, 2212–2226. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Klok, H.-A. Polymer-protein conjugates: An enzymatic activity perspective. Polym. Chem. 2010, 1, 1352–1373. [Google Scholar] [CrossRef]
- Vandermeulen, G.W.M.; Klok, H.-A. Peptide/protein hybrid materials: Enhanced control of structure and improved performance through conjugation of biological and synthetic polymers. Macromol. Biosci. 2004, 4, 383–398. [Google Scholar] [CrossRef]
- Theato, P. Chemical strategies for the synthesis of protein—Polymer conjugates. Adv. Polym. Sci. 2013, 253, 37–70. [Google Scholar]
- Heredia, K.L.; Bontempo, D.; Ly, T.; Byers, J.T.; Halstenberg, S.; Maynard, H.D. In situ preparation of protein-“smart” polymer conjugates with retention of bioactivity. J. Am. Chem. Soc. 2005, 127, 16955–16960. [Google Scholar]
- Boyer, C.; Bulmus, V.; Liu, J.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Well-defined protein-polymer conjugates via in situ RAFT polymerization. J. Am. Chem. Soc. 2007, 129, 7145–7154. [Google Scholar]
- Appel, E.A.; del Barrio, J.; Loh, X.J.; Scherman, O.A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 2012, 41, 6195–6214. [Google Scholar] [CrossRef]
- Smith, M.H.; Lyon, L.A. Tunable encapsulation of proteins within charged microgels. Macromolecules 2011, 44, 8154–8160. [Google Scholar] [CrossRef]
- Boyer, C.; Huang, X.; Whittaker, M.R.; Bulmus, V.; Davis, T.P. An overview of protein-polymer particles. Soft Matter 2011, 7, 1599–1614. [Google Scholar] [CrossRef]
- Morimoto, N.; Hirano, S.; Takahashi, H.; Loethen, S.; Thompson, D.H.; Akiyoshi, K. Self-assembled pH-sensitive cholesteryl pullulan nanogel as a protein delivery vehicle. Biomacromolecules 2013, 14, 56–63. [Google Scholar] [CrossRef]
- Koo, J.; Czeslik, C. Probing aggregation and fibril formation of insulin in polyelectrolyte multilayers. Colloids Surf. B 2012, 94, 80–88. [Google Scholar] [CrossRef]
- Mueller, A.; Eber, F.J.; Azucena, C.; Petershans, A.; Bittner, A.M.; Gliemann, H.; Jeske, H.; Wege, C. Inducible site-selective bottom-up assembly of virus-derived nanotube arrays on RNA-equipped wafers. ACS Nano 2011, 5, 4512–4520. [Google Scholar] [CrossRef]
- Seyrek, E.; Dubin, P. Glycosaminoglycans as polyelectrolytes. Adv. Colloid Interface Sci. 2010, 158, 119–129. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. 2002, 41, 391–412. [Google Scholar]
- Kayitmazer, A.B.; Seeman, D.; Minsky, B.B.; Dubin, P.L.; Xu, Y. Protein-polyelectrolyte interactions. Soft Matter 2013, 9, 2553–2583. [Google Scholar] [CrossRef]
- Coué, G.; Engbersen, J.F.J. Functionalized linear poly(amidoamine)s are efficient vectors for intracellular protein delivery. J. Control. Release 2011, 152, 90–98. [Google Scholar] [CrossRef]
- Tsiourvas, D.; Sideratou, Z.; Sterioti, N.; Papadopoulos, A.; Nounesis, G.; Paleos, C.M. Insulin complexes with PEGylated basic oligopeptides. J. Colloid Interface Sci. 2012, 384, 61–72. [Google Scholar] [CrossRef]
- Kurinomaru, T.; Tomita, S.; Kudo, S.; Ganguli, S.; Nagasaki, Y.; Shiraki, K. Improved complementary polymer pair system: Switching for enzyme activity by PEGylated polymers. Langmuir 2012, 28, 4334–4338. [Google Scholar] [CrossRef]
- Wilson, L.; Illanes, A.; Abián, O.; Pessela, B.C.C.; Fernández-Lafuente, R.; Guisán, J.M. Co-aggregation of penicillin g acylase and polyionic polymers: An easy methodology to prepare enzyme biocatalysts stable in organic media. Biomacromolecules 2004, 5, 852–857. [Google Scholar] [CrossRef]
- Kubiak-Ossowska, K.; Mulheran, P. Protein diffusion and long-term adsorption states at charged solid surfaces. Langmuir 2012, 28, 15577–15585. [Google Scholar] [CrossRef]
- Kubiak-Ossowska, K.; Mulheran, P. Mechanism of hen egg white lysozyme adsorption on a charged solid surface. Langmuir 2010, 26, 15954–15965. [Google Scholar] [CrossRef]
- Yang, Z.; Galloway, J.A.; Yu, H. Protein interactions with poly(ethylene glycol) self-assembled monolayers on glass substrates: Diffusion and adsorption. Langmuir 1999, 15, 8405–8411. [Google Scholar] [CrossRef]
- Wang, X.; Wu, G.; Lu, C.; Wang, Y.; Fan, Y.; Gao, H.; Ma, J. Synthesis of a novel zwitterionic biodegradable poly (α,β-L-aspartic acid) derivative with some L-histidine side-residues and its resistance to non-specific protein adsorption. Colloids Surf. B 2011, 86, 237–241. [Google Scholar] [CrossRef]
- Uhlmann, P.; Houbenov, N.; Brenner, N.; Grundke, K.; Burkert, S.; Stamm, M. In-situ investigation of the adsorption of globular model proteins on stimuli-responsive binary polyelectrolyte brushes. Langmuir 2007, 23, 57–64. [Google Scholar] [CrossRef]
- Becker, A.L.; Henzler, K.; Welsch, N.; Ballauff, M.; Borisov, O. Proteins and polyelectrolytes: A charged relationship. Curr. Opin. Colloid Interface Sci. 2012, 17, 90–96. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Sun, J. Layer-by-layer assembly for rapid fabrication of thick polymeric films. Chem. Soc. Rev. 2012, 41, 5998–6009. [Google Scholar] [CrossRef]
- Salloum, D.S.; Schlenoff, J.B. Protein adsorption modalities on polyelectrolyte multilayers. Biomacromolecules 2004, 5, 1089–1096. [Google Scholar] [CrossRef]
- Delcroix, M.F.; Huet, G.L.; Conard, T.; Demoustier-Champagne, S.; du Prez, F.E.; Landoulsi, J.; Dupont-Gillain, C.C. Design of mixed PEO/PAA brushes with switchable properties toward protein adsorption. Biomacromolecules 2013, 14, 215–225. [Google Scholar] [CrossRef]
- Kim, B.; Lam, C.N.; Olsen, B.D. Nanopatterned protein films directed by ionic complexation with water-soluble diblock copolymers. Macromolecules 2012, 45, 4572–4580. [Google Scholar] [CrossRef]
- Mukherjee, S.; Pfeifer, C.M.; Johnson, J.M.; Liu, J.; Zlotnick, A. Redirecting the coat protein of a spherical virus to assemble into tubular nanostructures. J. Am. Chem. Soc. 2006, 128, 2538–2539. [Google Scholar] [CrossRef]
- Cadena-Nava, R.D.; Hu, Y.; Garmann, R.F.; Ng, B.; Zelikin, A.N.; Knobler, C.M.; Gelbart, W.M. Exploiting fluorescent polymers to probe the self-assembly of virus-like particles. J. Phys. Chem. B 2011, 115, 2386–2391. [Google Scholar]
- Hu, Y.; Zandi, R.; Anavitarte, A.; Knobler, C.M.; Gelbart, W.M. Packaging of a polymer by a viral capsid: The interplay between polymer length and capsid size. Biophys. J. 2008, 94, 1428–1436. [Google Scholar] [CrossRef]
- Ng, B.C.; Chan, S.T.; Lin, J.; Tolbert, S.H. Using polymer conformation to control architecture in semiconducting polymer/viral capsid assemblies. ACS Nano 2011, 5, 7730–7738. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Hiekkataipale, P.; de la Torre, J.Á.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Electrostatic self-assembly of virus–polymer complexes. J. Mater. Chem. 2011, 21, 2112–2117. [Google Scholar] [CrossRef]
- Grover, G.N.; Maynard, H.D. Protein-polymer conjugates: Synthetic approaches by controlled radical polymerizations and interesting applications. Curr. Opin. Chem. Biol. 2010, 14, 818–827. [Google Scholar] [CrossRef]
- Discher, D.E.; Ahmed, F. Polymersomes. Ann. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef]
- Horn, A.; Hiltl, S.; Fery, A.; Böker, A. Ordering and printing virus arrays: A straightforward way to functionalize surfaces. Small 2010, 6, 2122–2125. [Google Scholar] [CrossRef]
- Chen, C.-H.; Yang, K.-L. Improving protein transfer efficiency and selectivity in affinity contact printing by using UV-modified surfaces. Langmuir 2011, 27, 5427–5432. [Google Scholar] [CrossRef]
- Kumar, N.; Hahm, J. Nanoscale protein patterning using self-assembled diblock copolymers. Langmuir 2005, 21, 6652–6655. [Google Scholar] [CrossRef]
- Kumar, N.; Parajuli, O.; Hahm, J.-I. Two-dimensionally self-arranged protein nanoarrays on diblock copolymer templates. J. Phys. Chem. B 2007, 111, 4581–4587. [Google Scholar] [CrossRef]
- Thomas, C.S.; Glassman, M.J.; Olsen, B.D. Solid-state nanostructured materials from self-assembly of a globular protein-polymer diblock copolymer. ACS Nano 2011, 5, 5697–5707. [Google Scholar] [CrossRef]
- Thomas, C.S.; Xu, L.; Olsen, B.D. Kinetically controlled nanostructure formation in self-assembled globular protein-polymer diblock copolymers. Biomacromolecules 2012, 13, 2781–2792. [Google Scholar] [CrossRef]
- Lam, C.N.; Olsen, B.D. Phase transitions in concentrated solution self-assembly of globular protein–polymer block copolymers. Soft Matter 2013, 9, 2393–2402. [Google Scholar] [CrossRef]
- Velonia, K. Protein-polymer amphiphilic chimeras: Recent advances and future challenges. Polym. Chem. 2010, 1, 944–952. [Google Scholar] [CrossRef]
- Velonia, K.; Rowan, A.E.; Nolte, R.J.M. Lipase polystyrene giant amphiphiles. J. Am. Chem. Soc. 2002, 124, 4224–4225. [Google Scholar] [CrossRef]
- Lavigueur, C.; García, J.G.; Hendriks, L.; Hoogenboom, R.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Thermoresponsive giant biohybrid amphiphiles. Polym. Chem. 2011, 2, 333–340. [Google Scholar] [CrossRef]
- le Droumaguet, B.; Mantovani, G.; Haddleton, D.M.; Velonia, K. Formation of giant amphiphiles by post-functionalization of hydrophilic protein-polymer conjugates. J. Mater. Chem. 2007, 17, 1916–1922. [Google Scholar] [CrossRef]
- Daskalaki, E.; le Droumaguet, B.; Gérard, D.; Velonia, K. Multifunctional Giant Amphiphiles via simultaneous copper(I)-catalyzed azide-alkyne cycloaddition and living radical polymerization. Chem. Commun. 2012, 48, 1586–1588. [Google Scholar]
- Boerakker, M.J.; Hannink, J.M.; Bomans, P.H.H.; Frederik, P.M.; Nolte, R.J.M.; Meijer, E.M.; Sommerdijk, N.A.J.M. Giant amphiphiles by cofactor reconstitution. Angew. Chem. Int. Ed. 2002, 41, 4239–4241. [Google Scholar] [CrossRef]
- Reynhout, I.C.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Self-assembled architectures from biohybrid triblock copolymers. J. Am. Chem. Soc. 2007, 129, 2327–2332. [Google Scholar] [CrossRef]
- Oohora, K.; Onoda, A.; Kitagishi, H.; Yamaguchi, H.; Harada, A.; Hayashi, T. A chemically-controlled supramolecular protein polymer formed by a myoglobin-based self-assembly system. Chem. Sci. 2011, 2, 1033–1038. [Google Scholar] [CrossRef]
- Yaşayan, G.; Saeed, A.O.; Fernández-Trillo, F.; Allen, S.; Davies, M.C.; Jangher, A.; Paul, A.; Thurecht, K.J.; King, S.M.; Schweins, R.; et al. Responsive hybrid block co-polymer conjugates of proteins-controlled architecture to modulate substrate specificity and solution behaviour. Polym. Chem. 2011, 2, 1567–1578. [Google Scholar] [CrossRef]
- Böker, A.; He, J.; Emrick, T.; Russell, T.P. Self-assembly of nanoparticles at interfaces. Soft Matter 2007, 3, 1231–1248. [Google Scholar] [CrossRef]
- Kaur, G.; He, J.; Xu, J.; Pingali, S.; Jutz, G.; Böker, A.; Niu, Z.; Li, T.; Rawlinson, D.; Emrick, T.; et al. Interfacial assembly of turnip yellow mosaic virus nanoparticles. Langmuir 2009, 25, 5168–5176. [Google Scholar] [CrossRef]
- He, J.; Niu, Z.; Tangirala, R.; Wang, J.-Y.; Wei, X.; Kaur, G.; Wang, Q.; Jutz, G.; Böker, A.; Lee, B.; et al. Self-assembly of tobacco mosaic virus at oil/water interfaces. Langmuir 2009, 25, 4979–4987. [Google Scholar] [CrossRef]
- Schulz, A.; Fioroni, M.; Linder, M.B.; Nessel, A.; Bocola, M.; Subkowski, T.; Schwaneberg, U.; Böker, A.; Rodríguez-Ropero, F. Exploring the mineralization of hydrophobins at a liquid interface. Soft Matter 2012, 8, 11343–11352. [Google Scholar] [CrossRef]
- Pai, S.S.; Przybycien, T.M.; Tilton, R.D. Protein PEGylation attenuates adsorption and aggregation on a negatively charged and moderately hydrophobic polymer surface. Langmuir 2010, 26, 18231–18238. [Google Scholar] [CrossRef]
- Hu, Y.; Samanta, D.; Parelkar, S.S.; Hong, S.W.; Wang, Q.; Russell, T.P.; Emrick, T. Ferritin-polymer conjugates: Grafting chemistry and integration into nanoscale assemblies. Adv. Funct. Mater. 2010, 20, 3603–3612. [Google Scholar] [CrossRef]
- Mougin, N.C.; van Rijn, P.; Park, H.; Müller, A.H.E.; Böker, A. Hybrid capsules via self-assembly of thermoresponsive and interfacially active bionanoparticle-polymer conjugates. Adv. Funct. Mater. 2011, 21, 2470–2476. [Google Scholar] [CrossRef]
- van Rijn, P.; Park, H.; Özlem Nazli, K.; Mougin, N.C.; Böker, A. Self-assembly process of soft ferritin-PNIPAAm conjugate bionanoparticles at polar-apolar interfaces. Langmuir 2013, 29, 276–284. [Google Scholar] [CrossRef]
- van Rijn, P.; Mougin, N.C.; Franke, D.; Park, H.; Böker, A. Pickering emulsion templated soft capsules by self-assembling cross-linkable ferritin-polymer conjugates. Chem. Commun. 2011, 47, 8376–8378. [Google Scholar]
- van Rijn, P.; Mougin, N.C.; Böker, A. Hierarchical structures via self-assembling protein-polymer hybrid building blocks. Polymer 2012, 53, 6045–6052. [Google Scholar] [CrossRef]
- Liu, Z.; Qiao, J.; Niu, Z.; Wang, Q. Natural supramolecular building blocks: From virus coat proteins to viral nanoparticles. Chem. Soc. Rev. 2012, 41, 6178–6194. [Google Scholar] [CrossRef]
- Lee, L.A.; Niu, Z.; Wang, Q. Viruses and virus-like protein assemblies—Chemically programmable nanoscale building blocks. Nano Res. 2010, 2, 349–364. [Google Scholar]
- de la Escosura, A.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Viruses and protein cages as nanocontainers and nanoreactors. J. Mater. Chem. 2009, 19, 2274–2278. [Google Scholar] [CrossRef]
- Holder, P.G.; Finley, D.T.; Stephanopoulos, N.; Walton, R.; Clark, D.S.; Francis, M.B. Dramatic thermal stability of virus-polymer conjugates in hydrophobic solvents. Langmuir 2010, 26, 17383–17388. [Google Scholar]
- Manzenrieder, F.; Luxenhofer, R.; Retzlaff, M.; Jordan, R.; Finn, M.G. Stabilization of virus-like particles with poly(2-oxazoline)s. Angew. Chem. Int. Ed. 2011, 50, 2601–2605. [Google Scholar] [CrossRef]
- Pokorski, J.K.; Breitenkamp, K.; Liepold, L.O.; Qazi, S.; Finn, M.G. Functional virus-based polymer-protein nanoparticles by atom transfer radical polymerization. J. Am. Chem. Soc. 2011, 133, 9242–9245. [Google Scholar] [CrossRef]
- Takeuchi, T.; Hishiya, T. Molecular imprinting of proteins emerging as a tool for protein recognition. Org. Biomol. Chem. 2008, 6, 2459–2467. [Google Scholar] [CrossRef]
- Gao, J.; Tian, H.; Wang, Y.; Yang, Q.; Liu, D.; Mi, H. The design of protein-imprinted polymers as antibody substitutes for investigating protein-protein interactions. Biomaterials 2012, 33, 3344–3352. [Google Scholar] [CrossRef]
- Ying, X.; Cheng, G.; Li, X. The imprinting induce-fit model of specific rebinding of macromolecularly imprinted polymer microspheres. J. Appl. Polym. Sci. 2011, 122, 1847–1856. [Google Scholar] [CrossRef]
- Bolisay, L.D.; Culver, J.N.; Kofinas, P. Molecularly imprinted polymers for tobacco mosaic virus recognition. Biomaterials 2006, 27, 4165–4168. [Google Scholar] [CrossRef]
- Verheyen, E.; Schillemans, J.P.; van Wijk, M.; Demeniex, M.-A.; Hennink, W.E.; van Nostrum, C.F. Challenges for the effective molecular imprinting of proteins. Biomaterials 2011, 32, 3008–3020. [Google Scholar] [CrossRef]
- Cumbo, A.; Lorber, B.; Corvini, P.F.-X.; Meier, W.; Shahgaldian, P. A synthetic nanomaterial for virus recognition produced by surface imprinting. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Zhang, X.; Smith, D.L.; Meriin, A.B.; Engemann, S.; Russel, D.E.; Roark, M.; Washington, S.L.; Maxwell, M.M.; Marsh, J.L.; Thompson, L.M.; et al. A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 892–897. [Google Scholar] [CrossRef]
- Karpuj, M.V.; Garren, H.; Slunt, H.; Price, D.L.; Gusella, J.; Becher, M.W.; Steinman, L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington’s disease brain nuclei. Proc. Natl. Acad. Sci. USA 1999, 96, 7388–7393. [Google Scholar]
- Hexapeptide, P.; Zheng, J.; Liu, C.; Sawaya, M.R.; Vadla, B.; Khan, S.; Woods, R.J.; Eisenberg, D.; Goux, W.J.; Nowick, J.S. Macrocyclic β-sheet peptides that inhibit the aggregation of a tau-protein-derived hexapeptide. J. Am. Chem. Soc. 2011, 133, 3144–3157. [Google Scholar] [CrossRef]
- Marotta, N.P.; Cherwien, C.A.; Abeywardana, T.; Pratt, M.R. O-GlcNAc modification prevents peptide-dependent acceleration of α-synuclein aggregation. ChemBioChem 2012, 13, 2665–2670. [Google Scholar] [CrossRef]
- Rühs, P.A.; Adamcik, J.; Bolisetty, S.; Sánchez-Ferrer, A.; Mezzenga, R. A supramolecular bottle-brush approach to disassemble amyloid fibrils. Soft Matter 2011, 7, 3571–3579. [Google Scholar] [CrossRef]
- Skaat, H.; Chen, R.; Grinberg, I.; Margel, S. Engineered polymer nanoparticles containing hydrophobic dipeptide for inhibition of amyloid-β fibrillation. Biomacromolecules 2012, 13, 2662–2670. [Google Scholar] [CrossRef]
- le Droumaguet, B.; Nicolas, J.; Brambilla, D.; Mura, S.; Maksimenko, A.; de Kimpe, L.; Salvati, E.; Zona, C.; Airoldi, C.; Canovi, M.; et al. Versatile and efficient targeting using a single nanoparticulate platform: Application to cancer and Alzheimer’s disease. ACS Nano 2012, 6, 5866–5879. [Google Scholar] [CrossRef]
- Tan, H.; Jin, H.; Mei, H.; Zhu, L.; Wei, W.; Wang, Q.; Liang, F.; Zhang, C.; Li, J.; Qu, X.; et al. PEG-urokinase nanogels with enhanced stability and controllable bioactivity. Soft Matter 2012, 8, 2644–2650. [Google Scholar] [CrossRef]
- Muszanska, A.K.; Busscher, H.J.; Herrmann, A.; van der Mei, H.C.; Norde, W. Pluronic-lysozyme conjugates as anti-adhesive and antibacterial bifunctional polymers for surface coating. Biomaterials 2011, 32, 6333–6341. [Google Scholar]
- Browning, M.B.; Dempsey, D.; Guiza, V.; Becerra, S.; Rivera, J.; Russell, B.; Höök, M.; Clubb, F.; Miller, M.; Fossum, T.; et al. Multilayer vascular grafts based on collagen-mimetic proteins. Acta Biomater. 2012, 8, 1010–1021. [Google Scholar] [CrossRef]
- Fischlechner, M.; Donath, E. Viruses as building blocks for materials and devices. Angew. Chem. Int. Ed. 2007, 46, 3184–3193. [Google Scholar] [CrossRef]
- Tseng, R.J.; Tsai, C.; Ma, L.; Ouyang, J.; Ozkan, C.S.; Yang, Y. Digital memory device based on tobacco mosaic virus conjugated with nanoparticles. Nat. Nanotechnol. 2006, 1, 72–77. [Google Scholar]
- Evans, D.J. The bionanoscience of plant viruses: Templates and synthons for new materials. J. Mater. Chem. 2008, 18, 3746–3754. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Van Rijn, P. Polymer Directed Protein Assemblies. Polymers 2013, 5, 576-599. https://doi.org/10.3390/polym5020576
Van Rijn P. Polymer Directed Protein Assemblies. Polymers. 2013; 5(2):576-599. https://doi.org/10.3390/polym5020576
Chicago/Turabian StyleVan Rijn, Patrick. 2013. "Polymer Directed Protein Assemblies" Polymers 5, no. 2: 576-599. https://doi.org/10.3390/polym5020576
APA StyleVan Rijn, P. (2013). Polymer Directed Protein Assemblies. Polymers, 5(2), 576-599. https://doi.org/10.3390/polym5020576