Physical Properties of Polypeptide Electrospun Nanofiber Cell Culture Scaffolds on a Wettable Substrate

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Polymers
2.2. Electrospinning
2.3. Crosslinking
2.4. Dye Labeling and Protein Adsorption
2.5. Spectroscopy and Microscopy
2.5.1. CD
2.5.2. FTIR
2.5.3. EDX
2.5.4. Microscopy
3. Results
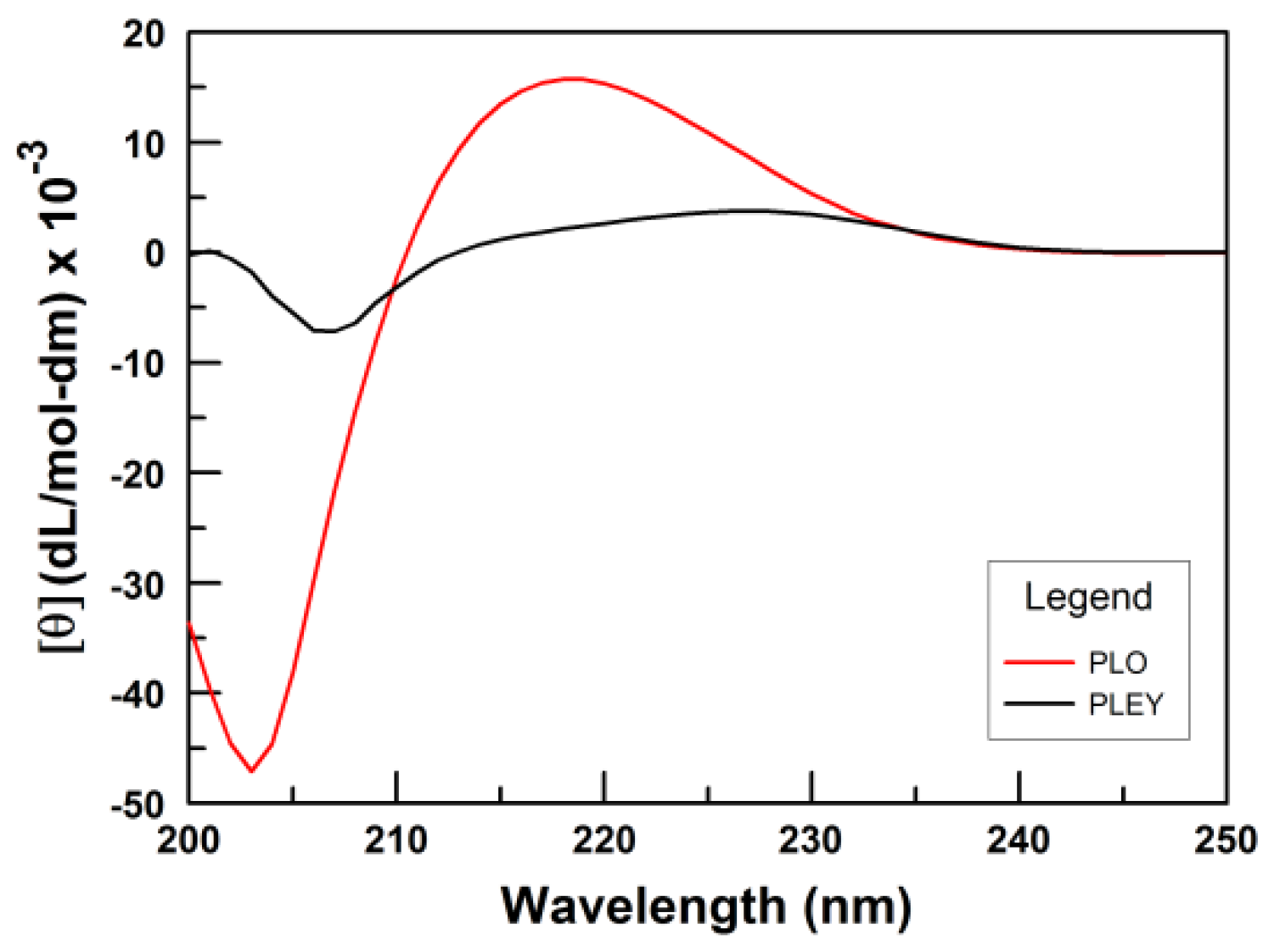
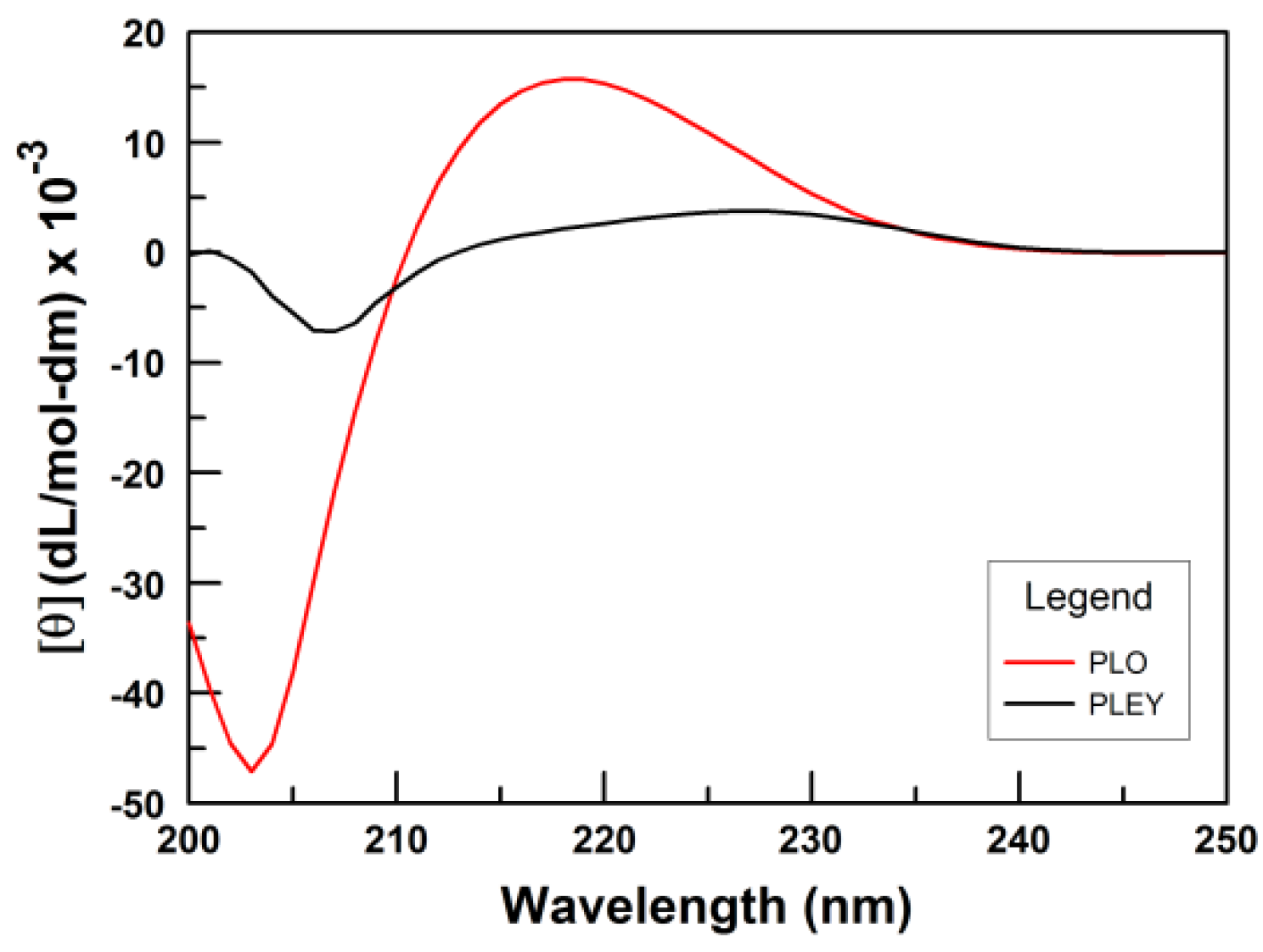
3.1. Polymer Conformation

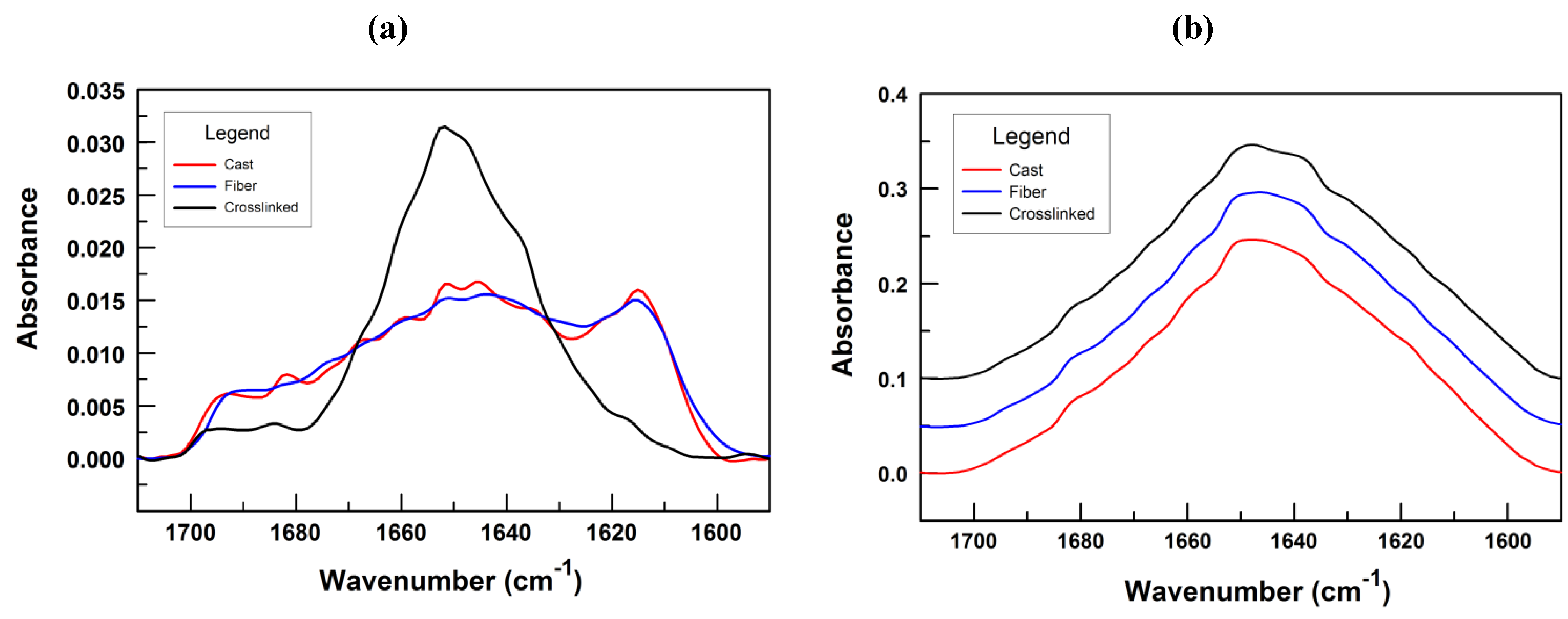
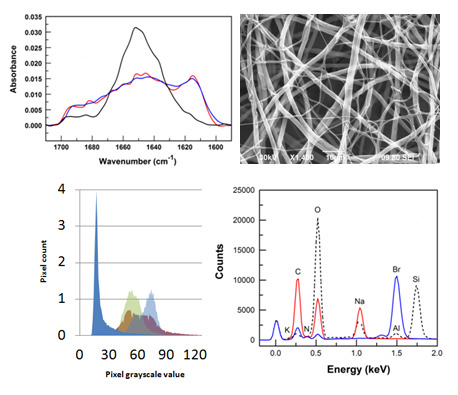
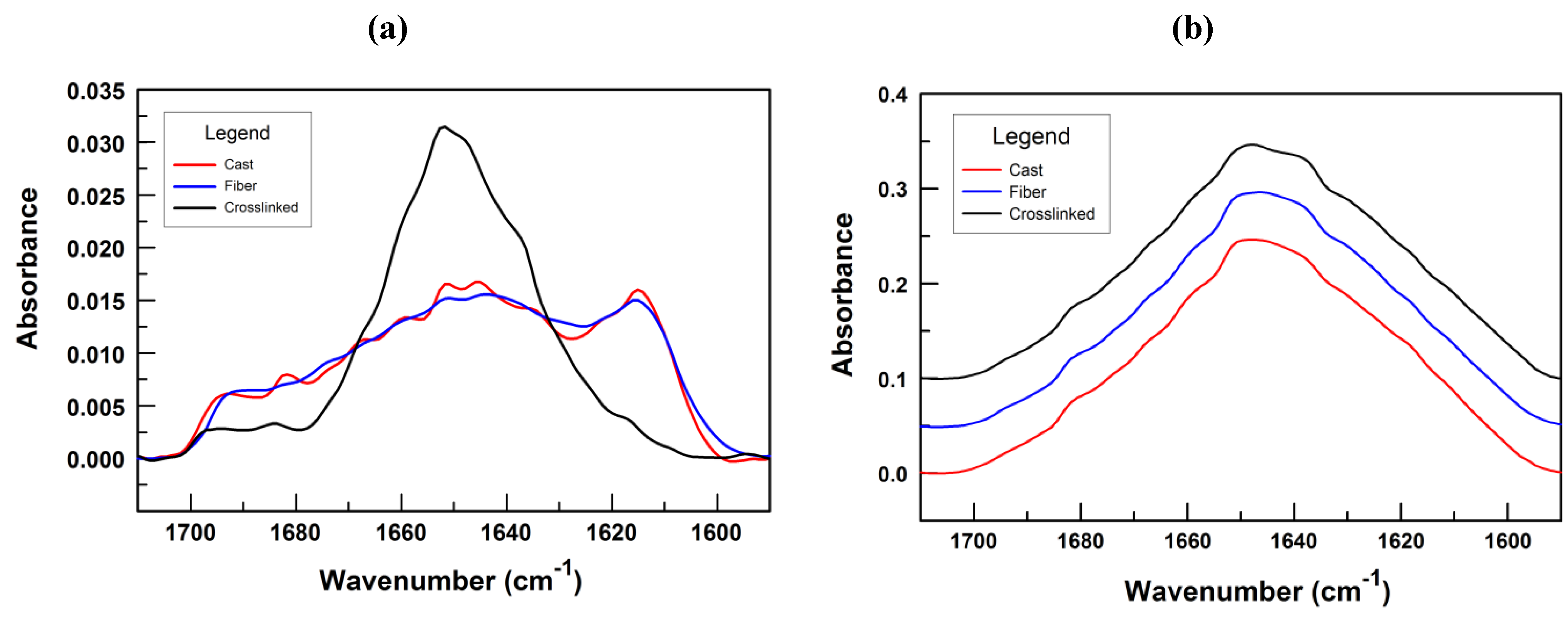
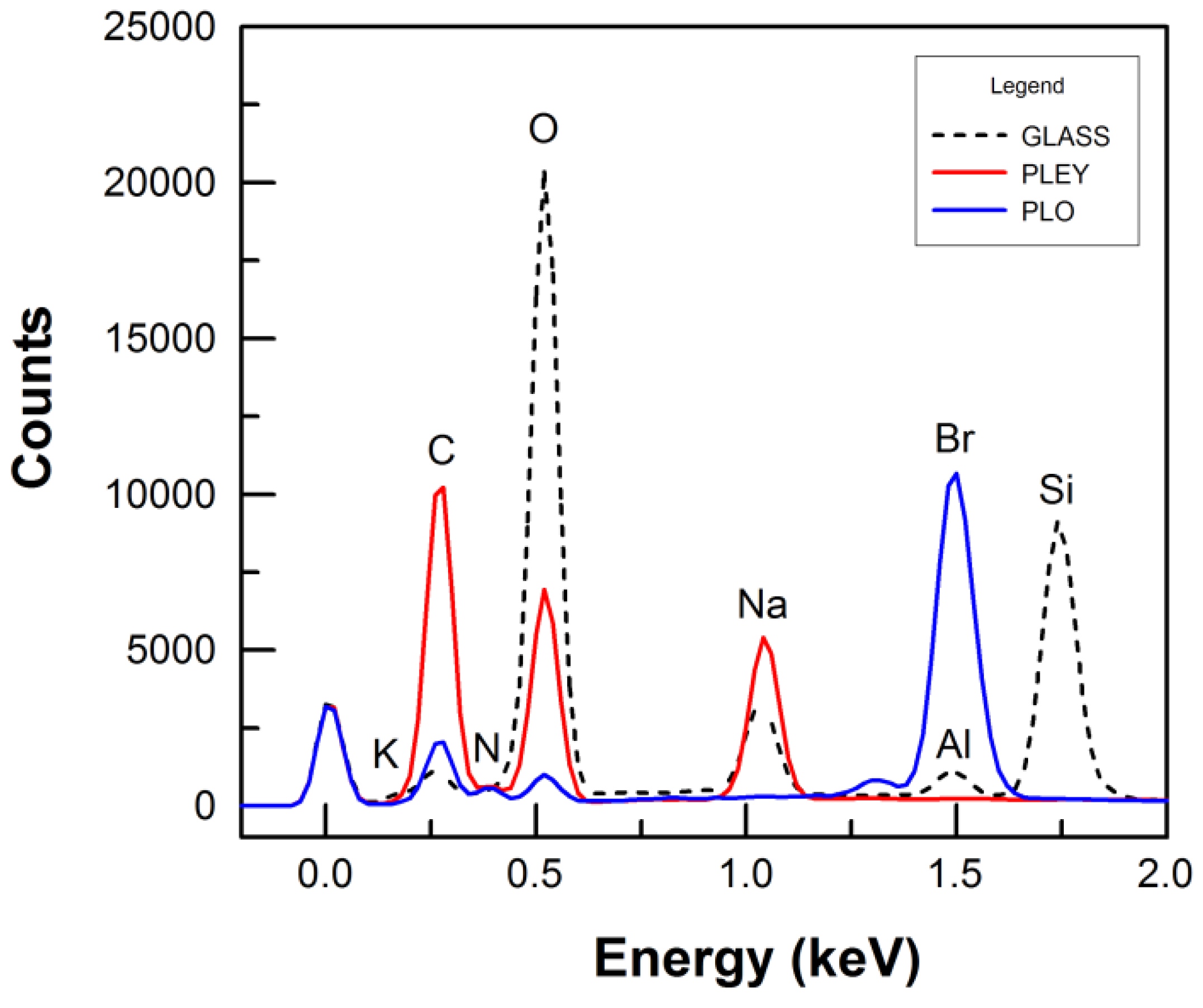
3.2. Fiber Stability and Composition
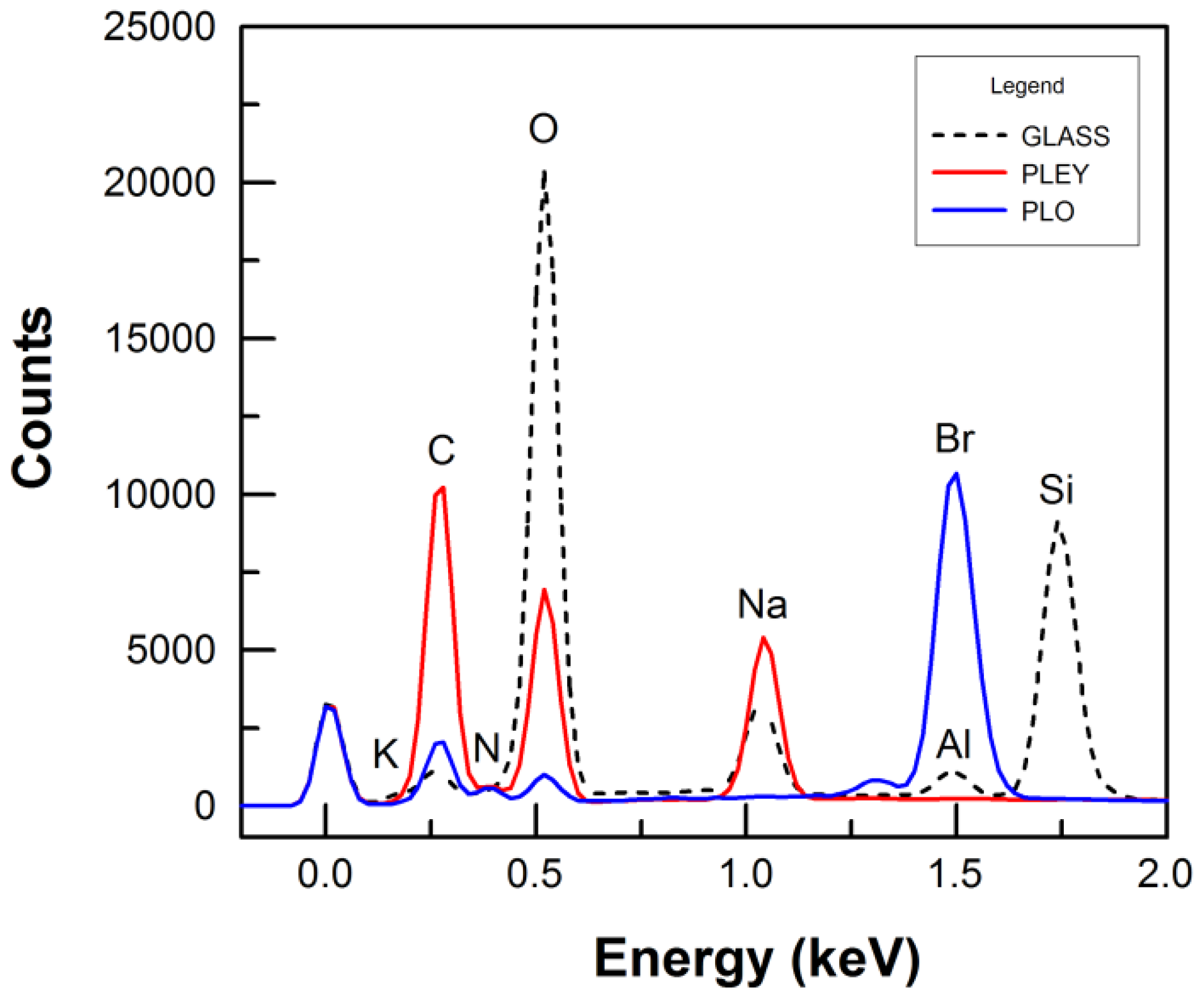
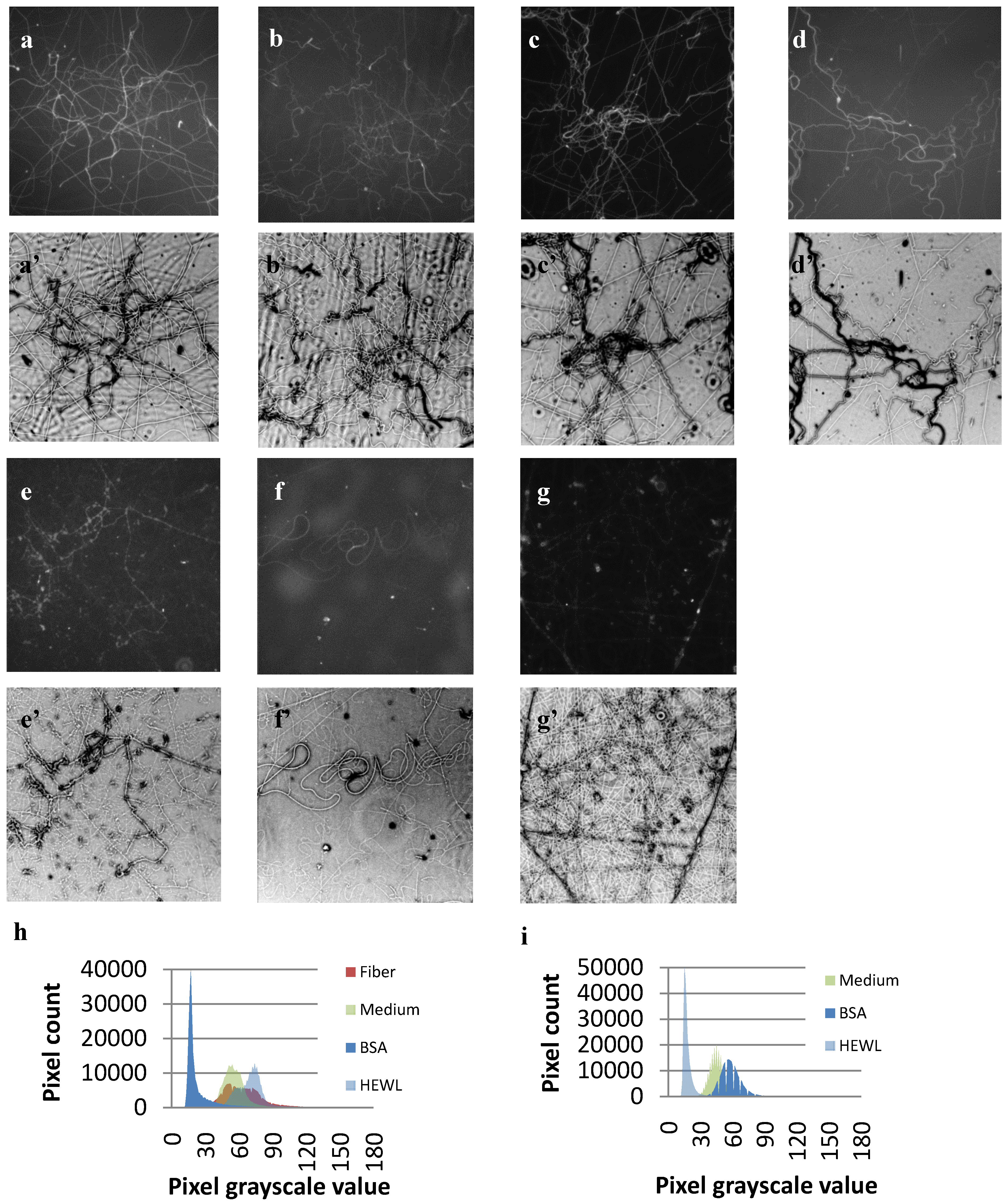
3.3. Protein Adsorption
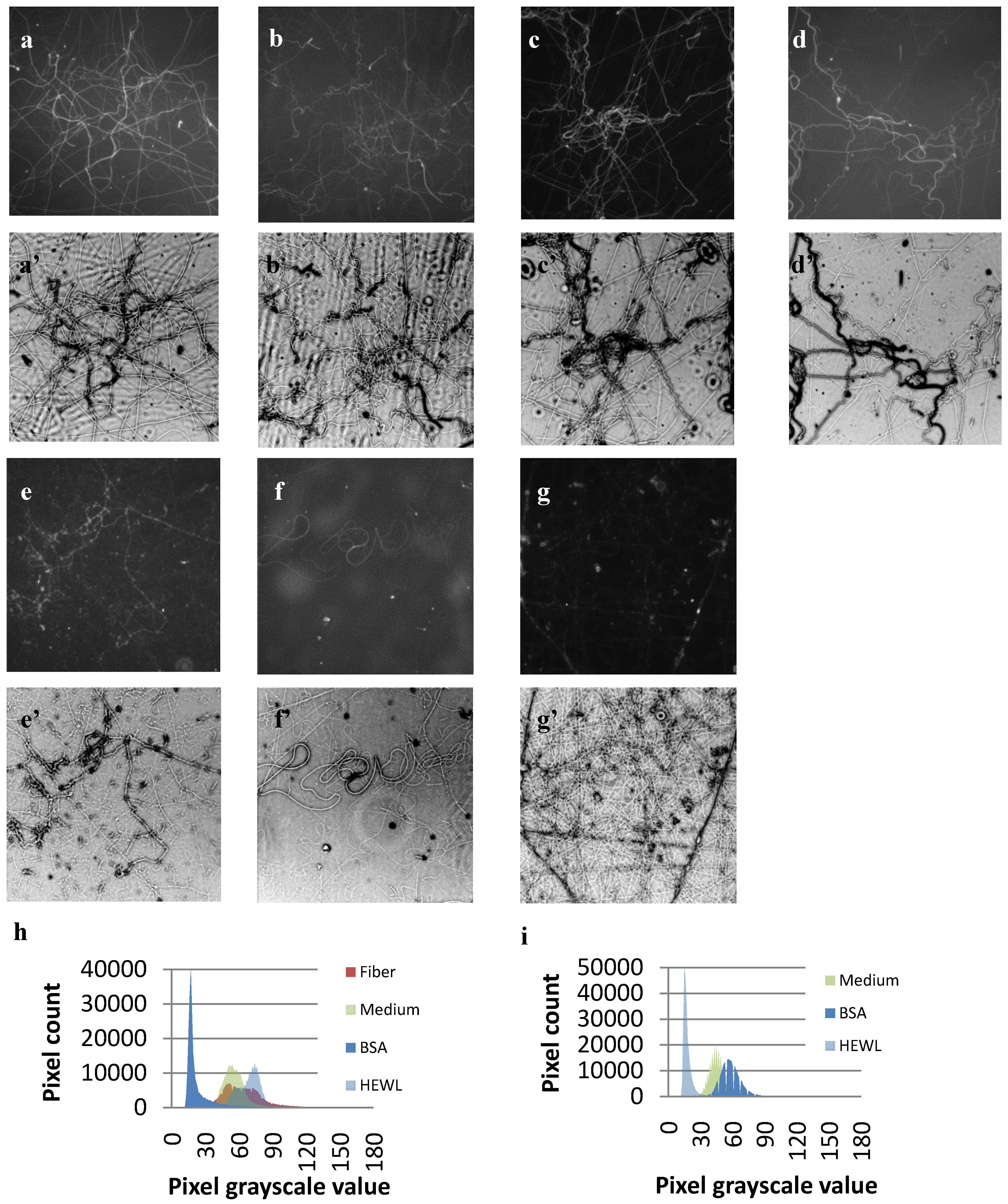
4. Discussion
4.1. Fiber Formation, Composition and Structure
4.2. Crosslinking
4.3. Electrical Properties
4.4. Maximum Charge Density on an Unmodified Protein Fiber
5. Conclusions
Acknowledgments
References
- Rutledge, G.C.; Fridrikh, S.V. Formation of fibers by electrospinning. Adv. Drug Deliv. Rev. 2007, 59, 1384–1391. [Google Scholar] [CrossRef]
- Sill, T.J.; von Recum, H.A. Electrospinning: Applications in drug delivery and tissue engineering. Biomaterials 2008, 29, 1989–2006. [Google Scholar] [CrossRef]
- Yoo, H.S.; Kim, T.G.; Park, T.G. Surface-functionalized electrospun nanofibers for tissue engineering and drug delivery. Adv. Drug Deliv. Rev. 2009, 61, 1033–1042. [Google Scholar] [CrossRef]
- Khadka, D.B.; Haynie, D.T. Insoluble synthetic polypeptide mats from aqueous solution by electrospinning. ACS Appl. Mater. Interfaces 2010, 2, 2728–2732. [Google Scholar]
- Agarwal, S.; Greiner, A. On the way to clean and safe electrospinning-green electrospinning: Emulsion and suspension electrospinning. Polym. Adv. Technol. 2011, 22, 372–378. [Google Scholar] [CrossRef]
- Khadka, D.B.; Cross, M.C.; Haynie, D.T. A synthetic polypeptide electrospun biomaterial. ACS Appl. Mater. Interfaces 2011, 3, 2994–3001. [Google Scholar]
- Khadka, D.B.; Haynie, D.T. Protein- and peptide-based electrospun nanofibers in medical biomaterials. Nanomedicine 2012, in press. [Google Scholar]
- Ner, Y.; Stuart, J.A.; Whited, G.; Sotzing, G.A. Electrospinning nanoribbons of a bioengineered silk-elastin-like protein (SELP) from water. Polymer 2009, 50, 5828–5836. [Google Scholar]
- Deitzel, J.M.; Kleinmeyer, J.; Harris, D.; Beck Tan, N.C. The effect of processing variables on the morphology of electrospun nanofibers and textiles. Polymer 2001, 42, 261–272. [Google Scholar]
- Yarin, A.L.; Koombhongse, S.; Reneker, D.H. Bending instability in electrospinning of nanofibers. J. Appl. Phys. 2001, 89, 3018–3026. [Google Scholar]
- Minato, K.I.; Ohkawa, K.; Yamamoto, H. Chain conformations of poly(γ-benzyl-L-glutamate) pre and post an electrospinning process. Macromol. Biosci. 2006, 6, 487–495. [Google Scholar] [CrossRef]
- Hohman, M.M.; Shin, M.; Rutledge, G.; Brenner, M.P. Electrospinning and electricity forced jets. I. Stability theory. Phys. Fluids 2001, 13, 2201–2220. [Google Scholar] [CrossRef]
- Fridrikh, S.V.; Yu, J.H.; Brenner, M.P.; Rutledge, G.C. Controlling the fiber diameter during electrospinning. Phys. Rev. Lett. 2003, 90, 114502:1–114502:4. [Google Scholar]
- He, J.H.; Xu, L.; Wu, Y.; Liu, Y. Mathematical models for continuous electrospun nanofibers and electrospun nanoporous microspheres. Polym. Int. 2007, 56, 1323–1329. [Google Scholar]
- Strobl, G. The Physics of Polymers; Springer: Berlin, Germany, 2007. [Google Scholar]
- Zheng, X.; Baker, H.; Hancock, W.S.; Fawaz, F.; McCaman, M.; Pungor, E. Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol. Prog. 2006, 22, 1294–1300. [Google Scholar]
- Chaudhuri, S.R.; Yang, J.T. Helix-coil transition of poly-L-ornithine in solution. Biochemistry 1968, 7, 1379–1383. [Google Scholar]
- Wada, A. Helix-coil transformation and titration curve of poly-L-glutamic acid. Mol. Phys. 1960, 3, 409–416. [Google Scholar]
- Greenfield, N.; Fasman, G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. [Google Scholar] [CrossRef]
- Wetlaufer, D. Ultraviolet spectra of proteins and amino acids. Adv. Protein. Chem. 1962, 17, 303–390. [Google Scholar] [CrossRef]
- Chakrabartty, A.; Kortemme, T.; Padmahabhan, S.; Baldwin, R.L. Aromatic side-chain contribution to far-ultraviolet circular dichroism of helical peptides and its effect on measurement of helix propensities. Biochemistry 1993, 32, 5560–5565. [Google Scholar]
- Chirgadze, Y.N.; Nevskaya, N.A. Infrared spectra and resonance interaction of amide-I vibration of the antiparallel-chain pleated sheet. Biopolymers 1976, 15, 637–648. [Google Scholar] [CrossRef]
- Jackson, M.; Mantsch, H.H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef]
- Imoto, T.; Johnson, L.N.; North, A.T.C.; Phillips, D.C.; Rupley, J.A. Vertebrate lysozymes. In The Enzymes; Boyer, P.D., Lardy, H., Myrback, K., Eds.; Academic: New York, NY, USA, 1972; pp. 665–868. [Google Scholar]
- Malamud, D.; Drysdale, J.W. Isoelectric points of proteins: A table. Anal. Biochem. 1978, 86, 620–647. [Google Scholar]
- Shenoy, S.L.; Bates, W.D.; Frisch, H.L.; Wnek, G.E. Role of chain entanglements on fiber formation during electrospinning of polymer solutions: Good solvent, non-specific polymer-polymer interaction limit. Polymer 2005, 46, 3372–3384. [Google Scholar]
- Doty, P.; Wada, A.; Yang, J.T.; Blout, E.R. Polypeptides. VII. Molecular configurations of poly-L-glutamic acid in water-dioxane solution. J. Polym. Sci. 1957, 23, 851–857. [Google Scholar] [CrossRef]
- Zandomeneghi, G.; Krebs, M.R.H.; McCammon, M.G.; Fändrich, M. FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 2004, 13, 3314–3321. [Google Scholar]
- Richardson, J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981, 34, 167–339. [Google Scholar] [CrossRef]
- Laurine, E.; Gregoire, E.; Fändrich, M.; Engemann, S.; Marchal, S.; Thion, L.; Mohr, M.; Monsarrat, B.; Michel, B.; Dobson, C.M.; et al. Lithostathine quadruple-helical filaments from proteinase K-resistant deposits in Creutzfeldt-Jakob disease. J. Biol. Chem. 2003, 278, 51770–51778. [Google Scholar]
- Nyquist, R.A.; Clark, T.D.; Streck, R. Infrared study of alkyl carboxylic acids in CCl4 and/or CHCl3 solutions. Vib. Spectrosc. 1994, 7, 275–286. [Google Scholar] [CrossRef]
- Baldwin, R.L. In search of the energetic role of peptide hydrogen bonds. J. Biol. Chem. 2003, 278, 17581–17588. [Google Scholar] [CrossRef]
- Poland, D.; Scheraga, H.A. Theory of Helix-Coil Transition Theory in Biopolymers; Academic: New York, NY, USA, 1970. [Google Scholar]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 1692, 103–119. [Google Scholar]
- Junge, K.; Binnebösel, M.; von Trotha, K.T.; Rosch, R.; Kling, U.; Neumann, U.P.; Jansen, P.L. Mesh biocompatibility: Effects of cellular inflammation and tissue remodelling. Langenbecks Arch. Surg. 2012, 397, 255–270. [Google Scholar]
- Squires, T.M.; Brenner, M.P. Like-charge attraction and hydrodynamic interaction. Phys. Rev. Lett. 2000, 85, 4976–4979. [Google Scholar]
- Behrens, S.H.; Grier, D.G. The charge of glass and silica surfaces. J. Chem. Phys. 2001, 115, 6716–6721. [Google Scholar] [CrossRef]
- Fischer, H.; Polikarpov, I.; Craievich, A.F. Average protein density is a molecular-weight-dependent function. Protein Sci. 2004, 13, 2825–2828. [Google Scholar]
- Curtis, A.S.G.; Forrester, J.V.; McInnes, C.; Lawrie, F. Adhesion of cells to polystyrene surfaces. J. Cell Biol. 1983, 97, 1500–1506. [Google Scholar] [CrossRef]
- Curtis, A.S.G. Cell adhesion. Prog. Biophys. Mol. Biol. 1973, 27, 317–375. [Google Scholar]
- Gingell, D.; Todd, I. Red blood cell adhesion. II. Interferometric examination of the interaction with hydrocarbon oil and glass. J. Cell Sci. 1980, 41, 135–149. [Google Scholar]
- Pernodet, N.; Rafailovich, M.; Sokolov, J.; Xu, D.; Yang, N.-L.; McLeod, K.J. Fibronectin fibrillogenesis on sulfonated polystyrene surfaces. J. Biomed. Mater. Res. A 2003, 64, 684–692. [Google Scholar]
- Harris, A.K.; Pryer, N.K.; Paydarfar, D. Effects of electric fields on fibroblast contractility and cytoskeleton. J. Exp. Zool. 1990, 253, 163–176. [Google Scholar] [CrossRef]
- Poo, M. In situ electrophoresis of membrane components. Annu. Rev. Biophys. Bioeng. 1981, 10, 245–276. [Google Scholar] [CrossRef]
- Sun, S.; Wise, J.; Cho, M. Human fibroblast migration in three-dimensional collagen gel in response to noninvasive electrical stimulus. I. Characterization of induced three-dimensional cell movement. Tissue. Eng. 2004, 10, 1548–1557. [Google Scholar]
- Nuccitelli, R. A role for endogenous electric fields in wound healing. Curr. Top. Dev. Biol. 2003, 58, 1–26. [Google Scholar] [CrossRef]
- Borgens, R.B.; Vanable, J.W., Jr.; Jaffe, L.F. Bioelectricity and regeneration: Large currents leave the stumps of regenerating newt limbs. Proc. Nat. Acad. Sci. USA 1977, 74, 4528–4532. [Google Scholar]
- Mir, L.M.; Bureau, M.F.; Gehl, J.; Rangara, R.; Rouy, D.; Caillaud, J.M.; Delaere, P.; Branellec, D.; Schwartz, B.; Scherman, D. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Nat. Acad. Sci. USA 1999, 96, 4262–4267. [Google Scholar]
- Hochmuth, R.M. Micropipette aspiration of living cells. J. Biomech. 2000, 33, 15–22. [Google Scholar] [CrossRef]
- Kinraide, T.B.; Wang, P. The surface charge density of plant cell membranes (σ): An attempt to resolve conflicting values for intrinsic σ. J. Exp. Bot. 2010, 61, 2507–2518. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Haynie, D.T.; Khadka, D.B.; Cross, M.C. Physical Properties of Polypeptide Electrospun Nanofiber Cell Culture Scaffolds on a Wettable Substrate. Polymers 2012, 4, 1535-1553. https://doi.org/10.3390/polym4031535
Haynie DT, Khadka DB, Cross MC. Physical Properties of Polypeptide Electrospun Nanofiber Cell Culture Scaffolds on a Wettable Substrate. Polymers. 2012; 4(3):1535-1553. https://doi.org/10.3390/polym4031535
Chicago/Turabian StyleHaynie, Donald T., Dhan B. Khadka, and Michael C. Cross. 2012. "Physical Properties of Polypeptide Electrospun Nanofiber Cell Culture Scaffolds on a Wettable Substrate" Polymers 4, no. 3: 1535-1553. https://doi.org/10.3390/polym4031535
APA StyleHaynie, D. T., Khadka, D. B., & Cross, M. C. (2012). Physical Properties of Polypeptide Electrospun Nanofiber Cell Culture Scaffolds on a Wettable Substrate. Polymers, 4(3), 1535-1553. https://doi.org/10.3390/polym4031535