Molecular Design and Property Prediction of Sterically Confined Polyimides for Thermally Stable and Transparent Materials

 ,
,  and
and
Abstract
:
1. Introduction
2. Methodology
2.1. Molecular Design
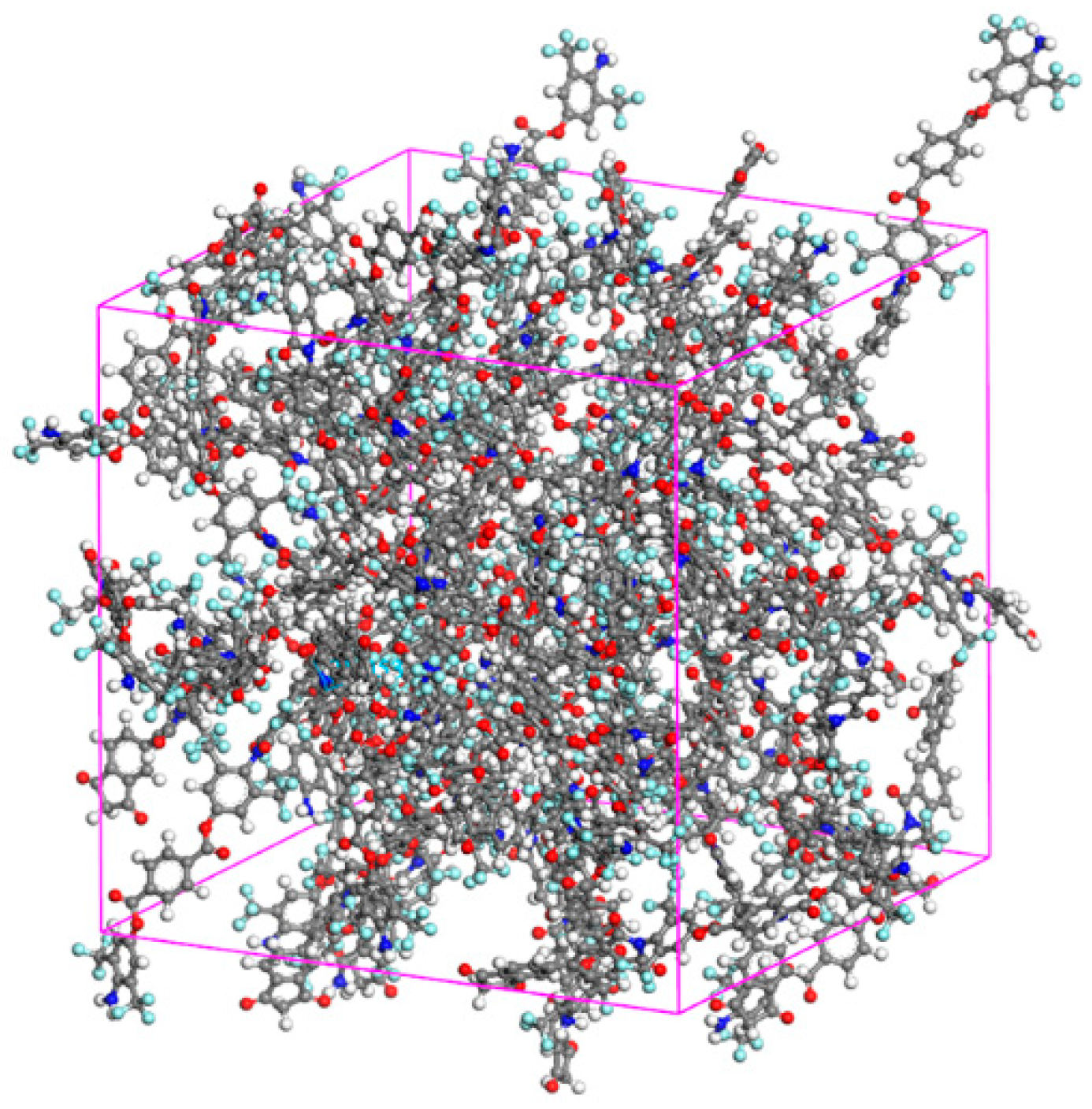
2.2. Simulation Protocol
2.3. Details of MD Calculations
3. Results and Discussion
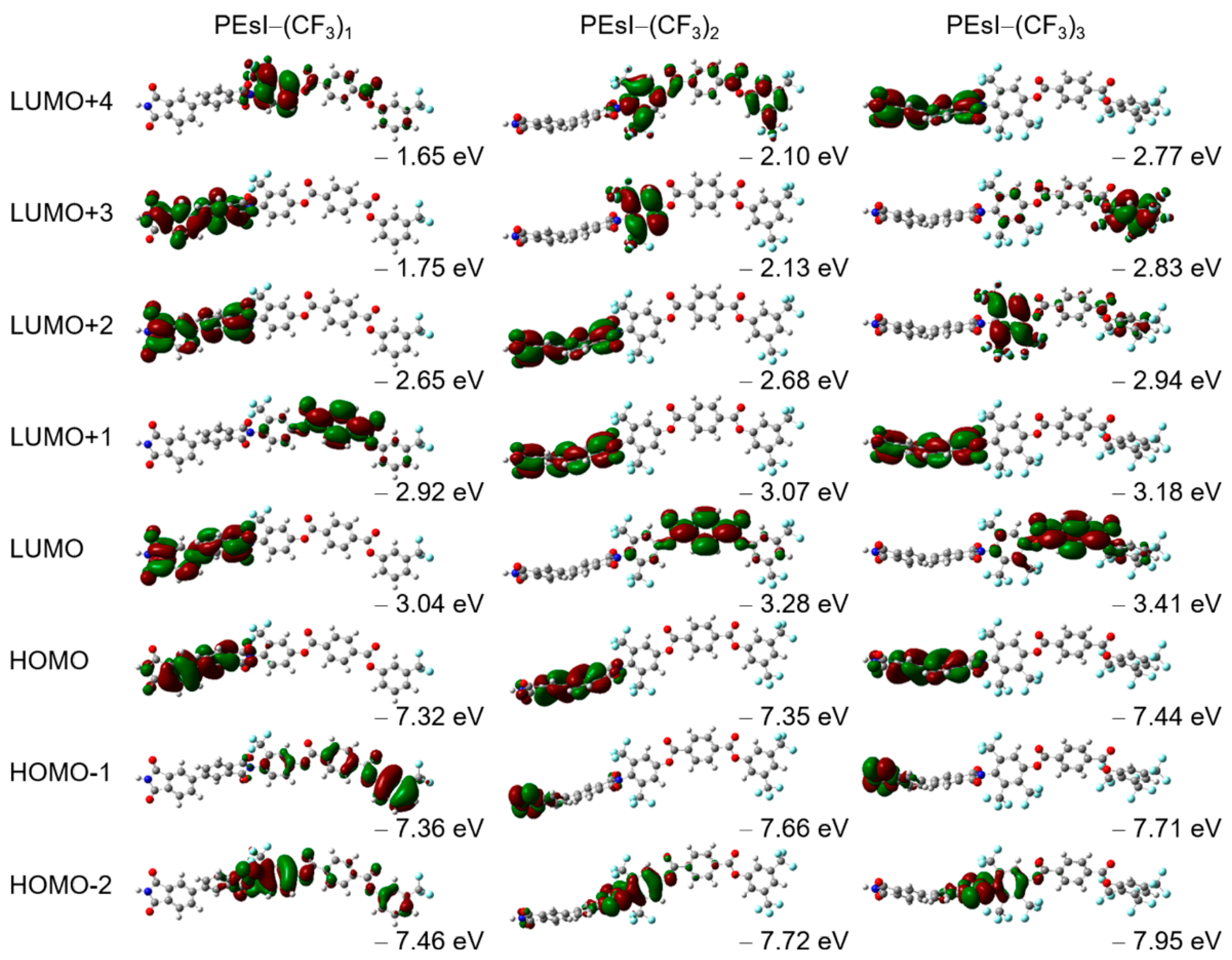
3.1. Molecular Orbital Calculation
3.2. Determination of the Tg and CTE
3.3. Mean-Square Displacement
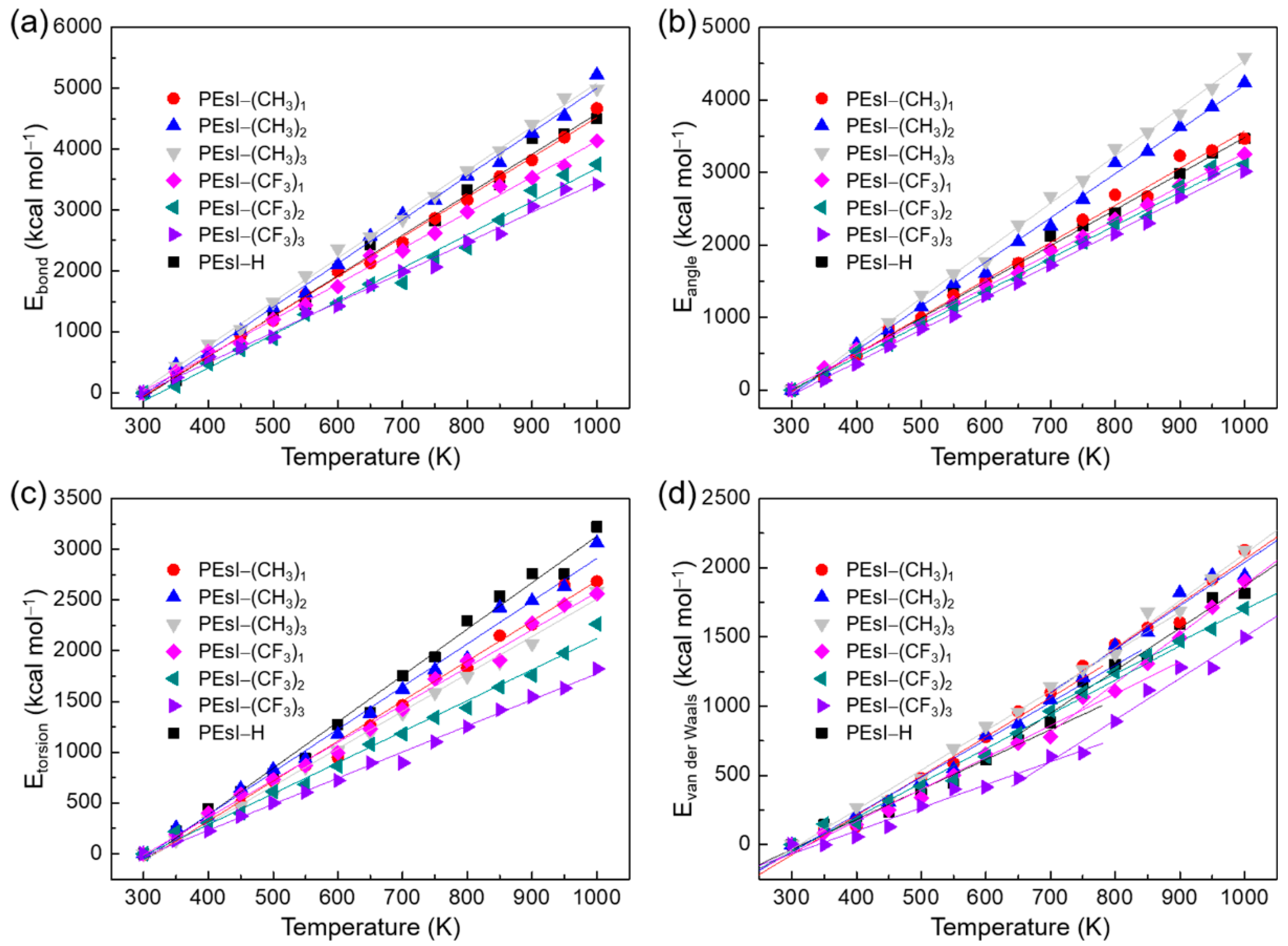
3.4. Role of the Energy Components
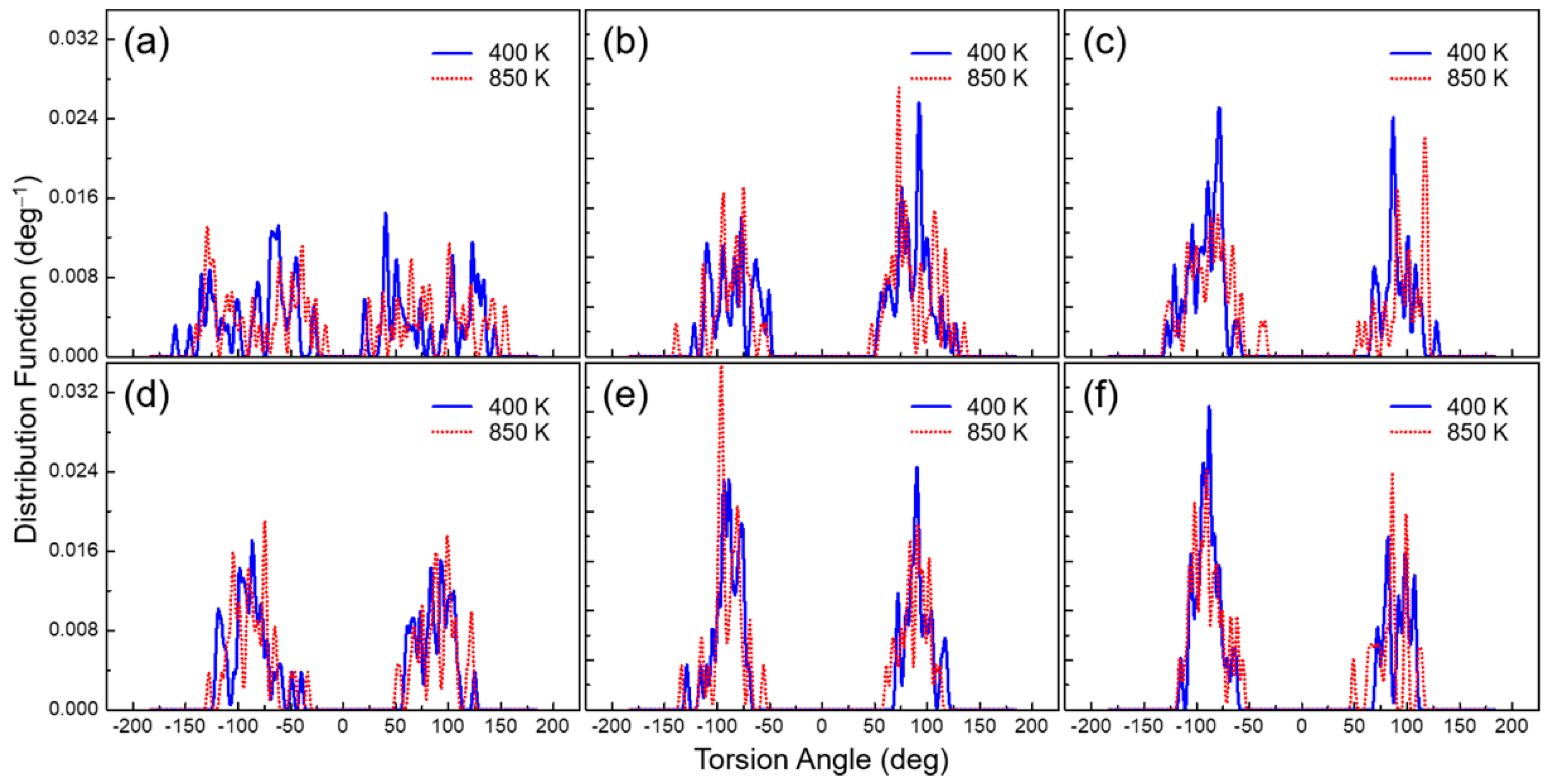
3.5. Torsion Angle Distribution
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- MacDonald, W.A. Engineered films for display technologies. J. Mater. Chem. 2004, 14, 4–10. [Google Scholar] [CrossRef]
- Lee, S.J.; Ko, J.; Nam, K.-H.; Kim, T.; Lee, S.H.; Kim, J.H.; Chae, G.S.; Han, H.; Kim, Y.S.; Myoung, J.-M. Fully solution-processed and foldable metal-oxide thin-film transistor. ACS Appl. Mater. Interfaces 2016, 8, 12894–12900. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-C.; Kim, Y.; Ha, C.-S. Polymers for flexible displays: From material selection to device applications. Prog. Polym. Sci. 2008, 33, 581–630. [Google Scholar] [CrossRef]
- Logothetidis, S. Flexible organic electronic devices: Materials, process and applications. Mater. Sci. Eng. B-Adv. 2008, 152, 96–104. [Google Scholar] [CrossRef]
- Hasegawa, M.; Hirano, D.; Fujii, M.; Haga, M.; Takezawa, E.; Yamaguchi, S.; Kshikawa, A.; Kagayama, T. Solution-processable colorless polyimides derived from hydrogenated pyromellitic dianhydride with controlled steric structure. J. Polym. Sci. Pol. Chem. 2013, 51, 575–592. [Google Scholar] [CrossRef]
- Ha, C.-S.; Choi, M.-C.; Wakita, J.; Ando, S. Highly transparent and refractive polyimides with controlled molecular structure by chlorine side groups. Macromolecules 2009, 42, 5112–5120. [Google Scholar]
- Yen, H.-J.; Chen, C.-J.; Liou, G.-S. Flexible multi-colored electrochromic and volatile polymer memory devices derived from starburst triarylamine-based electroactive polyimide. Adv. Funct. Mater. 2013, 23, 5307–5316. [Google Scholar] [CrossRef]
- Lim, J.; Shin, D.G.; Yeo, H.; Goh, M.; Ku, B.-C.; Yang, C.-M.; Lee, D.S.; Hwang, J.-Y.; Park, B.; You, N.-H. The mechanical and electrical properties of carbon nanotube-grafted polyimide nanocomposites. J. Polym. Sci. Part B-Polym. Phys. 2014, 52, 906–966. [Google Scholar] [CrossRef]
- Erhard, D.P.; Richter, F.; Bartz, C.B.A.; Schmidt, H.-W. Fluorinated aromatic polyimides as high-performance electret materials. Macromol. Rapid Commun. 2015, 36, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.-H.; Yu, J.; You, N.-H.; Han, H.; Ku, B.-C. Synergistic toughening of polymer nanocomposites by hydrogen-bond assisted three-dimensional network of functionalized graphene oxide and carbon nanotubes. Compos. Sci. Technol. 2017, 149, 228–234. [Google Scholar] [CrossRef]
- Lim, J.; Yeo, H.; Goh, M.; Ku, B.-C.; Kim, S.G.; Lee, H.S.; Park, B.; You, N.-H. Grafting of polyimide onto chemically-functionalized graphene nanosheets for mechanically-strong barrier membranes. Chem. Mater. 2015, 27, 2040–2047. [Google Scholar] [CrossRef]
- Zhai, L.; Yang, S.; Fan, L. Preparation and characterization of highly transparent and colorless semi-aromatic polyimide films derived from alicyclic dianhydride and aromatic diamines. Polymer 2012, 53, 3529–3539. [Google Scholar] [CrossRef]
- Banach, M.J.; Friend, R.H.; Sirringhaus, H. Influence of the molecular weight on the thermotropic alignment of thin liquid crystalline polyfluorene copolymer films. Macromolecules 2003, 36, 2838–2844. [Google Scholar] [CrossRef]
- Kurosawa, T.; Higashihara, T.; Ueda, M. Polyimide memory: A pithy guideline for future applications. Polym. Chem. 2013, 4, 16–30. [Google Scholar] [CrossRef]
- Takasaki, T.; Kuwana, Y.; Takahashi, T.; Hayashida, S. Synthesis and optical properties of polyimides. J. Polym. Sci. Pol. Chem. 2000, 38, 4832–4838. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, C.; Wang, D.; Dang, G.; Chen, C.; Zhou, H.; Zhao, X. High transparent polyimides containing pyridine and biphenyl units: Synthesis, thermal, mechanical, crystal and optical properties. Polymer 2015, 62, 1–10. [Google Scholar] [CrossRef]
- Rusanov, A.L.; Shifrina, Z.B. Preparation of soluble polyimides containing pendent imide groups. High Perform. Polym. 1993, 5, 107–121. [Google Scholar] [CrossRef]
- Lin, S.-H.; Li, F.; Cheng, S.Z.D.; Harris, F.W. Organo-soluble polyamides: Synthesis and polymerization of 2,2′-bis(trifluoromethyl)-4,4′,5,5′-biphenyltetracarboxylic dianhydride. Macromolecules 1998, 31, 2080–2086. [Google Scholar] [CrossRef]
- Yeo, H.; Goh, M.; Ku, B.-C.; You, N.-H. Synthesis and characterization of highly-fluorinated colorless polyimides derived from 4,4′-((perfluoro-[1,1′-biphenyl]-4,4′-diyl)bis(oxy))bis(2,6-dimethylaniline) and aromatic dianhydrides. Polymer 2015, 76, 280–286. [Google Scholar] [CrossRef]
- Hasegawa, M.; Ishigami, T.; Ishii, J. Optically transparent aromatic poly(ester imide)s with low coefficients of thermal expansion (1). Self-orientation behavior during solution casting process and substituent effect. Polymer 2015, 74, 1–15. [Google Scholar] [CrossRef]
- Hasegawa, M.; Hirai, T.; Ishigami, T.; Takahashi, S.; Ishii, J. Optically transparent aromatic poly(ester imide)s with low coefficients of thermal expansion. 2: Effect of the introduction of alkyl-substituted p-biphenylene units. Polym. Int. 2018, 67, 431–444. [Google Scholar] [CrossRef]
- Nam, K.-H.; Kim, H.; Choi, H.K.; Yeo, H.; Goh, M.; Yu, J.; Hahn, J.R.; Han, H.; Ku, B.-C.; You, N.-H. Thermomechanical and optical properties of molecularly controlled polyimides derived from ester derivatives. Polymer 2017, 108, 502–512. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, X.; He, Z.; Lan, F.; Liu, H. The glass transition temperature measurements of polyethylene: Determined by using molecular dynamic method. RSC Adv. 2016, 6, 12053–12060. [Google Scholar] [CrossRef]
- Li, C.; Strachan, A. Molecular dynamics predictions of thermal and mechanical properties of thermoset polymer EPON862/DETDA. Polymer 2011, 52, 2920–2928. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Gurtovenko, A.A.; Larin, S.V.; Nazarychev, V.M.; Lyulin, A.V. Microsecond Atomic-Scale Molecular Dynamics Simulations of Polyimides. Macromolecules 2013, 46, 6357–6363. [Google Scholar] [CrossRef]
- Liang, T.; Yang, X.; Zhang, X. Prediction of polyimide materials with high glass-transition temperatures. J. Polym. Sci. Part B-Polym. Phys. 2001, 39, 2243–2251. [Google Scholar] [CrossRef]
- Mohammadi, M.; Fazli, H.; Karevan, M.; Davoodi, J. The glass transition temperature of PMMA: A molecular dynamics study and comparison of various determination methods. Eur. Polym. J. 2017, 91, 121–133. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Wang, W.-H.; Zhang, Z.; Xu, L.; Li, P. Study of the glass transition temperature and the mechanical properties of PET/modified silica nanocomposite by molecular dynamics simulation. Eur. Polym. J. 2016, 75, 36–45. [Google Scholar] [CrossRef]
- Sun, H. Compass: An ab initio force-field optimized for condensed-phase applications—Overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Zheng, Q.; Xue, Q.; Yan, K.; Gao, X.; Li, Q.; Hao, L. Effect of chemisorption on the interfacial bonding characteristics of carbon nanotube-polymer composites. Polymer 2008, 49, 800–808. [Google Scholar] [CrossRef]
- Yani, Y.; Lamm, M.H. Molecular dynamics simulation of mixed matrix nanocomposites containing polyimide and polyhedral oligomeric silsesquioxane (POSS). Polymer 2009, 50, 1324–1332. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03 (Revision C.01); Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Nogi, M.; Kim, C.; Sugahara, T.; Inui, T.; Takahashi, T.; Suganuma, K. High thermal stability of optical transparency in cellulose nanofiber paper. Appl. Phys. Lett. 2013, 102, 181911. [Google Scholar] [Green Version]
- Wu, C.; Xu, W. Atomistic molecular simulations of structure and dynamics of crosslinked epoxy resin. Polymer 2007, 48, 5802–5812. [Google Scholar] [CrossRef]
- Morita, H.; Tanaka, K.; Kajiyama, T.; Nishi, T.; Doi, M. Study of the glass transition temperature of polymer surface by coarse-grained molecular dynamics simulation. Macromolecules 2006, 39, 6233–6237. [Google Scholar] [CrossRef]
- Yang, H.; Li, Z.-S.; Qian, H.-J.; Yang, Y.-B.; Zhang, X.-B.; Sun, C.-C. Molecular dynamics simulation studies of binary blend miscibility of poly(3-hydroxybutyrate) and poly(ethylene oxide). Polymer 2004, 45, 453–457. [Google Scholar] [CrossRef]
- Yu, K.-Q.; Li, Z.-S.; Sun, J. Polymer structures and glass transition: A molecular dynamics simulation study. Macromol. Theory Simul. 2001, 10, 624–633. [Google Scholar] [CrossRef]
- Luo, Z.; Jiang, J. Molecular dynamics and dissipative particle dynamics simulations for the miscibility of poly(ethylene oxide)/poly(vinyl chloride) blends. Polymer 2010, 51, 219–299. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample code | Tgexp (K) | Tg (K) | αexp (ppm·K−1) | αglassy (ppm·K−1) | αrubbery (ppm·K−1) |
|---|---|---|---|---|---|
| PEsI–(CH3)1 | – | 701.5 | NA | 11.2 | 21.4 |
| PEsI–(CH3)2 | >623 | 720.8 | 11.3 | 11.3 | 21.7 |
| PEsI–(CH3)3 | – | 737.4 | – | 12.6 | 20.8 |
| PEsI–(CF3)1 | – | 730.9 | – | 12.8 | 22.3 |
| PEsI–(CF3)2 | – | 714.9 | – | 13.4 | 22.3 |
| PEsI–(CF3)3 | – | 696.9 | – | 9.5 | 21.3 |
| PEsI–H | >623 | 676.9 | 8.2 | 9.7 | 19.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, K.-H.; Choi, H.K.; Yeo, H.; You, N.-H.; Ku, B.-C.; Yu, J. Molecular Design and Property Prediction of Sterically Confined Polyimides for Thermally Stable and Transparent Materials. Polymers 2018, 10, 630. https://doi.org/10.3390/polym10060630
Nam K-H, Choi HK, Yeo H, You N-H, Ku B-C, Yu J. Molecular Design and Property Prediction of Sterically Confined Polyimides for Thermally Stable and Transparent Materials. Polymers. 2018; 10(6):630. https://doi.org/10.3390/polym10060630
Chicago/Turabian StyleNam, Ki-Ho, Hoi Kil Choi, Hyeonuk Yeo, Nam-Ho You, Bon-Cheol Ku, and Jaesang Yu. 2018. "Molecular Design and Property Prediction of Sterically Confined Polyimides for Thermally Stable and Transparent Materials" Polymers 10, no. 6: 630. https://doi.org/10.3390/polym10060630