Photoinduced Phase Transition in Strongly Electron-Lattice and Electron–Electron Correlated Molecular Crystals

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
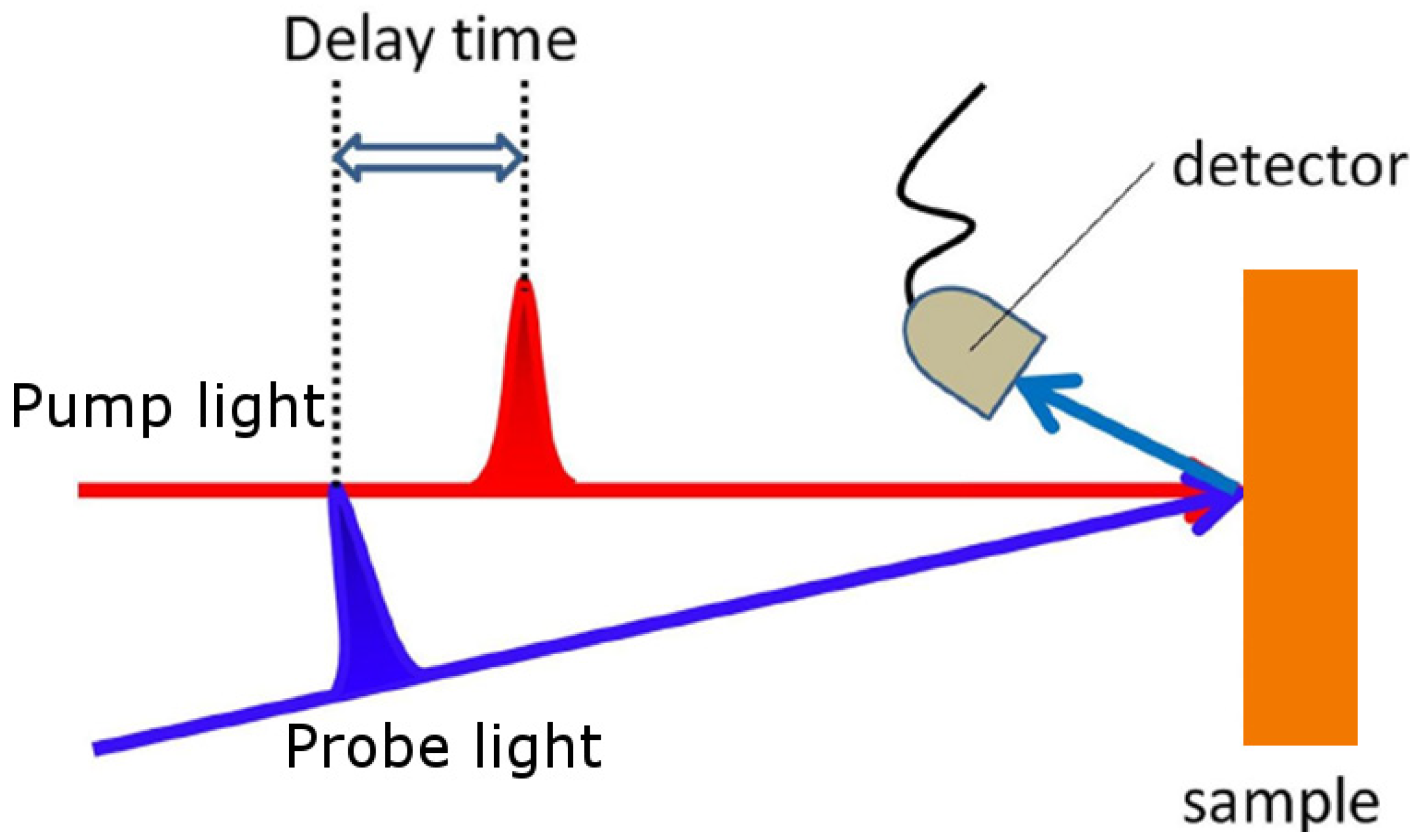
2. Experimental Setup

3. Results and Discussion
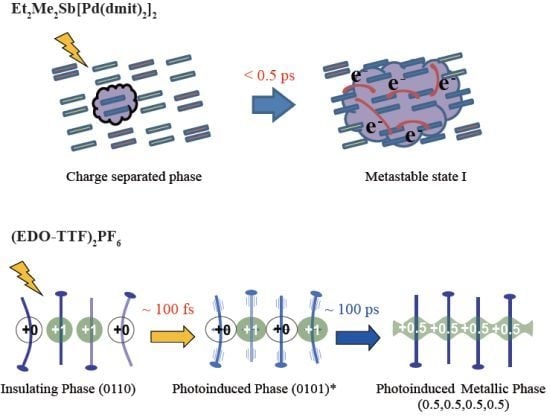
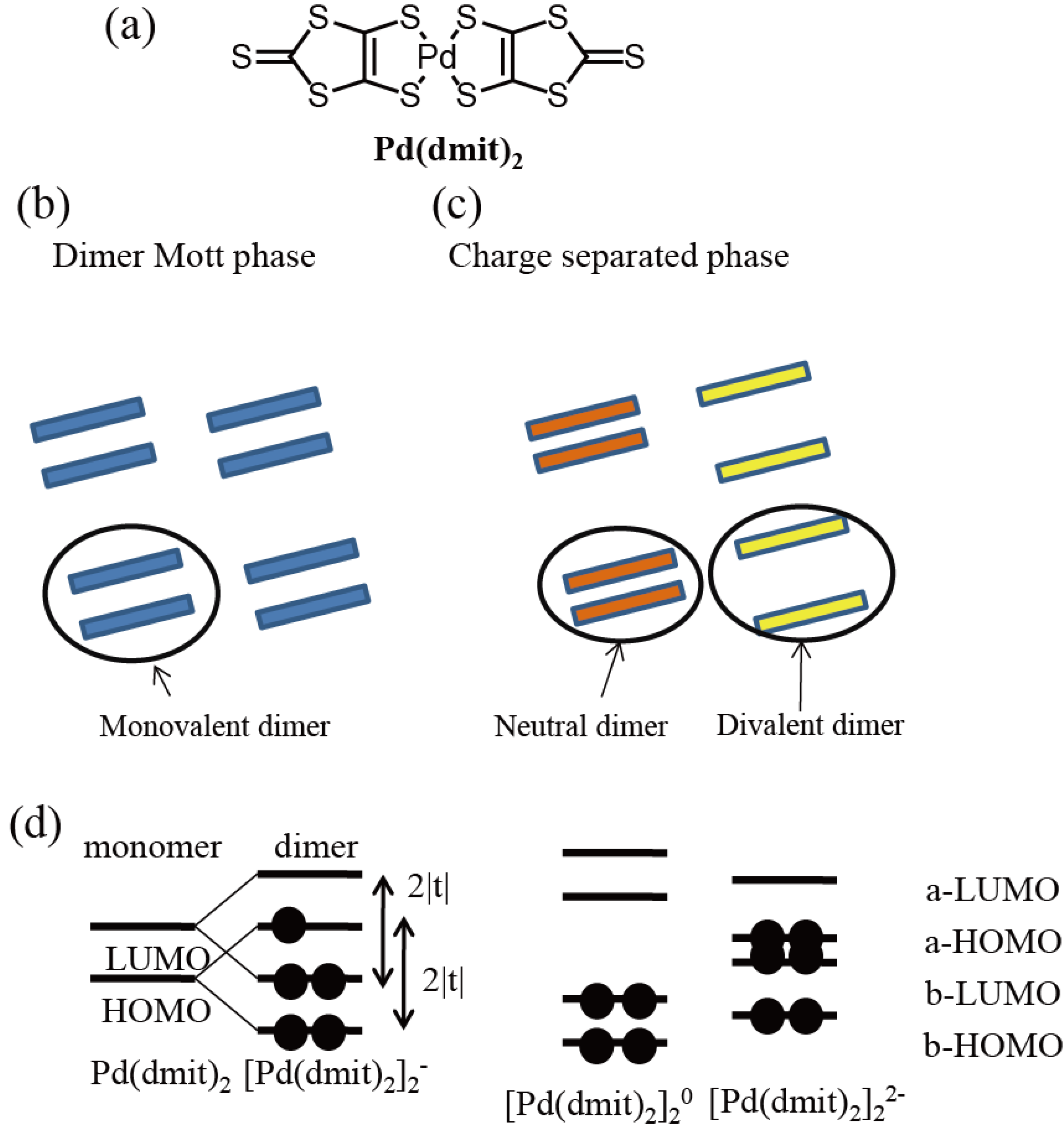
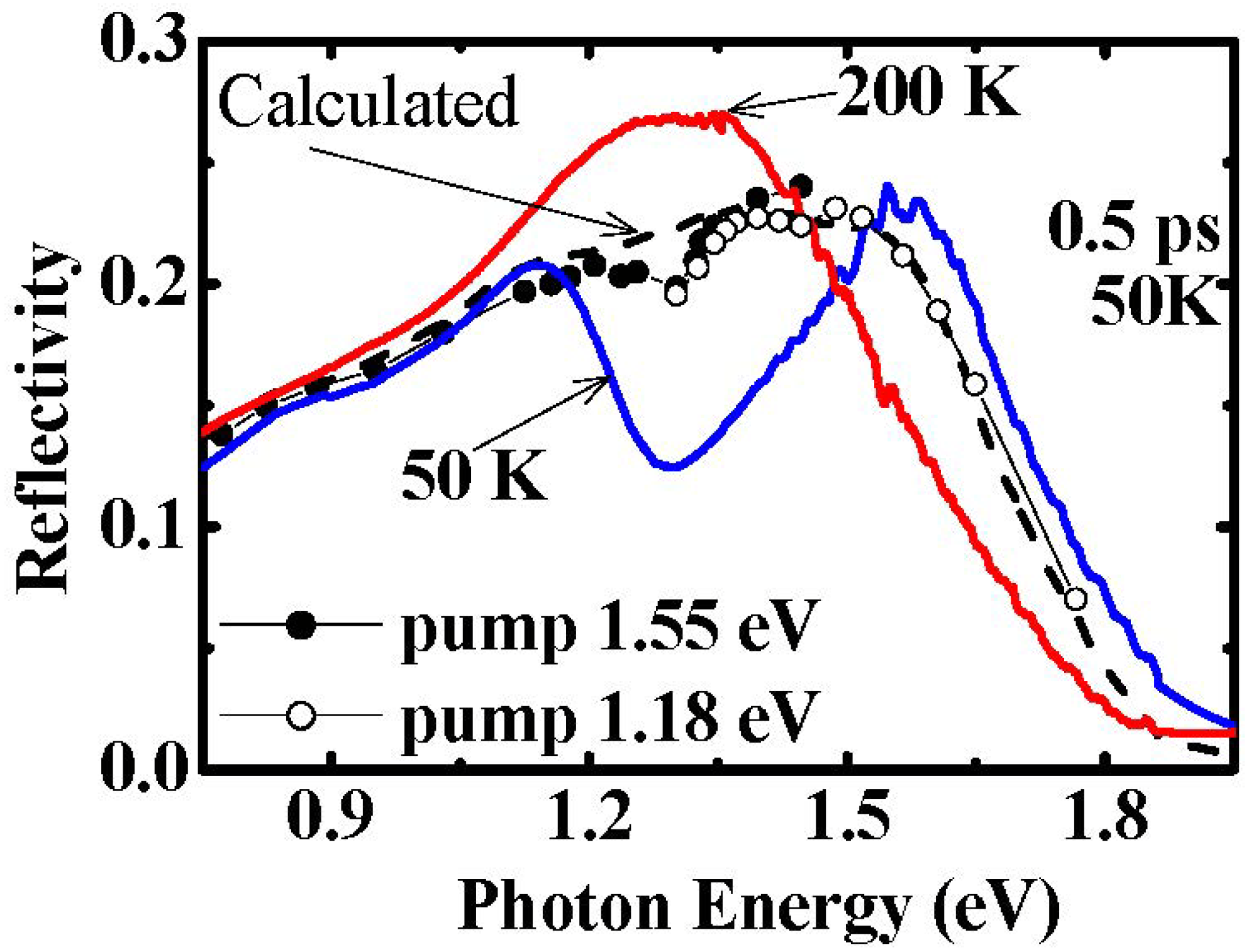
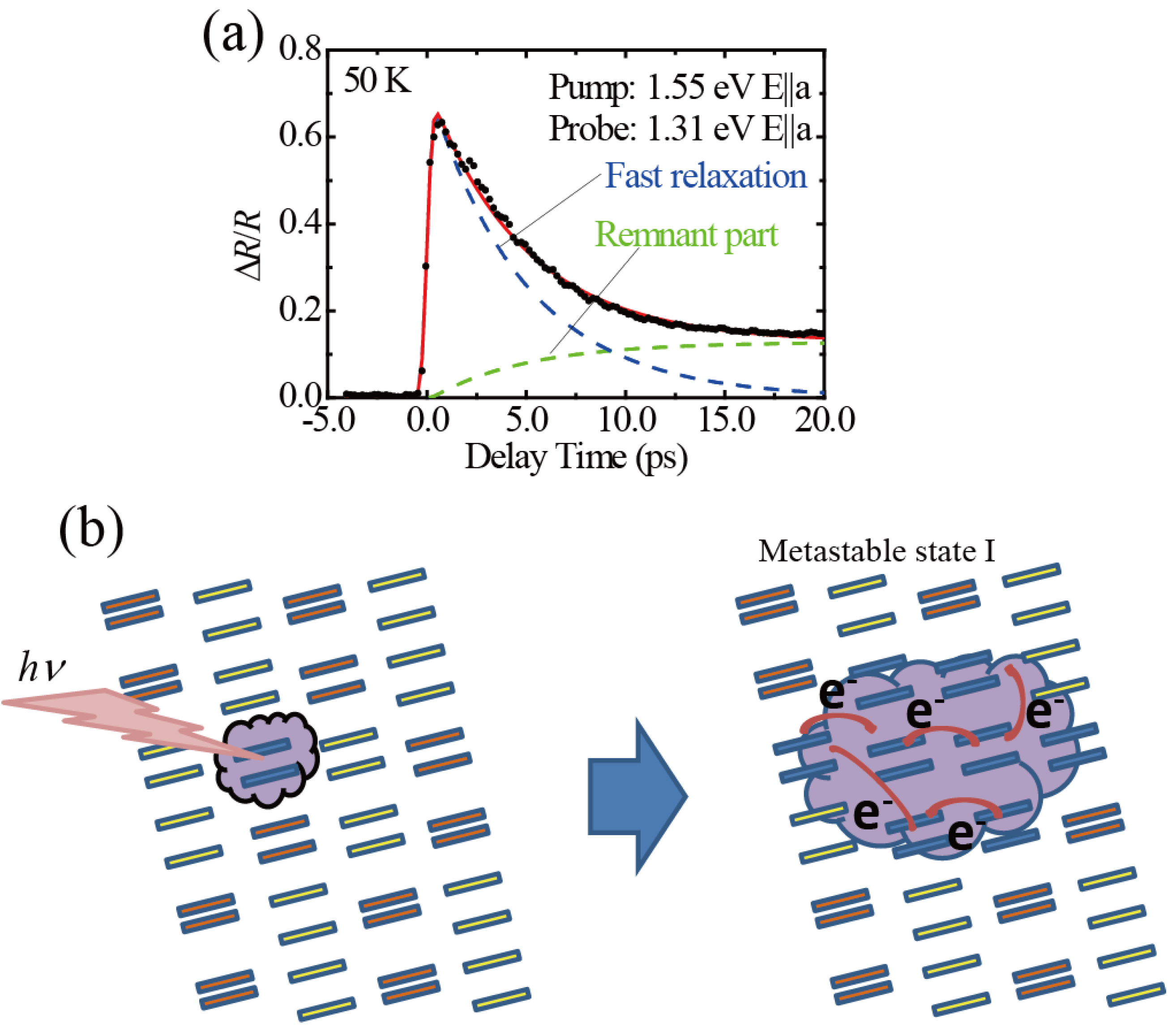
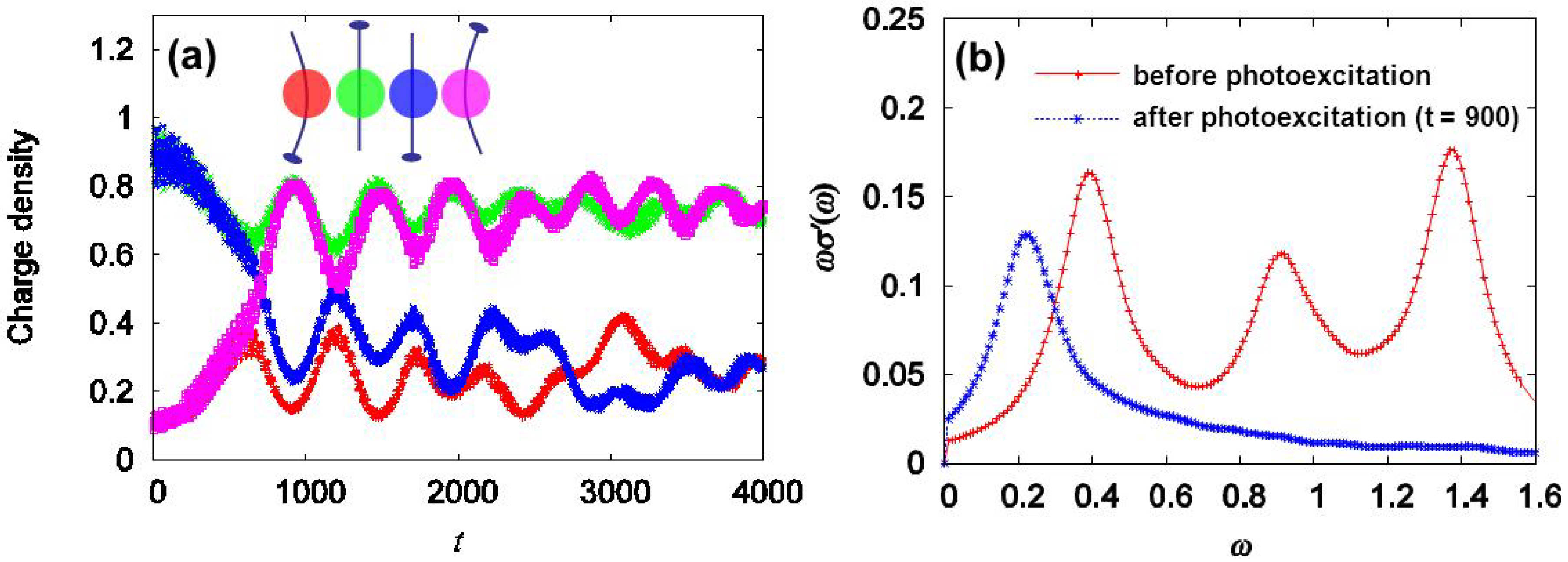
3.1. Photoinduced Phase Transition from Charge Separated Phase to Dimer Mott Insulating Phase in Et2Me2Sb[Pd(dmit)2]2 (dmit = 1,3-Dithiol-2-thione-4,5-dithiolate)


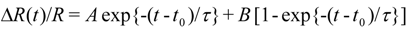
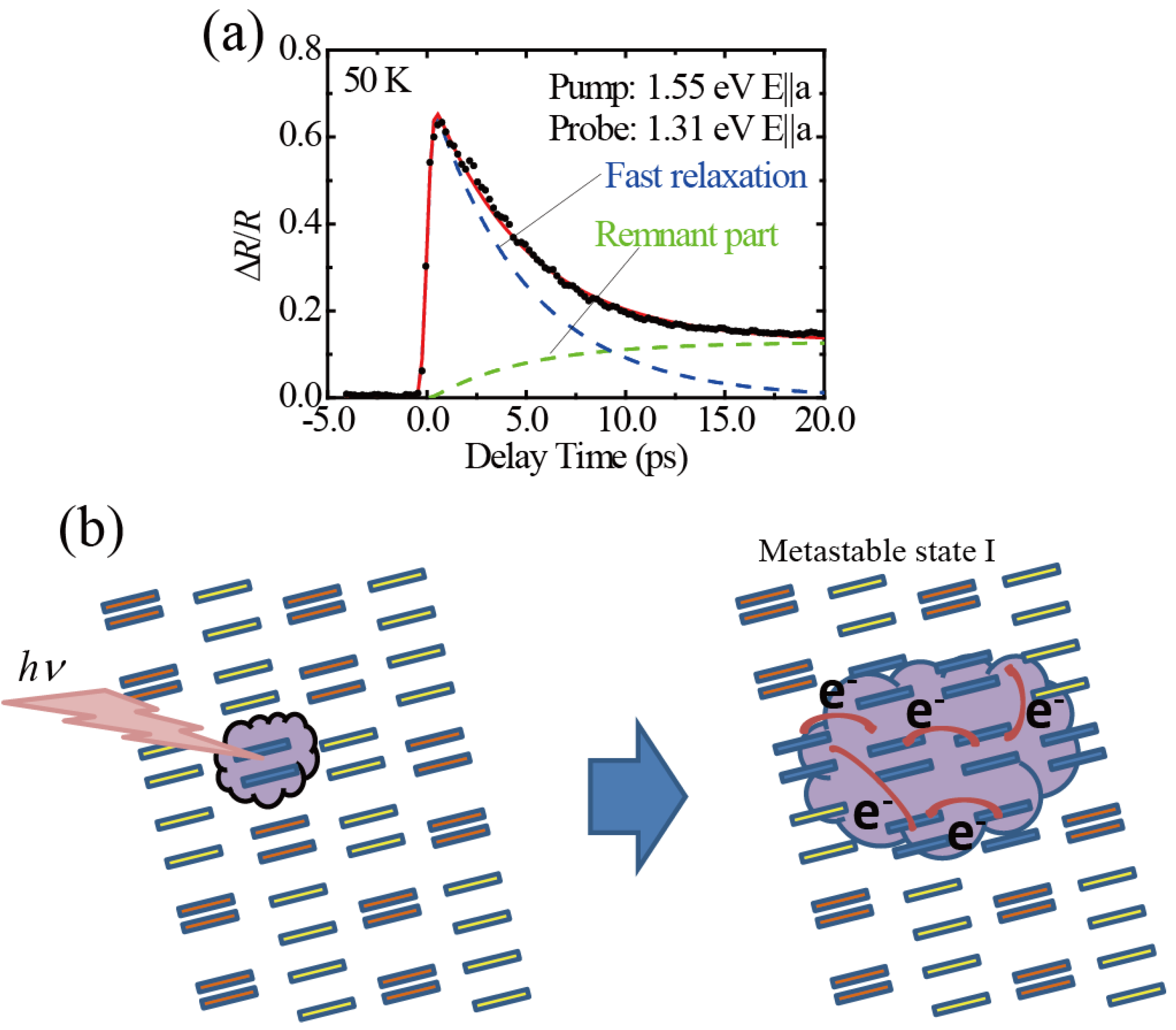
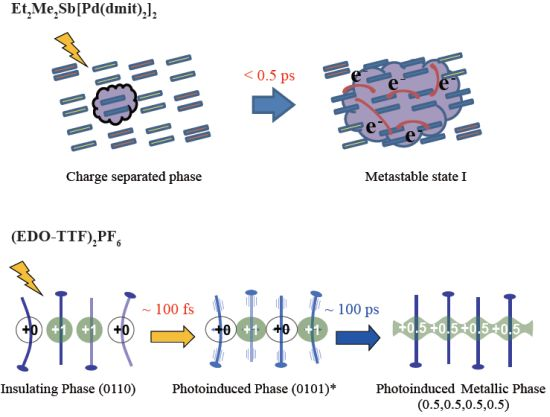
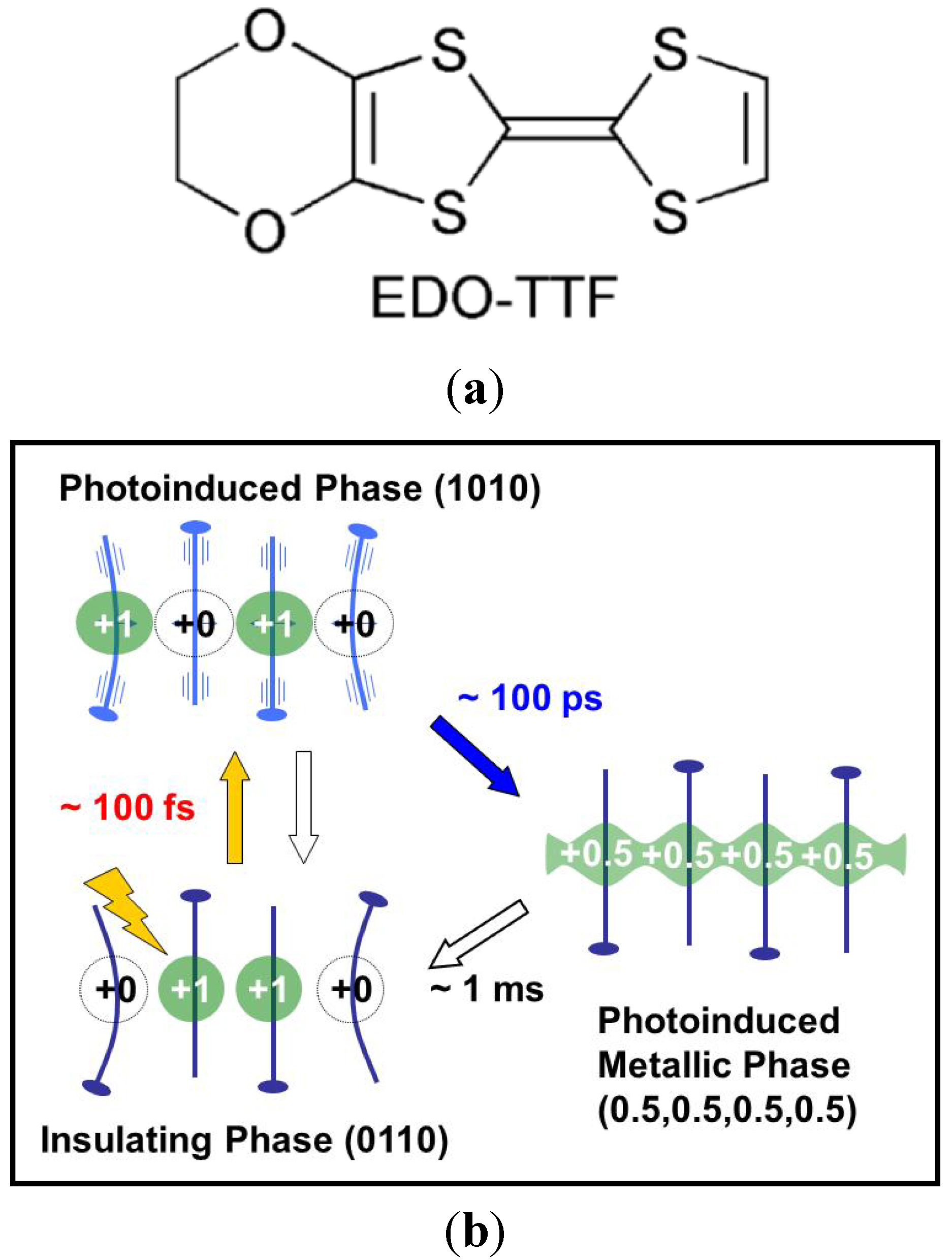
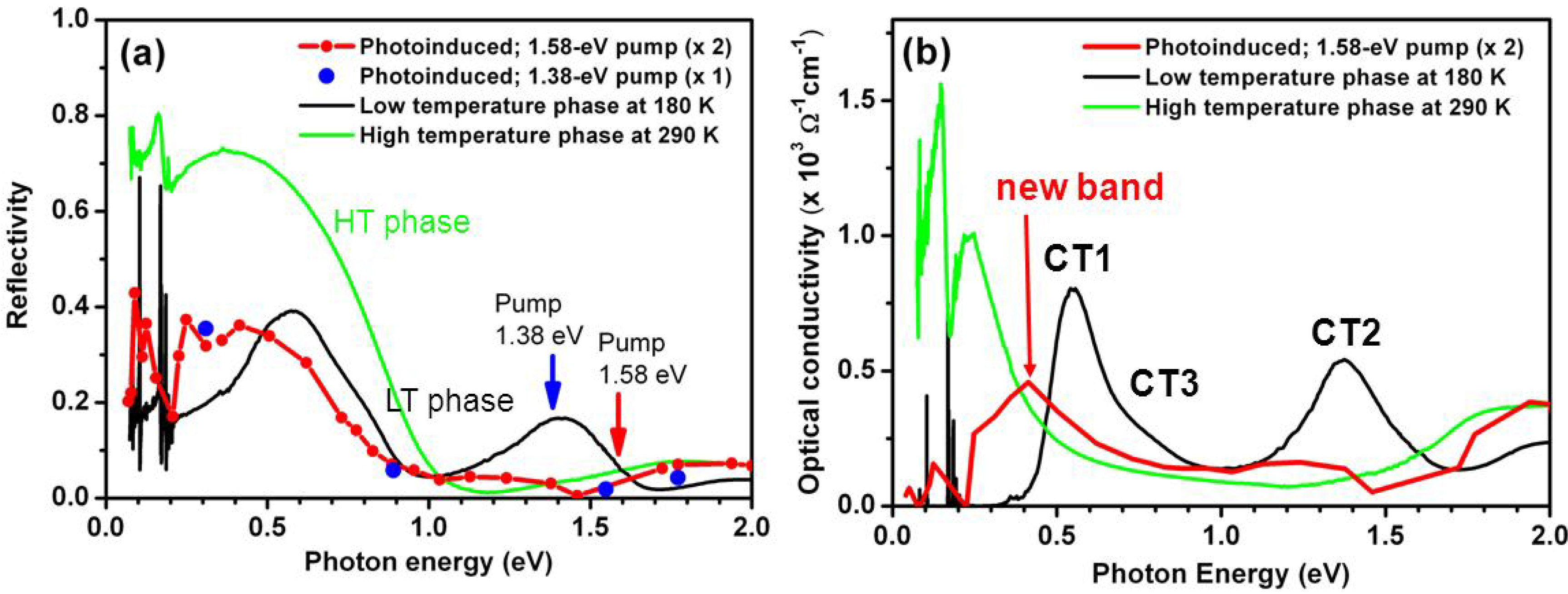
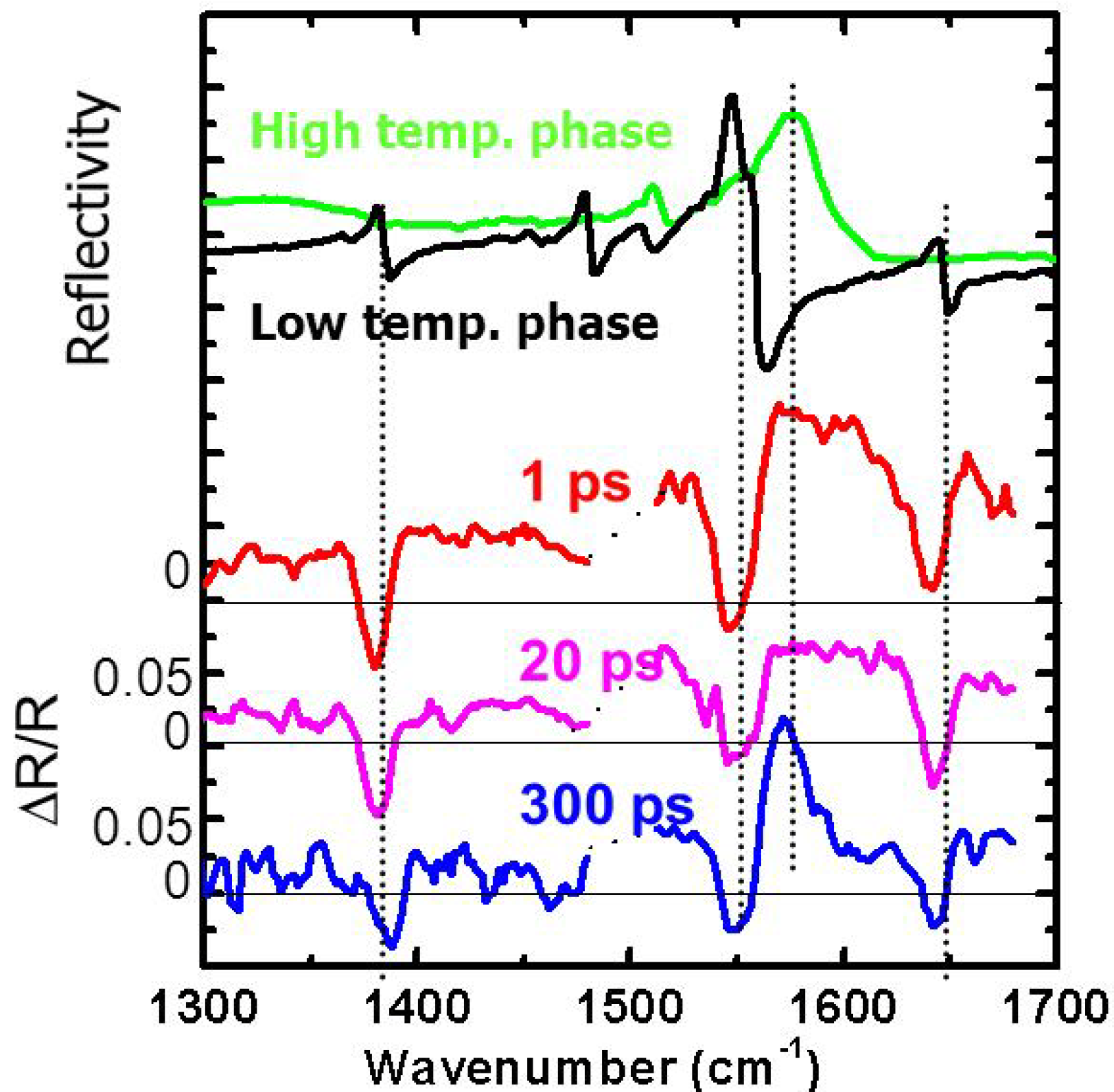
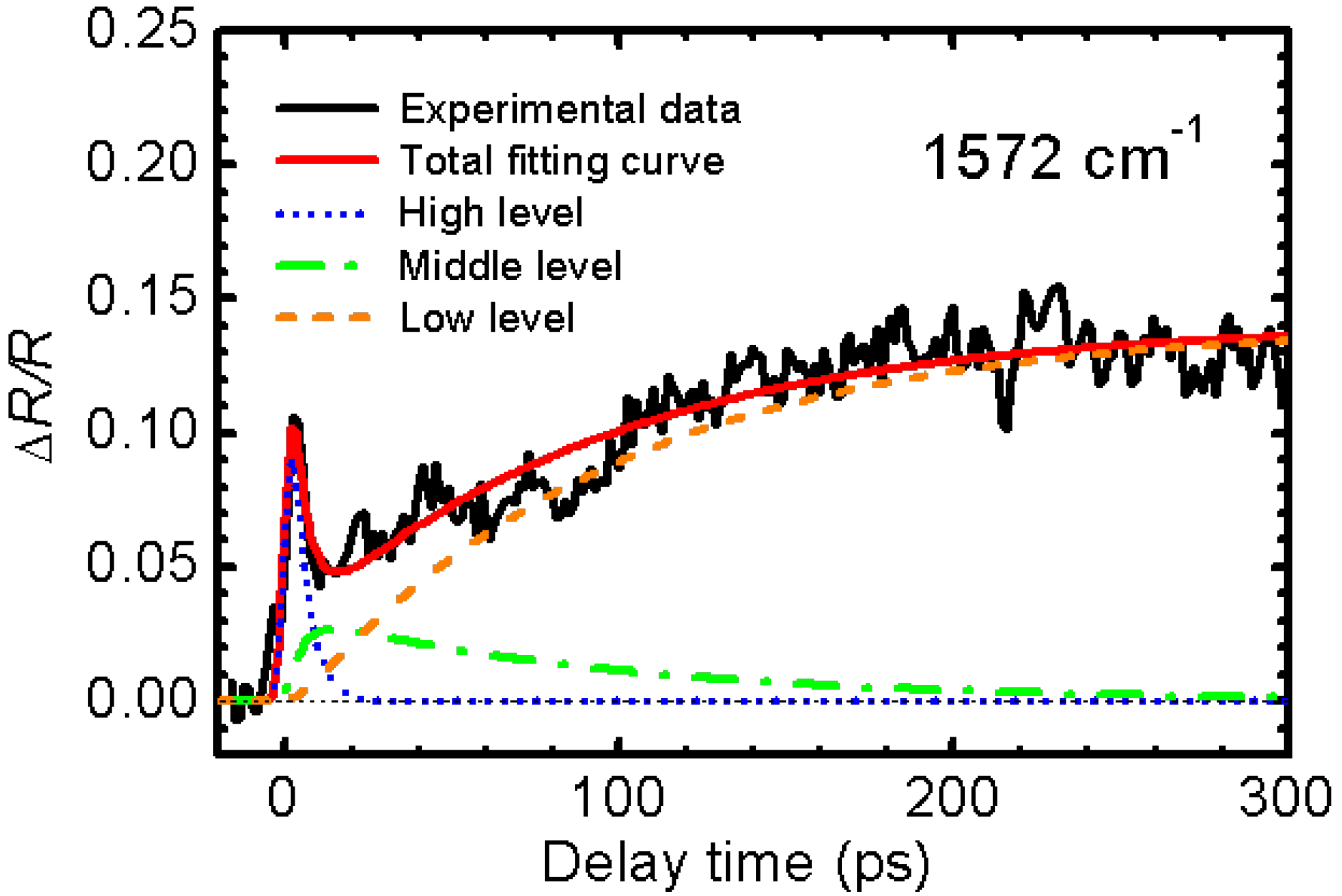
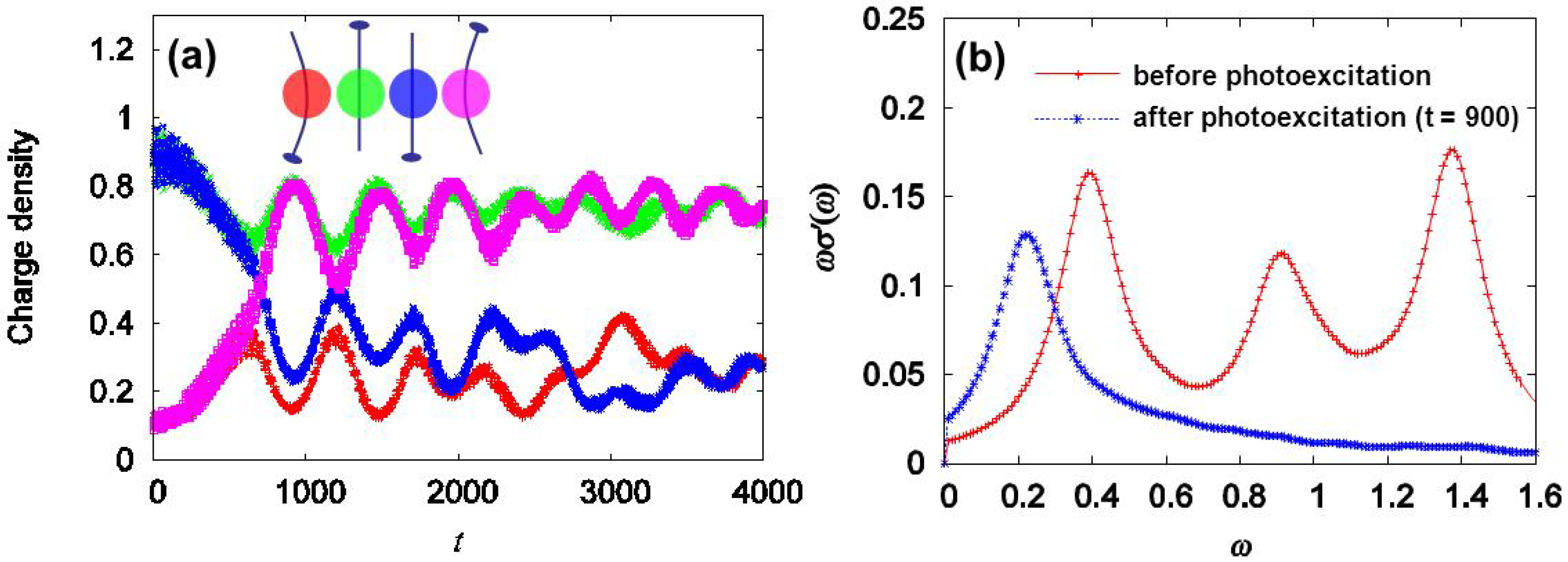
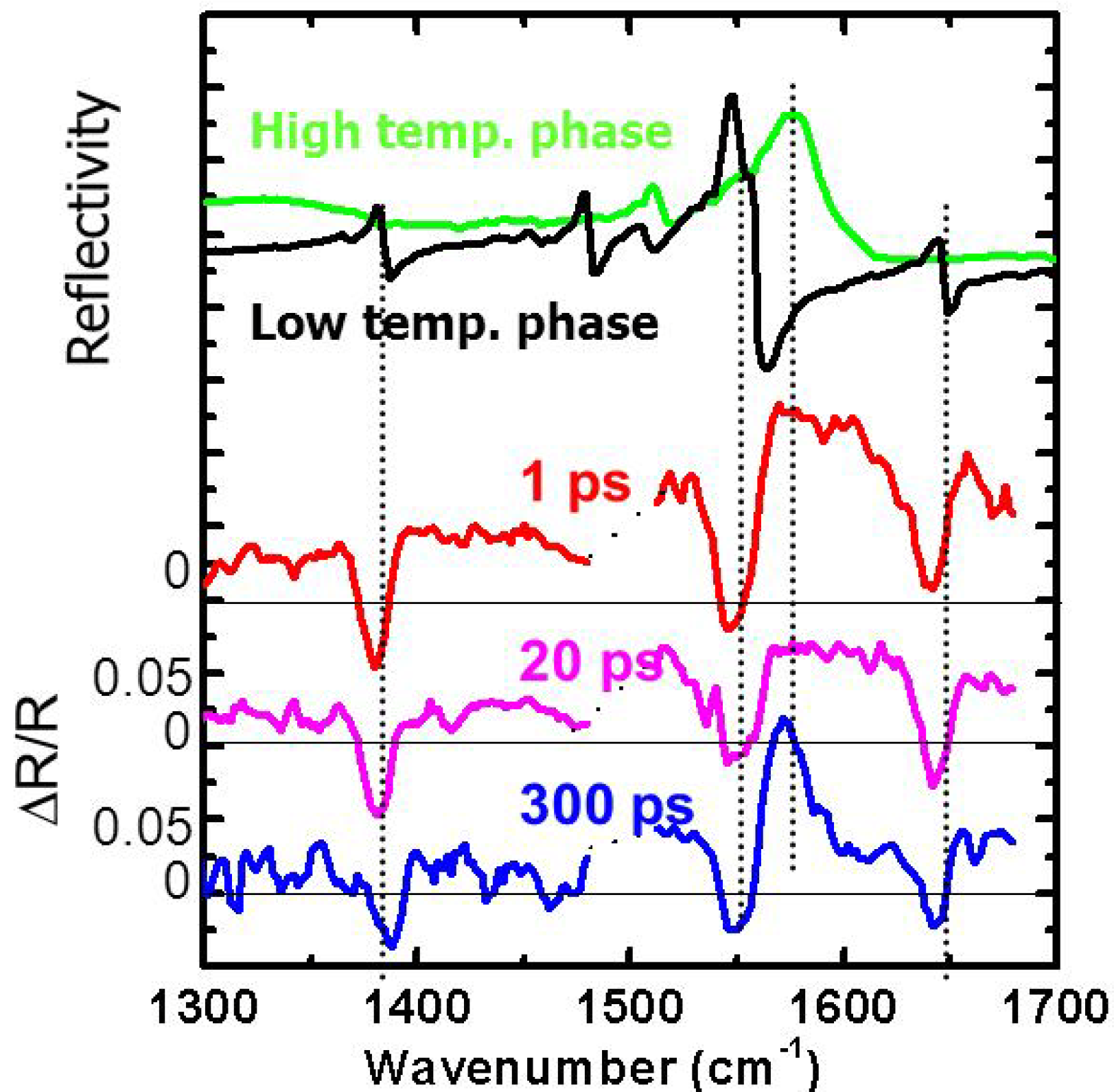
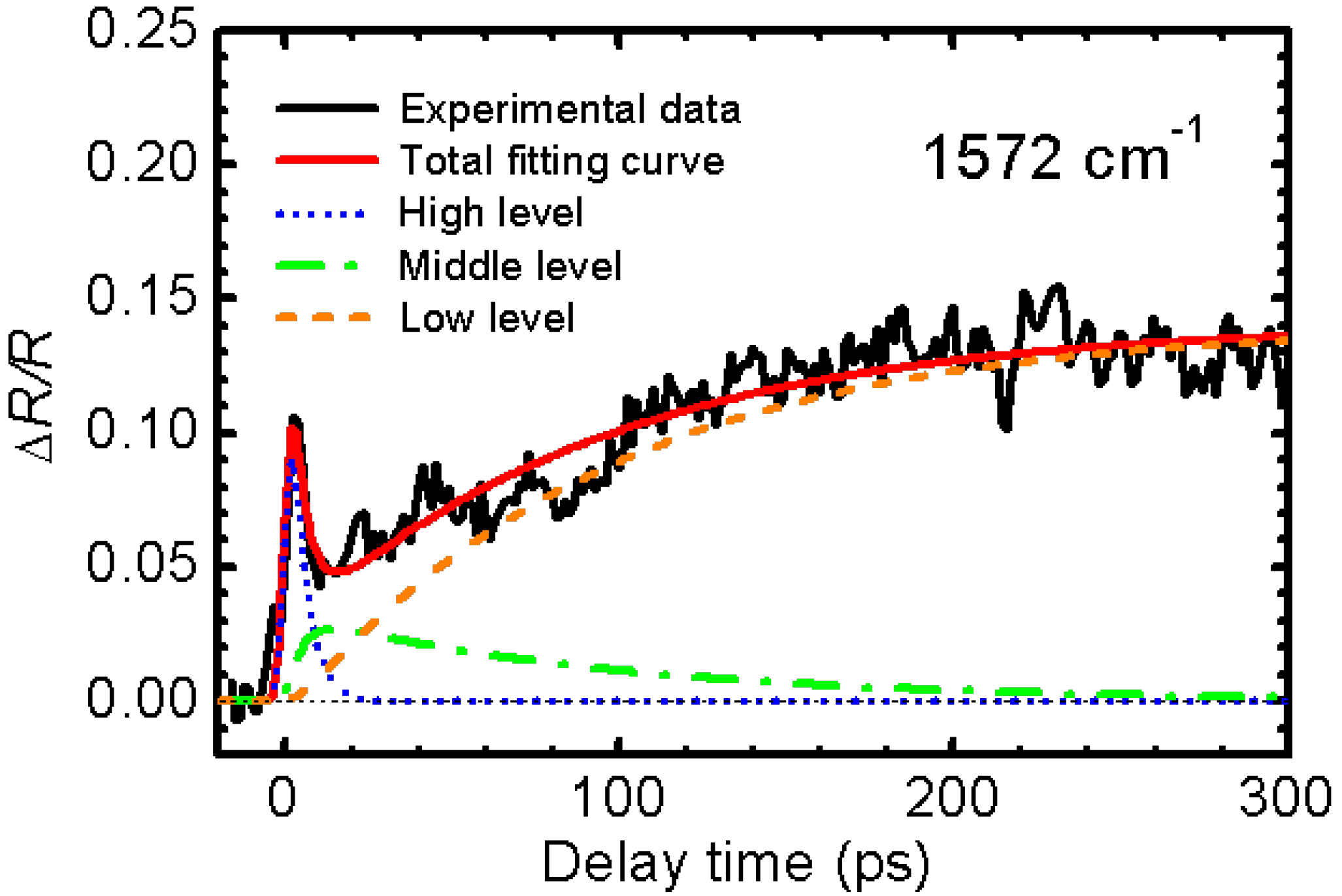
3.2. Photoinduced Phase Transition in Strong Electron-Lattice Interaction System: (EDO-TTF)2PF6


4. Summary
Acknowledgments
Conflict of Interest
References
- Dagotto, E. Complexity in strongly correlated electronic systems. Science 2005, 309, 257–262. [Google Scholar] [CrossRef]
- Photoinduced Phase Transitions; Nasu, K. (Ed.) World Scientific Pub Co Inc: Singapore, 2004.
- Koshihara, S.; Adachi, S. Photo-Induced phase transition in an electron-lattice correlated system—Future role of a time-resolved X-ray measurement for materials science. J. Phys. Soc. Jpn. 2006, 75, 011005:1–011005:10. [Google Scholar]
- Koshihara, S.; Tokura, Y.; Mitani, T.; Saito, G.; Koda, T. Photoinduced valence instability in the organic molecular compound tetrathiafulvalene-p-chloranil (TTF-CA). Phys. Rev. 1990, 42, 6853–6856. [Google Scholar] [CrossRef]
- Suzuki, T.; Sakamaki, T.; Tanimura, K.; Koshihara, S.; Tokura, Y. Ionic-to-neutral phase transformation induced by photoexcitation of the charge-transfer band in tetrathiafulvalene-p-chloranil crystals. Phys. Rev. 1999, 60, 6191–6193. [Google Scholar] [CrossRef]
- Collet, E.; Lemée-Cailleau, M.-H.; Buron-Le Cointe, M.; Cailleau, H.; Wulff, M.; Luty, T.; Koshihara, S.-Y.; Meyer, M.; Toupet, L.; Rabiller, P.; et al. Laser-induced ferroelectric structural order in an organic charge-transfer crystal. Science 2003, 300, 612–615. [Google Scholar]
- Okamoto, H.; Ishige, Y.; Tanaka, S.; Kishida, H.; Iwai, S.; Tokura, Y. Photoinduced phase transition in tetrathiafulvalene-p-chloranil observed in femtosecond reflection spectroscopy. Phys. Rev. 2004, 70, 165202:1–165202:18. [Google Scholar]
- Matsubara, Y.; Okimoto, Y.; Yoshida, T.; Ishikawa, T.; Koshihara, S.; Onda, K. Photoinduced neutral-to-ionic phase transition in tetrathiafulvalene-p-chloranil studied by time-resolved vibrational spectroscopy. J. Phys. Soc. Jpn. 2011, 80, 124711:1–124711:5. [Google Scholar]
- Matsubara, Y.; Yoshida, T.; Ishikawa, T.; Okimoto, Y.; Koshihara, S.; Onda, K. Photoinduced ionic to neutral phase transition in TTF-CA studied by time-resolved infrared vibrational spectroscopy. Acta Phys. Pol. 2012, 121, 340–342. [Google Scholar]
- Koshihara, S.; Tokura, Y.; Iwasa, Y.; Koda, T. Inverse Peierls transition induced by photoexcitation in potassium tetracyanoquinodimethane crystals. Phys. Rev. 1991, 44, 431–434. [Google Scholar] [CrossRef]
- Tanimura, K.; Akimoto, I. Femtosecond time-resolved spectroscopy of photoinduced ionic-to-neutral phase transition in tetrathiafulvalen-p-chloranil crystals. J. Luminesc. 2001, 94–95, 483–488. [Google Scholar] [CrossRef]
- Iwai, S.; Tanaka, S.; Fujinuma, K.; Kishida, H.; Okamoto, H.; Tokura, Y. Ultrafast optical switching from an ionic to a neutral state in tetrathiafulvalene-p-chloranil (TTF-CA) observed in femtosecond reflection spectroscopy. Phys. Rev. Lett. 2002, 88, 057402:1–057402:4. [Google Scholar]
- Uemura, H.; Okamoto, H. Direct detection of the ultrafast response of charges and molecules in the photoinduced neutral-to-ionic transition of the organic tetrathiafulvalene-p-chloranil solid. Phys. Rev. Lett. 2010, 105. [Google Scholar]
- Itatani, J.; Rini, M.; Cavalleri, A.; Onda, K.; Ishikawa, T.; Koshihara, S.; Shao, X.; Yamochi, H.; Saito, G.; Shoenlein, R.W. Ultrafast Gigantic Photo-Response in (EDO-TTF)2PF6 Initiated by 10-fs Laser Pulses. In Ultrafast Phenomena XV; Corkum, P., Jonas, D.M., Miller, D.R., Weiner, A.M., Eds.; Springer-Verlag: Berlin, Germany, 2007; pp. 621–623. [Google Scholar]
- Kawakami, Y.; Iwai, S.; Fukatsu, T.; Miura, M.; Yoneyama, N.; Sasaki, T.; Kobayashi, N. Optical modulation of effective on-site coulomb energy for the mott transition in an organic dimer insulator. Phys. Rev. Lett. 2009, 103, 066403:1–066403:4. [Google Scholar]
- Fukazawa, N.; Shimizu, M.; Ishikawa, T.; Okimoto, Y.; Koshihara, S.; Hiramatsu, T.; Nakano, Y.; Yamochi, H.; Saito, G.; Onda, K. Charge and structural dynamics in photoinduced phase transition of (edo-ttf)2pf6 examined by picosecond time-resolved vibrational spectroscopy. J. Phys. Chem. 2012, 116, 5892–5899. [Google Scholar]
- Demsar, J.; Podobnik, B.; Kabanov, V.; Wolf, T.; Mihailovic, D. Superconducting Gap Δc, the Pseudogap Δp, and Pair Fluctuations above Tc in Overdoped Y1-xCaxBa2Cu3O7-δ from Femtosecond Time-Domain Spectroscopy. Phys. Rev. Lett. 1999, 82, 4918–4921. [Google Scholar] [CrossRef]
- Gedik, N.; Yang, D.-S.; Logvenov, G.; Bozovic, I.; Zewail, A.H. Nonequilibrium phase transitions in cuprates observed by ultrafast electron crystallography. Science 2007, 316, 425–429. [Google Scholar]
- Tomeljak, A.; Schäfer, H.; Städter, D.; Beyer, M.; Biljakovic, K.; Demsar, J. Dynamics of photoinduced charge-density-wave to metal phase transition in K0.3MoO3. Phys. Rev. Lett. 2009, 102. [Google Scholar]
- Miyano, K.; Tanaka, T.; Tomioka, Y.; Tokura, Y. Photoinduced insulator-to-metal transition in a perovskite manganite. Phys. Rev. Lett. 1997, 78, 4257–4260. [Google Scholar]
- Cavalleri, A.; Tóth, C.; Siders, C.W.; Squier, J.A.; Ráksi, F.; Forget, P.; Kieffer, J.C. Femtosecond structural dynamics in Vo2 during an ultrafast solid-solid phase transition. Phys. Rev. Lett. 2001, 87, 237401:1–237401:4. [Google Scholar]
- Perfetti, L.; Loukakos, P.; Lisowski, M.; Bovensiepen, U.; Berger, H.; Biermann, S.; Cornaglia, P.; Georges, A.; Wolf, M. Time evolution of the electronic structure of 1T-TaS2 through the insulator-metal transition. Phys. Rev. Lett. 2006, 97, 067402:1–067402:4. [Google Scholar]
- Watanabe, S.; Kondo, R.; Kagoshima, S.; Shimano, R. Observation of ultrafast photoinduced closing and recovery of the spin-density-wave gap in (TMTSF)2PF6. Phys. Rev. 2009, 80, 220408(R):1–220408(R):4. [Google Scholar]
- Guérin, L.; Hébert, J.; Buron-Le Cointe, M.; Adachi, S.; Koshihara, S.; Cailleau, H.; Collet, E. Capturing one-dimensional precursors of a photoinduced transformation in a material. Phys. Rev. Lett. 2010, 105, 31–34. [Google Scholar]
- Sciaini, G.; Miller, R.J.D. Femtosecond electron diffraction: Heralding the era of atomically resolved dynamics. Rep. Prog. Phys. 2011, 74. [Google Scholar]
- Kato, R. Conducting metal dithiolene complexes: Structural and electronic properties. Chem. Rev. 2004, 104, 5319–5346. [Google Scholar] [CrossRef]
- Ota, A.; Yamochi, H.; Saito, G. A novel metal-insulator phase transition observed in (EDO-TTF)2PF6. J. Mater. Chem. 2002, 12, 2600–2602. [Google Scholar]
- Ishikawa, T.; Fukazawa, N.; Matsubara, Y.; Nakajima, R.; Onda, K.; Okimoto, Y.; Koshihara, S.; Lorenc, M.; Collet, E.; Tamura, M.; et al. Large and ultrafast photoinduced reflectivity change in the charge separated phase of Et2Me2Sb[Pd(1,3-dithiol-2-thione-4,5-dithiolate)2]2. Phys. Rev. 2009, 80, 115108:316–115108:318. [Google Scholar]
- Onda, K.; Ogihara, S.; Yonemitsu, K.; Maeshima, N.; Ishikawa, T.; Okimoto, Y.; Shao, X.; Nakano, Y.; Yamochi, H.; Saito, G.; et al. Photoinduced change in the charge order pattern in the quarter-filled organic conductor (EDO-TTF)2PF6 with a strong electron-phonon interaction. Phys. Rev. Lett. 2008, 101, 067403:1–067403:4. [Google Scholar]
- Tamura, M.; Takenaka, K.; Takagi, H.; Sugai, S.; Tajima, A.; Kato, R. Spectroscopic evidence for the low-temperature charge-separated state of [Pd(dmit)2] salts. Chem. Phys. Lett. 2005, 411, 133–137. [Google Scholar] [CrossRef]
- Nakao, A.; Kato, R. Structural study of low temperature charge-separated phases of Pd(dmit)2-based molecular conductors. J. Phys. Soc. Jpn. 2005, 74, 2754–2763. [Google Scholar] [CrossRef]
- Tamura, M.; Kato, R. Variety of valence bond states formed of frustrated spins on triangular lattices based on a two-level system Pd(dmit)2. Sci. Technol. Adv. Mater. 2009, 10. [Google Scholar] [CrossRef]
- Iwai, S.; Yamamoto, K.; Kashiwazaki, A.; Hiramatsu, F.; Nakaya, H.; Kawakami, Y.; Yakushi, K.; Okamoto, H.; Mori, H.; Nishio, Y. Photoinduced melting of a stripe-type charge-order and metallic domain formation in a layered BEDT-TTF-Based organic salt. Phys. Rev. Lett. 2007, 98. [Google Scholar] [CrossRef]
- Fukazawa, N.; Tanaka, T.; Ishikawa, T.; Okimoto, Y.; Koshihara, S.; Yamamoto, T.; Tamura, M.; Kato, R.; Onda, K. Photoinduced Phase Transition of the Pd(dmit)2 Salts Having Different Order of Phase Transition Examined by Time-Resolved Vibrational Spectroscopy. 2012. in preparation. [Google Scholar]
- Underhill, A.E.; Clark, R.A.; Marsden, I.; Allan, M.; Friend, R.H.; Tajima, H.; Naito, T.; Tamura, M.; Kuroda, H.; Kobayashi, A.; Kobayashi, H.; et al. Structural and electronic properties of Cs[Pd(dmit)2]2. J. Phys. Condens. Matter 1991, 3, 933–954. [Google Scholar]
- Tajima, H.; Naito, T.; Tamura, M.; Kobayashi, A.; Kuroda, H.; Kato, R.; Kobayashi, H.; Clark, R.A.; Underhill, A.E. Energy level inversion in strongly dimerized [Pd(dmit)2] salts. Solid State Commun. 1991, 79, 337–341. [Google Scholar] [CrossRef]
- Ishikawa, T.; Tanaka, T.; Fukazawa, N.; Matsubara, Y.; Okimoto, Y.; Onda, K.; Koshihara, S.; Tamura, M.; Kato, R. Slow dynamics of the photoinduced phase transition in Pd(dmit)2 salts. Acta Phys. Pol. 2012, 121, 316–318. [Google Scholar]
- Ota, A.; Yamochi, H.; Saito, G. A novel metal-insulator transition in (EDO-TTF)2X (X = PF6, AsF6). Synth. Met. 2003, 133–134, 463–465. [Google Scholar] [CrossRef]
- Drozdova, O.; Yakushi, K.; Ota, A.; Yamochi, H.; Saito, G. Spectroscopic study of the [0110] charge ordering in (EDO-TTF)2PF6. Synth. Met. 2003, 133–134, 277–279. [Google Scholar] [CrossRef]
- Drozdova, O.; Yakushi, K.; Yamamoto, K.; Ota, A.; Yamochi, H.; Saito, G.; Tashiro, H.; Tanner, D. Optical characterization of 2kF bond-charge-density wave in quasi-one-dimensional 3/4-filled (EDO-TTF)2X (X = PF6 and AsF6). Phys. Rev. 2004, 70, 075107:1–075107:21. [Google Scholar]
- Aoyagi, S.; Kato, K.; Ota, A.; Yamochi, H.; Saito, G.; Suematsu, H.; Sakata, M.; Takata, M. Direct observation of bonding and charge ordering in (EDO-TTF)2PF6. Angew. Chem. Int. Ed. 2004, 43, 3670–3673. [Google Scholar]
- Chollet, M.; Guerin, L.; Uchida, N.; Fukaya, S.; Shimoda, H.; Ishikawa, T.; Matsuda, K.; Hasegawa, T.; Ota, A.; Yamochi, H.; et al. Gigantic photoresponse in 1/4-filled-band organic salt (EDO-TTF)2PF6. Science 2005, 307, 86–89. [Google Scholar]
- Onda, K.; Ishikawa, T.; Chollet, M.; Shao, X.; Yamochi, H.; Saito, G.; Koshihara, S. Ultrafast infrared spectroscopic study of the photo-induced phase transition in (EDO-TTF)2PF6. J. Phys. Conf. Ser. 2005, 21, 216–220. [Google Scholar] [CrossRef]
- Onda, K.; Ogihara, S.; Ishikawa, T.; Okimoto, Y.; Shao, X.; Nakano, Y.; Yamochi, H.; Saito, G.; Koshihara, S. Anomalous photo-induced response by double-pulse excitation in the organic conductor (EDO-TTF)2PF6. J. Phys. Conf. Ser. 2009, 148. [Google Scholar] [CrossRef]
- Onda, K.; Shimizu, M.; Sakaguchi, F.; Ogihara, S.; Ishikawa, T.; Okimoto, Y.; Koshihara, S.; Shao, X.F.; Nakano, Y.; Yamochi, H.; et al. Ultrafast and large reflectivity change by ultraviolet excitation of the metallic phase in the organic conductor (EDO-TTF)2PF6. Physica 2010, 405, S350–S352. [Google Scholar] [CrossRef]
- Yonemitsu, K.; Maeshima, N. Photoinduced melting of charge order in a quarter-filled electron system coupled with different types of phonons. Phys. Rev. 2007, 76. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kitayama, M.; Chono, A.; Onda, K.; Okimoto, Y.; Koshihara, S.; Nakano, Y.; Yamochi, H.; Morikawa, T.; Shirahata, T.; et al. Probing the metal-insulator phase transition in the (DMEDO-EBDT)2PF6 single crystal by optical measurements. J. Phys. Condens. Matter 2012, 24, 195501. [Google Scholar] [CrossRef]
- Itatani, J.; Rini, M.; Cavalleri, A.; Onda, K.; Ishikawa, T.; Ogihara, S.; Koshihara, S.; Shao, X.F.; Nakano, Y.; Yamochi, H.; et al. Ultrafast Gigantic Photo-Response in Charge-Ordered Organic Salt (EDO-TTF)2PF6 on 10-fs time scales. In Ultrafast Phenomena XVI; Corkum, P., Silvestri, S.D., Nelson, K.A., Riedle, E., Schoenlein, R.W., Eds.; Springer-Verlag: Berlin, Germany, 2009; Volume 92, pp. 185–187. [Google Scholar]
- Onda, K.; Ogihara, S.; Itatani, J.; Ishikawa, T.; Okimoto, Y.; Koshihara, S.; Shao, X.; Nakano, Y.; Hideki, Y.; Saito, G. Photoinduced Dynamics of a Quasi-1-D Organic Conductor over a Range from 10 fs to 100 ps. In Ultrafast Phenomena XVII; Chergu, M., Jonas, D.M., Riedle, E., Schoenlein, R.W., Taylor, A.J., Eds.; Oxford University Press: New York, NY, USA, 2011; pp. 188–190. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishikawa, T.; Onda, K.; Koshihara, S.-y. Photoinduced Phase Transition in Strongly Electron-Lattice and Electron–Electron Correlated Molecular Crystals. Crystals 2012, 2, 1067-1083. https://doi.org/10.3390/cryst2031067
Ishikawa T, Onda K, Koshihara S-y. Photoinduced Phase Transition in Strongly Electron-Lattice and Electron–Electron Correlated Molecular Crystals. Crystals. 2012; 2(3):1067-1083. https://doi.org/10.3390/cryst2031067
Chicago/Turabian StyleIshikawa, Tadahiko, Ken Onda, and Shin-ya Koshihara. 2012. "Photoinduced Phase Transition in Strongly Electron-Lattice and Electron–Electron Correlated Molecular Crystals" Crystals 2, no. 3: 1067-1083. https://doi.org/10.3390/cryst2031067