A New BEDT-TTF-Based Organic Charge Transfer Salt with a New Anionic Strong Acceptor, N,N'-Disulfo-1,4-benzoquinonediimine

Abstract
:1. Introduction
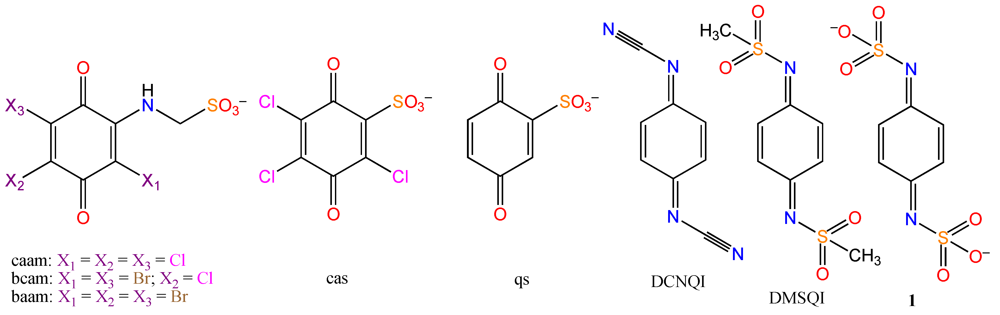
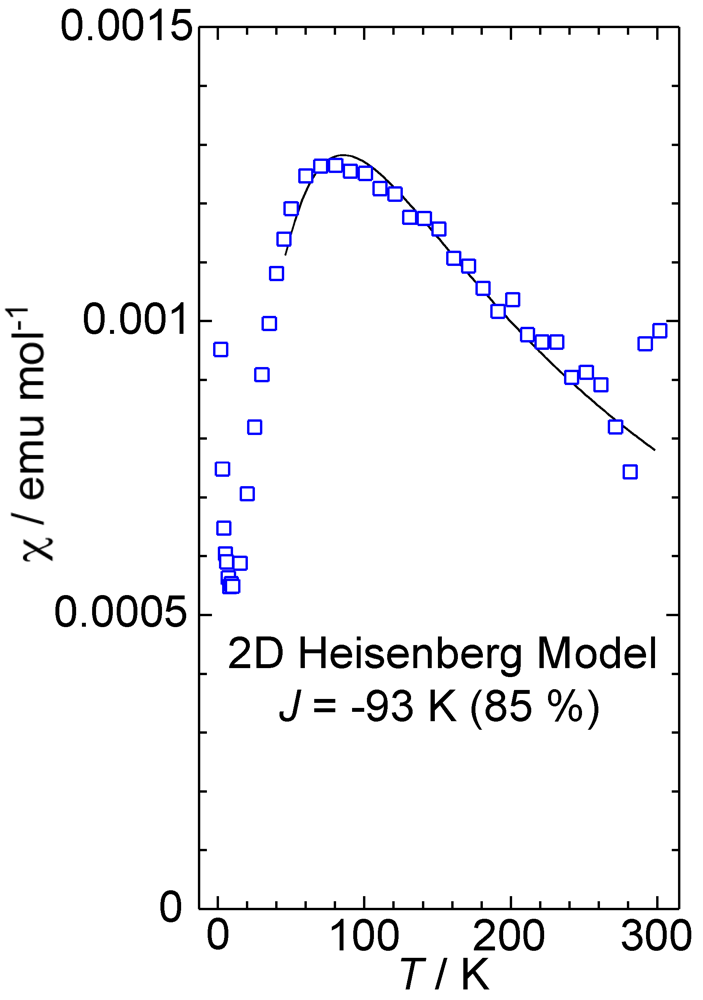
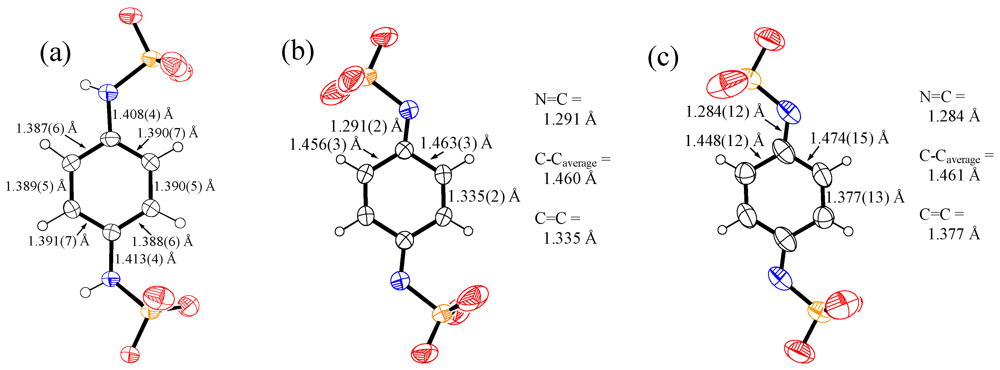
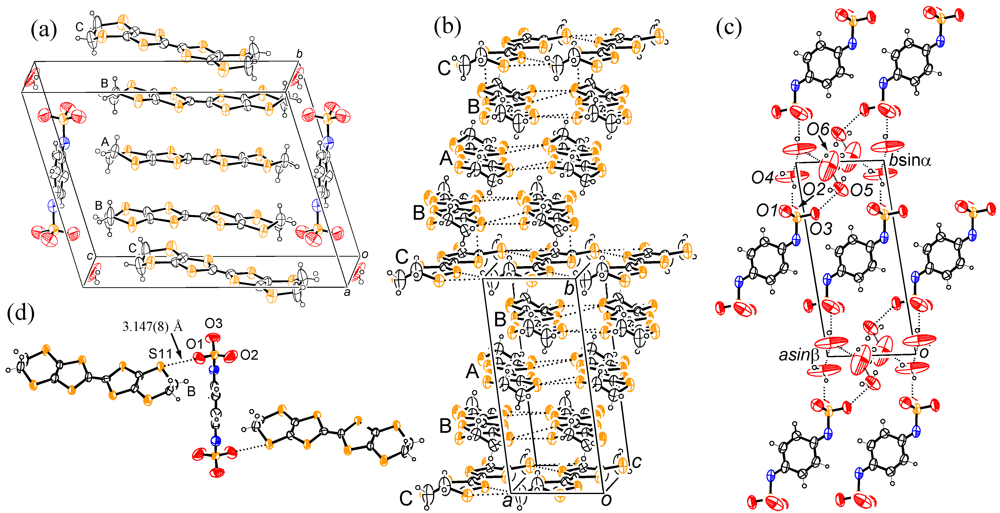
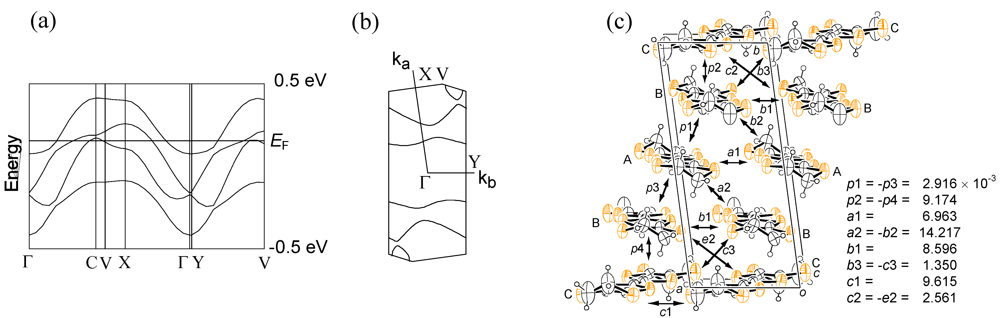
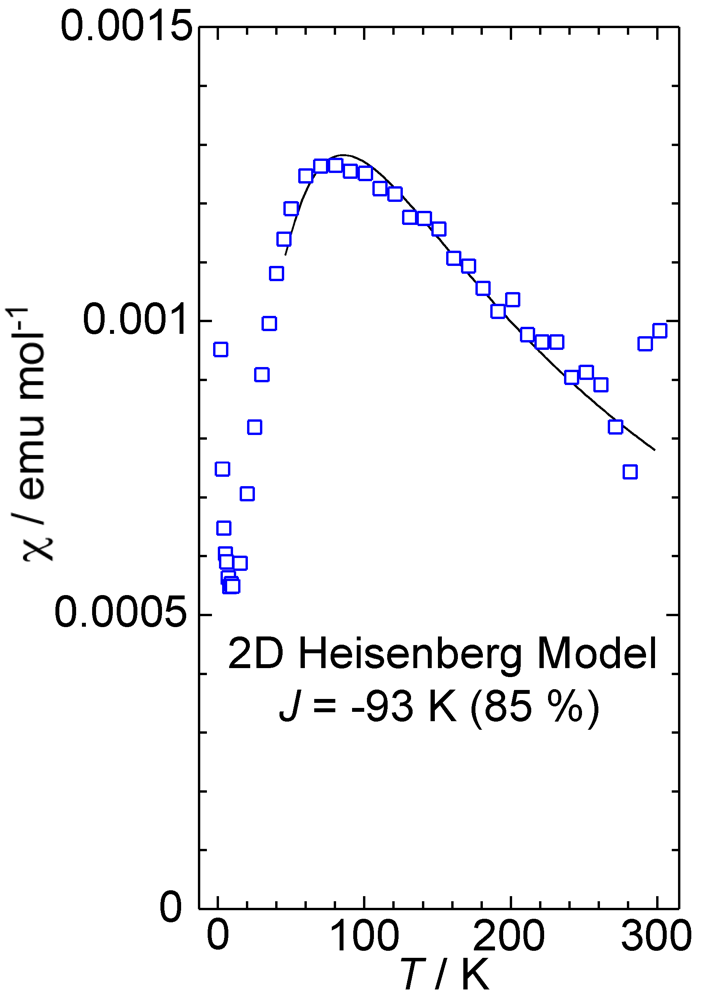
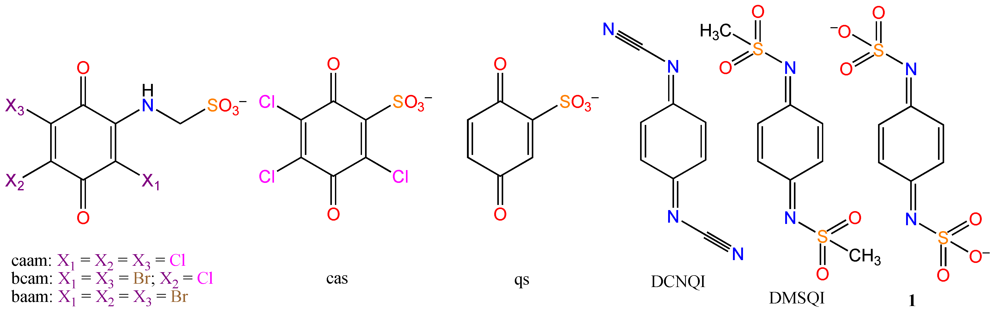
2. Results and Discussion
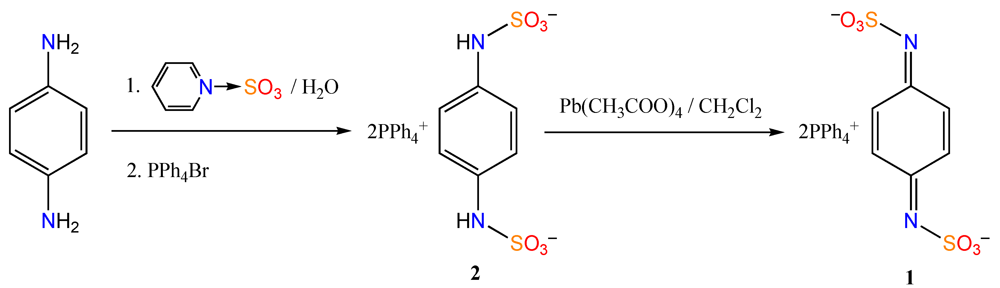
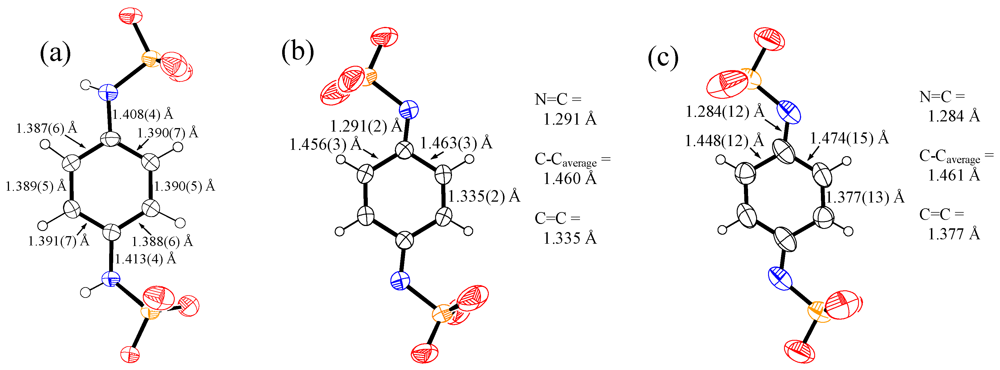
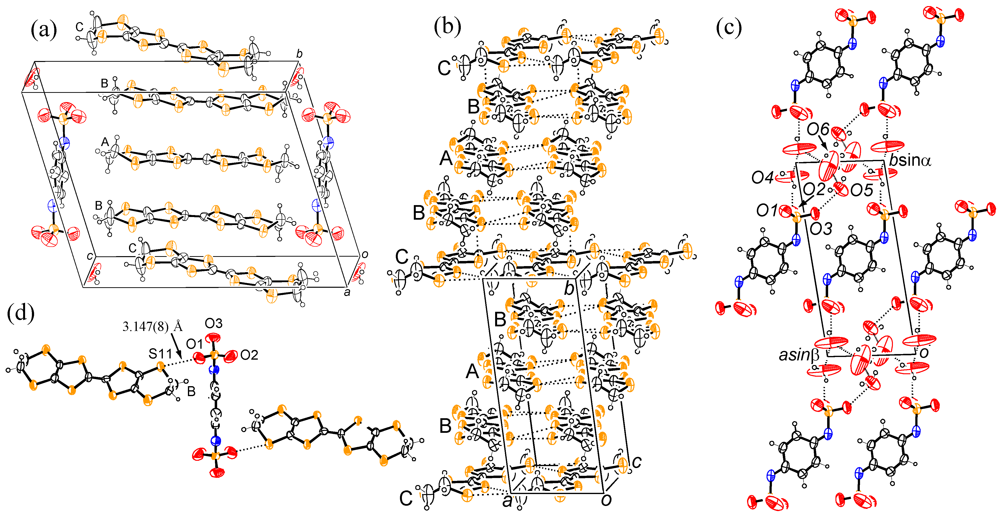

3. Experimental Section
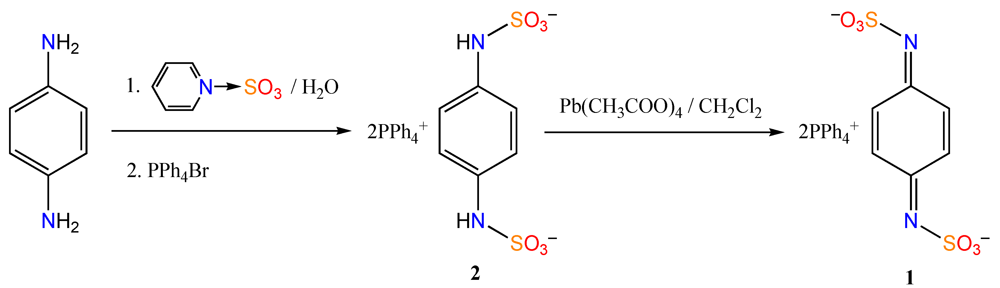
3.1. Synthesis of Ditetraphenylphosphonium N,N'-Disulfo-1,4-benzoquinonediimine [(PPh4)21]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | (PPh4)22·dichloroethane | (PPh4)21 | (ET)41·3H2O |
|---|---|---|---|
| Empirical formula | C56H50N2O6P2S2Cl2 | C54H44N2O6P2S2 | C46H42N2O9S34 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group | Pī | Pī | Pī |
| a (Å) | 13.849(3) | 9.859(2) | 6.742(3) |
| b (Å) | 14.817(3) | 13.683(4) | 15.518(7) |
| c (Å) | 13.542(3) | 9.664(2) | 18.034(7) |
| α (°) | 108.410(14) | 105.04(2) | 74.29(3) |
| β (°) | 102.833(14) | 93.651(18) | 82.16(3) |
| γ (°) | 98.057(15) | 110.735(20) | 80.35(3) |
| V (Å3) | 2503.4(9) | 1159.8(5) | 1782.2(14) |
| Z | 2 | 1 | 1 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 |
| Dcalc (mg m−3) | 1.39 | 1.35 | 1.73 |
| µ (mm−1) | 0.3312 | 0.2384 | 1.065 |
| No. of ref. collected | 12358 | 5645 | 14244 |
| No. of independent ref. | 11515 | 5333 | 7763 |
| No. of ref. used | 5746 [I > 1.5σ(I)] | 3987 [I > 1.0σ(I)] | 3129 [I > 2.5σ(I)] |
| No. of parameters | 691 | 386 | 424 |
| 2 θmax (°) | 27.5 | 27.5 | 27.5 |
| R | 0.066 [I > 1.5σ(I)] | 0.051 [I > 1.0σ(I)] | 0.067 [I > 2.5σ(I)] |
| Rw | 0.069 [I > 1.5σ(I)] | 0.055 [I > 1.0σ(I)] | 0.057 [I > 2.5σ(I)] |
3.2. Preparation of ET Salt with N,N´-Disulfo-1,4-benzoquinonediimine
4. Conclusions
Acknowledgments
References and Notes
- Mizuno, M.; Garito, A.F.; Cava, M.P. “Organic metals”: alkylthio substitution effects in tetrathiafulvalene–tetracyanoquinodimethane charge-transfer complexes. J. Chem. Soc. Chem. Commun. 1978, 18–19. [Google Scholar]
- Williams, J.M.; Ferraro, J.R.; Thorn, R.J.; Carlson, K.D.; Geiser, U.; Wang, H.H.; Kini, A.M.; Whangbo, M.-H. Organic Superconductor (Including Fullerenes) Synthesis, Structure, Properties and Theory; Prentice-Hall: New York, NY, USA, 1992. [Google Scholar]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors I. Parallel Molecules: β and β'' Phases. Bull. Chem. Soc. Jpn. 1998, 71, 2509–2526. [Google Scholar]
- Mori, T.; Mori, H.; Tanaka, S. Structural Genealogy of BEDT-TTF-Based Organic Conductors II. Inclined Molecules: θ, α, and κ Phases. Bull. Chem. Soc. Jpn. 1999, 72, 179–197. [Google Scholar]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors III. Twisted Molecules: δ and α' Phases. Bull. Chem. Soc. Jpn. 1999, 72, 2011–2027. [Google Scholar]
- Kini, A.M.; Geiser, U.; Wang, H.H.; Carlson, K.D.; Williams, J.M.; Kwok, W.K.; Vandervoort, K.G.; Thompson, J.E.; Stupka, D.L.; Jung, D.; Whangbo, M.-H. A new ambient-pressure organic superconductor, κ-(ET)2Cu[N(CN)2]Br, with the highest transition temperature yet observed (inductive onset Tc = 11.6 K, resistive onset = 12.5 K). Inorg. Chem. 1990, 29, 2555–2557. [Google Scholar]
- Williams, J.M.; Kini, A.M.; Wang, H.H.; Carlson, K.D.; Geiser, U.; Montgomery, L.K.; Pyrka, G.J.; Watkins, D.M.; Kommers, J.M.; Boryschuk, S.J.; et al. From semiconductor-semiconductor transition (42 K) to the highest-Tc organic superconductor, κ-(ET)2Cu[N(CN)2]Cl (Tc = 12.5 K). Inorg. Chem. 1990, 29, 3274–3282. [Google Scholar]
- Bednorz, J.G.; Müller, K.A. Possible high Tc superconductivity in the Ba−La−Cu−O system. Z. Phys. B 1986, 64, 189–193. [Google Scholar]
- Yamamoto, T.; Tajima, H.; Yamaura, J.; Aonuma, S.; Kato, R. Reflectance Spectra and Electrical Resistivity of (Me2-DCNQI)2Li1-xCux. J. Phys. Soc. Jpn. 1999, 68, 1384–1391. [Google Scholar] [CrossRef]
- Mori, H.; Kamiya, M.; Haemori, M.; Suzuki, H.; Tanaka, S.; Nishio, Y.; Kajita, K.; Moriyama, H. First Systematic Band-Filling Control in Organic Conductors. J. Am. Chem. Soc. 2002, 124, 1251–1260. [Google Scholar]
- Katsuhara, M.; Kimura, S.; Mori, T.; Misaki, Y.; Tanaka, K. Band Filling Control by Chemical Approach in Molecular Conductors, (TTM-TTP)MxM'1-xCl4 [M, M´ = Fe, Ga, Co, and Mn]. Chem. Mater. 2002, 14, 458–462. [Google Scholar]
- Sasaki, T.; Oizumi, H.; Yoneyama, N.; Kobayashi, N.; Toyota, N. X-ray Irradiation-Induced Carrier Doping Effects in Organic Dimer–Mott Insulators. J. Phys. Soc. Jpn. 2007, 76, 123701:1–123701:4. [Google Scholar]
- Akutsu, H.; Yamashita, S.; Yamada, J.; Nakatsuji, S.; Hosokoshi, Y.; Turner, S.S. A Purely Organic Paramagnetic Metal, κ-β''-(BEDT-TTF)2(PO-CONHC2H4SO3), Where PO = 2,2,5,5-Tetramethyl-3-pyrrolin-1-oxyl Free Radical. Chem. Mater. 2010, 23, 762–764. [Google Scholar]
- Akutsu, H.; Kawamura, A.; Yamada, J.; Nakatsuji, S.; Turner, S.S. Anion polarity-induced dual oxidation states in a dual-layered purely organic paramagnetic charge-transfer salt, (TTF)3(PO-CON(CH3)C2H4SO3)2, where PO = 2,2,5,5-tetramethyl-3-pyrrolin-1-oxyl free radical. CrystEngComm 2011, 13, 5281–5284. [Google Scholar] [CrossRef] [Green Version]
- Akutsu, H.; Yamada, J.; Nakatsuji, S.; Turner, S.S. An anionic weak acceptor 2-aminomethylsulfo-3,5,6-trichloro-1,4-benzoquinone and its BEDT-TTF-based charge-transfer salts. Solid State Commun. 2007, 144, 144–147. [Google Scholar] [CrossRef]
- Akutsu, H.; Yamada, J.; Nakatsuji, S.; Turner, S.S. A new anionic acceptor, 2-sulfo-3,5,6-trichloro-1,4-benzoquinone and its charge-transfer salts. CrystEngComm 2009, 144, 2588–2592. [Google Scholar]
- Akutsu, H.; Sasai, T.; Yamada, J.; Nakatsuji, S.; Turner, S.S. New anionic acceptors Br2XQNHCH2SO3− [X = Br, BryCl1−y (y≈0.5), and Cl; Q=1,4-benzoquinone] and their charge-transfer salts. Physica B 2010, 405, S2–S5. [Google Scholar]
- Akutsu, H.; Maruyama, Y.; Yamada, J.; Nakatsuji, S.; Turner, S.S. A new BEDT-TTF-based organic metal with an anionic weak acceptor 2-sulfo-1,4-benzoquinone. Synth. Met. 2011, 161, 2339–2343. [Google Scholar]
- Hünig, S.; Herberth, E. N,N´-Dicyanoquinone Diimines (DCNQIs): Versatile Acceptors for Organic Conductors. Chem. Rev. 2004, 104, 5535–5563. [Google Scholar] [CrossRef]
- Adams, R.; Nagarkatti, A.S. Quinone Imides. I. p-Quinone Disulfonimides. J. Am. Chem. Soc. 1950, 72, 4601–4606. [Google Scholar]
- Spillane, W.J.; Sheahan, M.B.; Ryder, C.A. Synthesis and taste properties of sodium disubstituted phenylsulfamates. Structure-taste relationships for sweet and bitter/sweet sulfamates. Food Chem. 1993, 47, 363–369. [Google Scholar]
- Voltage versus saturated calomel electrode in CH3CN with 0.1 M Bu4NClO4, glassy carbon electrode, at room temperature, under nitrogen (scan rate = 50 mV·s−1).
- All water molecules are located close to the inversion centres and refined with multiplicity 0.5.
- Guionneau, P.; Kepert, C.J.; Bravic, G.; Chasseau, D.; Truter, M.R.; Kurmoo, M.; Day, P. Determining the charge distribution in BEDT-TTF salts. Synth. Met. 1997, 86, 1973–1974. [Google Scholar] [CrossRef]
- Mori, T.; Kobayashi, A.; Sasaki, Y.; Kobayashi, H.; Saito, G.; Inokuchi, H. The Intermolecular Interaction of Tetrathiafulvalene and Bis(ethylenedithio)tetrathiafulvalene in Organic Metals. Calculation of Orbital Overlaps and Models of Energy-band Structures. Bull. Chem. Soc. Jpn. 1984, 57, 627–633. [Google Scholar]
- Lines, M.E. The quadratic-layer antiferromagnet. J. Phys. Chem. Solids 1970, 31, 101–116. [Google Scholar] [CrossRef]
- Sugano, T.; Saito, G.; Kinoshita, M. Spin relaxation and diffusion in quasi-two-dimensional organic metals: The bis(ethylenedithiolo)tetrathiafulvalene compounds β-(BEDT-TTF)2X (X = I3 and IBr2). Phys. Rev. B 1987, 35, 6554–6559. [Google Scholar]
- Akutsu, H.; Yamada, J.; Nakatsuji, S.; Turner, S.S. A novel BEDT-TTF-based purely organic magnetic conductor, α-(BEDT-TTF)2(TEMPO-N(CH3)COCH2SO3)·3H2O. Solid State Commun. 2006, 140, 256–260. [Google Scholar] [CrossRef]
- Akutsu, H.; Ohnishi, R.; Yamada, J.; Nakatsuji, S.; Turner, S.S. Novel Bis(ethylenedithio)tetrathiafulvalene-Based Organic Conductor with 1,1'-Ferrocenedisulfonate. Inorg. Chem. 2007, 46, 8472–8474. [Google Scholar]
- Akutsu, H.; Yamashita, S.; Yamada, J.; Nakatsuji, S.; Turner, S.S. Novel Purely Organic Conductor with an Aminoxyl Radical, α-(BEDT-TTF)2(PO–CONHCH2SO3)·2H2O (PO = 2,2,5,5-Tetramethyl-3-pyrrolin-1-oxyl Free Radical). Chem. Lett. 2008, 37, 882–883. [Google Scholar] [CrossRef]
- Different recrystallisation conditions of (PPh4)21 sometimes gave the other two shapes of crystals. One (yellow needle) was assigned as (PPh4)21·2H2O, in which 1 is an anti isomer. The other (blown plate) was obtained as a trace which assigned as (PPh4)21, in which 1 was disordered and seemed to be a syn isomer. Here we used only the yellow blocks of (PPh4)21 for the characterisation of 1 and electrocrystallisation with ET.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Akutsu, H.; Yamada, J.-i.; Nakatsuji, S.; Turner, S.S. A New BEDT-TTF-Based Organic Charge Transfer Salt with a New Anionic Strong Acceptor, N,N'-Disulfo-1,4-benzoquinonediimine. Crystals 2012, 2, 182-192. https://doi.org/10.3390/cryst2020182
Akutsu H, Yamada J-i, Nakatsuji S, Turner SS. A New BEDT-TTF-Based Organic Charge Transfer Salt with a New Anionic Strong Acceptor, N,N'-Disulfo-1,4-benzoquinonediimine. Crystals. 2012; 2(2):182-192. https://doi.org/10.3390/cryst2020182
Chicago/Turabian StyleAkutsu, Hiroki, Jun-ichi Yamada, Shin’ichi Nakatsuji, and Scott S. Turner. 2012. "A New BEDT-TTF-Based Organic Charge Transfer Salt with a New Anionic Strong Acceptor, N,N'-Disulfo-1,4-benzoquinonediimine" Crystals 2, no. 2: 182-192. https://doi.org/10.3390/cryst2020182
APA StyleAkutsu, H., Yamada, J.-i., Nakatsuji, S., & Turner, S. S. (2012). A New BEDT-TTF-Based Organic Charge Transfer Salt with a New Anionic Strong Acceptor, N,N'-Disulfo-1,4-benzoquinonediimine. Crystals, 2(2), 182-192. https://doi.org/10.3390/cryst2020182