Abstract
A series of arylalkylcarbinol derivatives were deracemized through sequential combination of Candida antarctica lipase B (CAL-B) catalyzed resolution by hydrolysis and Mitsunobu stereoinversion. The (S)-acetates were obtained in 71%–99% ee and 76%–89% yields. An enantiocomplementarity was established for the hydrolysis and acylation reactions with CAL-B lipase. Thus, the (S) and (R) enantiomers of Indan-1-yl acetate, 1,2,3,4-tetrahydro-1-naphthalenol acetate and 1-(2-naphthyl) ethyl acetate were obtained in 91%–99% ee and 76%–89% yield.
1. Introduction
The development of efficient methods for the synthesis of enantiomerically pure alcohols is of tremendous importance [1,2,3]. They are important intermediates for the synthesis of pharmaceuticals, agrochemicals, pheromones, flavors, liquid crystals and chiral auxiliaries in asymmetric synthesis [4,5]. As an option for sustainability, chemists have continued to be attracted to the area of biocatalysts that have been increasingly used to attain enantiomerically enriched or pure secondary alcohols [6,7]. High stability, enantioselectivity and good commercial availability in free and immobilized forms have made lipases (EC 3.1.1.3) especially attractive kinetic resolution catalysts via the acylation [8,9]. Unlike other enzymes, lipases can also exhibit catalytic activity in biphasic media [10].
However, even in the case where the enantioselectivity factor is high (E > 100), the most striking limitation, common to strategies relying on kinetic resolution, is the maximum theoretical yield of 50% for a single enantiomer. In order to circumvent this constraint associated with the yield, several approaches have been developed, which render an enantio-convergent process delivering a single enantiomeric product in 100% theoretical yield [11,12,13]. Methods based on a dynamic kinetic resolution imply in situ transition-metal-catalyzed racemization of the slowly reacting enantiomer combined with kinetic resolution process [14,15,16]. Another strategy has been performed that allows selective inversion of configuration of one enantiomer via a microbial process [17,18] or a chemo-enzymatic protocol [19,20]. In the latter case, special emphasis is devoted to the deracemization of sec-alcohols and their derivatives [21]. In this field, one of the most versatile stereo chemical transformations is the Mitsunobu reaction [22]. It was found that the inversion of the configuration of chiral secondary alcohols following an enzymatic kinetic resolution step was of fruitful application [23,24].
In our previous study [25], we applied a lipase-catalyzed acylation, in presence of an immobilized lipase from Candida antarctica B, followed by Mitsunobu chemical stereoinversion to a series of arylalkylcarbinols. The corresponding (R)-acetates were obtained in good yields and enantiomeric excesses. These results urged us to explore the possibility of extending such a process to kinetic resolution by hydrolysis in order to obtain the (S)-acetates (Scheme 1). Our study has been realized mainly for the sake of comparison and also to emphasize the enantiocomplementarity of the two biocatalyzed reactions, acylation and hydrolysis, in presence of the same lipase. This comparison offers an additional catalysis tool; the strategy (acylation/inversion or hydrolysis/inversion) can be adapted to the desired enantiomer.
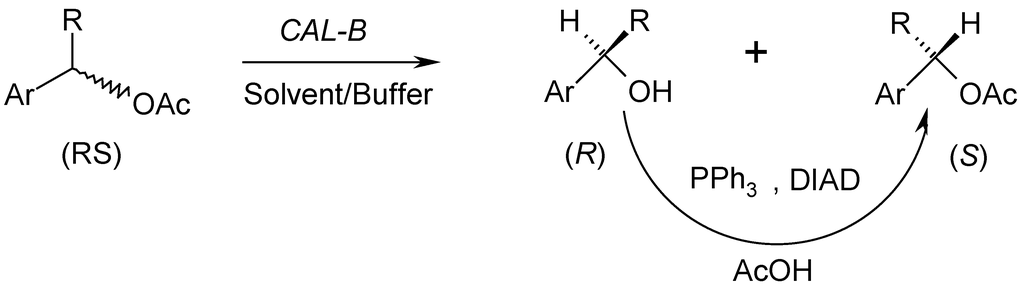
Scheme 1.
Lipase hydrolysis combined to Mitsunobu stereoinversion.
Few lipases showed excellent enantiocomplementary hydrolysis and acylation due to lower enantioselectivities disclosed generally after hydrolysis reaction in presence of the same enzyme [26,27]. Accordingly, enantiocomplementaritry between the two reactions is finding scant attention in the literature [28,29]. This efficient pathway has been reported previously in presence of Pseudomonas fluorescens lipase (PFL) [30].
Enzymatic hydrolysis in a biphasic medium is generally described with moderate selectivity in the literature. The amount of lipase is an important parameter for improving the enzymatic selectivity when studying the kinetic acylation [25,31]. The influence of this parameter was not examined previously in hydrolysis in biphasic media.
2. Results and Discussion
2.1. Optimization of the Lipase Amount
Before starting the deracemization process, we thought useful to study the effect of the amount of enzyme on activity and selectivity of hydrolysis reaction. In a recent report, our data confirmed the importance of decreasing the enzyme amount in order to enhance the enantioselectivity in the kinetic transesterification of a series of arylalkylcarbinols [31].
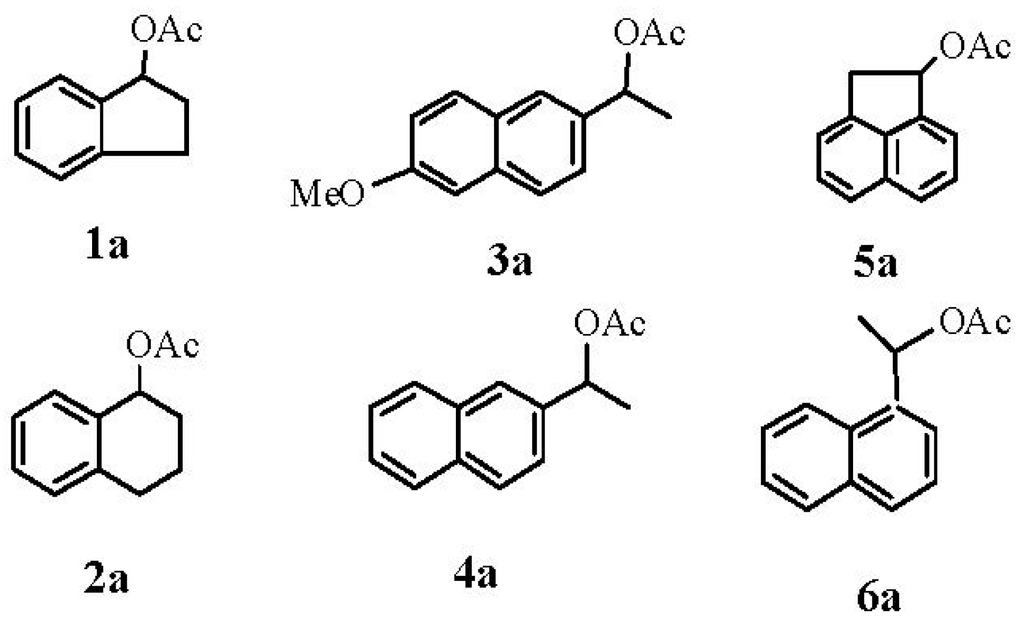
Scheme 2.
Substrate models.

Table 1.
Enzymatic hydrolysis of acetates 1a–6a. The shadowed lines emphasize the best results with high reactivities and selectivities (C and E).
| Entry | Substrate a | CAL-B (mg) | eeS (%) b-(S) d | eeP (%) b-(R) d | C % c | Ec |
|---|---|---|---|---|---|---|
| 1 | 1a | 150 | 44 | 96 | 32 | 76 |
| 2 | 12 | 98 | 99 | 50 | >500 | |
| 3 | 6 | 79 | 93 | 46 | 67 | |
| 4 | 2a | 150 | 83 | 98 | 46 | >200 |
| 5 | 50 | 99 | 99 | 50 | >500 | |
| 6 | 20 | 28 | 96 | 23 | 64 | |
| 7 | 3a | 150 | 98 | 99 | 50 | >500 |
| 8 | 20 | 99 | 98 | 50 | >500 | |
| 9 | 4a | 150 | 89 | 99 | 47 | >500 |
| 10 | 20 | 99 | 97 | 50 | >500 | |
| 11 | 5a | 150 | 7 | 99 | 6 | >200 |
| 12 | 60 | 10 | 96 | 9 | 54 | |
| 13 | 20 | - | - | - | - | |
| 14 | 6a | 150 | 75 | 87 | 46 | 32 |
| 15 | 60 | 51 | 99 | 34 | 70 | |
| 16 | 20 | 44 | 99 | 30 | >200 |
a Reactions were carried out with 1mmol of racemic acetate in diethyl ether/buffer solution pH = 7: (1/4) at 25 °C, in the presence of a catalytic amount of CAL-B (4500 U/g), for 48 h (72 h entries 12–16); b Measured by HPLC using Chiracel OD-H column; c Conversion C = ees /ees + eep; selectivity factor E = ln [(1 − C) (1 − ees)]/ln[(1 − C) (1 + ees)]; d Absolute configuration was determined by comparison of the sign of the specific rotation of the isolated product with the literature (see experimental).
The enzymatic hydrolysis of racemic secondary aromatic acetates 1a–6a (Scheme 2), which are also our study models, is carried out in biphasic system: phosphate buffer/diethyl ether (v/v) (1:4) pH = 7, in the presence of variable amounts of Candida antarctica lipase fraction B immobilized on acrylic resin (CAL-B) used previously [25]. After the appropriate time; a mixture of unreacted (S) acetate and formed (R) alcohol was obtained. The results are shown in Table 1.
The results from (Table 1) indicate the important influence of CAL-B on both reactivity and selectivity depending directly on the enzyme loading in the cases of substrates 1a and 2a (entries 1–6). Thus, decreasing the amount of the lipase from 150 mg to 12 mg for 1a and to 50 mg for 2a improves both reactivity and selectivity and the (R)-alcohols are obtained with excellent enantiomeric excesses (entries 2 and 5). Reducing the amount of CAL-B to lower values (entries 3 and 6) displays a negative effect on both reactivity and selectivity which drop dramatically, especially in the case of substrate 2a (entry 6).
On the other hand, we did not observe any change in activity or selectivity of the lipase by decreasing its amount to eight times its initial value with acetates 3a and 4a (entries 8 and 10), excellent enantioselectivities were reached in all cases (E > 500). For substrate 5a, the hydrolysis reaction did not afford good results. Despite the observed high enantioselectivity by using 150 mg of enzyme (entry 11), the conversion was very low and no reaction was detected when using 20 mg of lipase. The hydrolysis of acetate 6a proceeded with a good selectivity E > 200 by using 20 mg of CAL-B (entry 16) and the selectivity diminished by increasing the lipase amount (entries 14 and 15). The CAL-B displays (R)-enantiopreference since the (R)-acetates were selectively hydrolysed in all cases. On the basis of these results, we have shown a noticeable impact of the CAL-B amount on the activity and selectivity in the hydrolysis reaction. Thus, a threshold has been established for each substrate in order to use the minimum and effective amount of CAL-B. This study shows the important influence of the amount of lipase on the kinetic resolution by hydrolysis.
Few reports dealt with the impact of the enzyme loading and an elucidation of the mechanism is not an easy task since many parameters could be taken into account, such as the enzyme aggregates that might interfere and hinder the selectivity especially in the case of a large quantity [32], or the problems of mass transport limitation that probably hinder diffusion of the enzyme in the reaction medium [33].
2.2. Deracemization by Combined Enzymatic Hydrolysis/Mitsunobu Stereoinversion
It is well recognized that a deracemization process needs to fulfill, in a first step of kinetic resolution, the requirement of reaching a satisfactory enantioselectivity at C = 50% substrate conversion. For this reason, we limited our further study of the Mitsunobu inversion combined with hydrolysis in optimal conditions, to substrates 1a–4a. At the appropriate conversion and after removal of the enzyme by filtration, the crude mixture of unreacted (S)-acetate and the formed (R)-alcohol underwent Mitsunobu reaction. Triphenylphosphine and acetic acid were added to the mixture followed by the addition of diisopropylazodicarboxylate (DIAD) at 0 °C to convert the formed (R)-1–4 alcohols into the corresponding (S)-1a–4a acetates. The results are collected in Table 2.
Table 2.
Deracemization of acetates 1a–4a through a resolution/inversion process. The shadowed line emphasize the best result in terms of enantiomeric excess and yield.
| Substrate a | CAL-B catalyzed hydrolysis b | Mitsunobu inversion | ||||
|---|---|---|---|---|---|---|
| CAL-B (mg/mmole) | (R)-Alcohol (%ee) c | (S)-Acetate (%ee) c | E d | (S)-Acetate (%ee) | Yield e (%) | |
| 1a | 25 | 99 | 98 | >500 | 94 | 78 |
| 2a | 100 | 99 | 99 | >500 | 99 | 89 |
| 3a | 40 | 98 | 99 | >500 | 71 | 82 |
| 4a | 40 | 97 | 99 | >500 | 92 | 76 |
a 2 mmol substrate subjected to hydrolysis, in presence of the optimized amounts of lipase, then Mitsunobu reagents; b Conversion C = 49%–50% (see Table 1); c ee determined by HPLC using Chiracel OD-H; d Calculated from eealcohol and eeacetate and conversion C; e Isolated yield.
All hydrolysis reactions proceeded with an excellent enantioselectivity (E > 500). At 50% conversion, both the produced alcohol and the unreacted acetate showed an excellent enantiomeric excess (>99% from 2a) or good (97%–99% from 1a, 3a, 4a). This was achieved in presence of the adequate amount of CAL-B lipase. After the Mitsunobu reaction, no residual alcohol was detected. Except for substrate 3a, the other acetates showed either an excellent (>99% from 2a) or good with a slight drop in the enantiomeric excess (92%–94% from 4a and 1a respectively). A racemization-induced process under Mitsunobu reagents might give an explanation to the loss of stereoselectivity [34,35]. Both the inversion efficiency and yield depended on the aromatic substituents [36]. Recently, we observed almost full racemization during the nucleophilic substitution reaction due to the presence of an electron donating substituent ring [37].
A comparison between the deracemization process via acylation [25] and the current process combining hydrolysis and Mitsunobu inversion furnished important data (Table 3). Although the significant difference between the optical purities of acetates (S)-3a (ee = 71%) and (R)-3a (ee > 99%), our study pointed out that it is feasible to afford both the enantiomers of acetates 1a, 2a and 4a with good enantiomeric excesses (eeR = 91%–99%, eeS = 92%–99%) and satisfying yields (74%–89%).
Table 3.
Comparison of deracemization through acylation/inversion and hydrolysis/inversion processes.
| Substrate a | CAL-B Catalyzed Acylation/Mitsunobu Stereoinversion | Substrate b | CAL-B Catalyzed Hydrolysis/Mitsunobu Stereoinversion | ||||
|---|---|---|---|---|---|---|---|
| CAL-B (mg) | (R)-Acetate (%ee) | Yield (%) | CAL-B (mg) | (S)-Acetate (%ee) | Yield (%) | ||
| 1 | 12 | >99 | 82 | 1a | 25 | 94 | 78 |
| 2 | 100 | >99 | 79 | 2a | 100 | 99 | 89 |
| 3 | 40 | >99 | 74 | 3a | 40 | 71 | 82 |
| 4 | 50 | 91 | 76 | 4a | 40 | 92 | 76 |
a 2 mmol substrate, 4 mmol isopropenyl acetate, 10 mL diethyl ether, 24 h, room temperature; b 2 mmol substrate, diethyl ether/buffer solution pH = 7: (1/4), 48 h, 25 °C.
It is worth mentioning that an interesting enantiocomplementarity has been established between CAL-B-catalyzed acylation and the reaction of hydrolysis. Thus, the two enantiomers of 1,2,3,4-tetrahydronaphthalen-1-yl acetate 2a and Indan-1-yl acetate 1a were provided optically pure (ee > 99% for 2a) and (ee = 94%–99% for 1a) with high yields (Scheme 3). These results show the importance of the enantiocomplementarity concept.
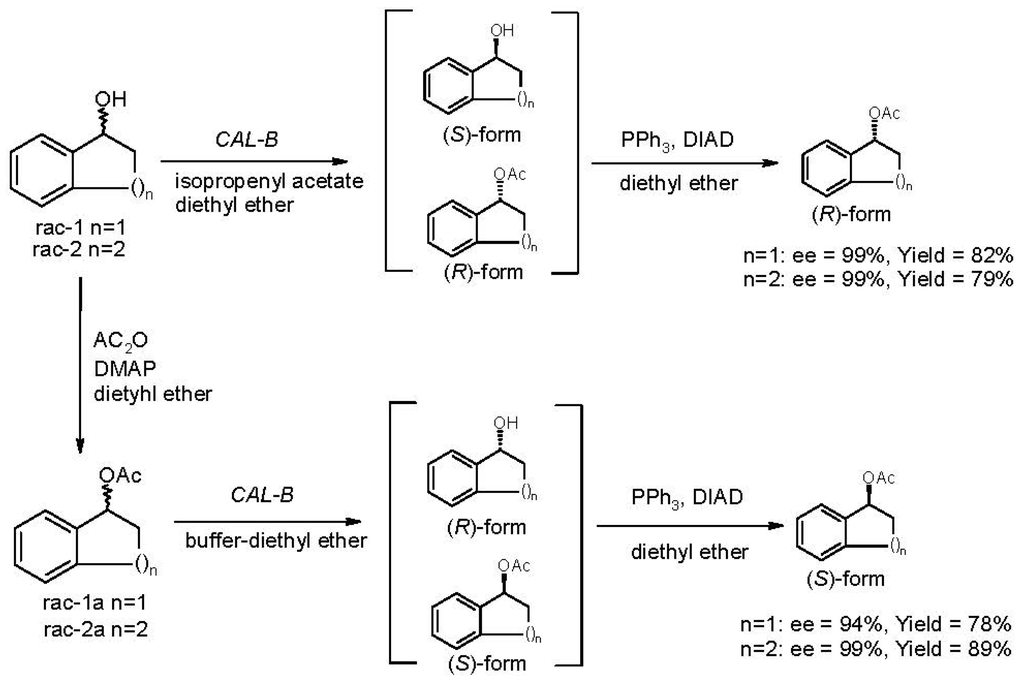
Scheme 3.
Enantiocomplementary preparation of both enantiomers of acetates 1a and 2a.
3. Experimental Section
3.1. General
Optical rotations were determined using a Perkin–Elmer (Waltham, MA, USA) 241 Polarimeter at room temperature using a cell of 1 dm length and k = 589 nm. The enantiomeric excesses were measured by a chiral stationary phase HPLC on Chiralcel® OD-H column (Chiral Technologies. Europe, Illkirch-Graffenstaden, France). Retention times are reported in minutes.
3.2. General Procedure for the Synthesis of Racemic Acetates 1a–6a
The acetates were synthesized by classical chemical acetylations via the corresponding racemic alcohol (1 equivalent), using 1.5 equivalent of anhydride acetic, 1.2 equivalent of Et3N, and a catalytic amount of 4-dimethylaminopyridine (0.1 equivalent) in 4 mL of ether. The acetates were obtained pure after standard work-up. The 1H NMR spectra of these products were in good agreement with the literature.
3.3. General Procedure for the Hydrolysis of Racemic Acetates 1a–6a with CAL-B
Racemic acetates 1a-6a (1 mmol), dissolved in 1.5 mL of diethyl ether, were added to phosphate buffer (6 mL) pH 7 and followed by a variable amount of immobilized lipase from Candida antarctica B. The reaction mixture was mechanically stirred at 25 °C for the indicated time (see Table 1). The hydrolysis was stopped by filtering off the enzyme. Diethyl ether was removed under vacuum and the resulting aqueous solution was extracted with EtOAc. The organic phase was dried over Na2SO4 and evaporated under reduced pressure.
3.4. Typical Procedure for Synthesis of Enantiomerically Pure Acetates (S)-1a–4a
All attempts failed to reach the adequate conversion for acetates 5a and 6a. The deracemization process mentioned above was applied only to acetates 1a–4a. Racemic acetates 1a–4a (2 mmol), dissolved in 3 mL of diethylic ether, were added to phosphate buffer (12 mL) pH7 followed by the adjusted amount of immobilized lipase from Candida antarctica B. The reaction mixture was then stirred at 25 °C for the indicated time until the conversion reached C = 50% (see Table 2). The reaction was stopped by filtering off the solid enzyme on Celite and the solvent evaporated under reduced pressure. The aqueous solution was extracted with EtOAc. The organic phase was dried over Na2SO4 and evaporated under reduced pressure. The crude mixture of the (S)-acetate and unreacted (R)-alcohol was dissolved in 4 mL of diethyl ether. To this solution were added AcOH (0.144 g, 2.4 mmol) and PPh3 (0.628 g, 2.4 mmol). The reaction mixture was immediately cooled to 0 °C and a solution of diisopropyl azodicarboxylate (DIAD) (0.48 g, 2.4 mmol) was added dropwise, under vigorous stirring during 20 min. The mixture was allowed to warm to room temperature and stirred for 24 h. Concentration of reaction mixture in vacuo followed by silica gel column chromatographic purification of the residue using hexane and ethyl acetate (8:2) gave only the acetates (S)-1a–4a in 76%–89% yields.
3.5. The Conditions for the Analysis of Alcohols (R)-1–6 Are Reported Below
- 1:
- (R)-(−)-Indan-1-ol: HPLC (Chiralcel® OD-H), tR = 37.02 min; tS = 43.24 min; (Hexane/i-PrOH 98:2 flow: 0.5 mL/min). [α]D = −16.7 (c 1, MeOH).
- 2:
- (R)-(−)-1,2,3,4-Tetrahydro-1-naphthalenol: HPLC (Chiralcel® OD-H), tR = 15.82 min; tS = 17.64 min; (hexane/i-PrOH 95:5, flow: 0.5 mL/min). [α]D = −28.1(c 2, MeOH).
- 3:
- (R)-(+)-1-(6-Methoxy-2-naphthyl) ethanol: HPLC (Chiralcel® OD-H), tR = 19.47 min; tS = 26.69 min; (hexane/i-PrOH 90:10, flow: 0.5 mL/min). [α]D = +36.4 (c 0.8, EtOH).
- 4:
- (R)-(+)-1-(2-Naphthyl) ethanol: HPLC (Chiralcel® OD-H), tR = 30.54 min; tS = 33.71 min; (hexane/i-PrOH 95:5, flow: 0.5 mL/min). [α]D = +36.5 (c 1, MeOH).
- 5:
- (R)-(−)-1-Acenaphthenol: HPLC (Chiralcel® OD-H), tR = 28.80 min; tS = 34.67 min; (hexane/i-PrOH 95:5, flow: 0.5 mL/min). [α]D = −1.4 (c 2.6, CHCL3).
- 6:
- (R)-(+)-1-(1-Naphthyl) ethanol: HPLC (Chiralcel® OD-H), tR = 20.05 min; tS = 28.90; (hexane/i-PrOH 90:10, flow: 0.5 mL/min). [α]D = +66.5 (c 1, MeOH).
3.6. The Conditions for the Analysis of Acetates (S)-1a–6a Are Reported Below
- 1a:
- (S)-(+)- Indan-1-yl acetate: HPLC (Chiralcel® OD-H), tS = 10.14 min; tR = 11.18 min; (Hexane/i-PrOH 98:2 flow: 0.5 mL/min). [α]D = −110.1 (c 2, CHCl3).
- 2a:
- (S)-(+)-1,2,3,4-Tetrahydro-1-naphthalenol acetate: HPLC (Chiralcel® OD-H), tS = 8.62 min; tR = 9.05 min; (hexane/i-PrOH 95:5, flow: 0.5 mL/min). [α]D = −112.8 (c 2, CHCl3).
- 3a:
- (S)-(−)-1-[2-(6-Methoxynaphthyl)] ethyl acetate: HPLC (Chiralcel® OD-H), tS = 10.78 min; tR = 12.05 min; (hexane/i-PrOH 90:10, flow: 0.5 mL/min). [α]D = −110 (c 1, EtOH).
- 4a:
- (S)-(−)-1-(2-Naphthyl) ethyl acetate: HPLC (Chiralcel® OD-H), tS = 10.47 min; tR = 11.98 min; (hexane/i-PrOH 95:5, flow: 0.5 mL/min). [α]D = −110.2 (c 1, CHCl3).
- 5a:
- (S)-(+)-1-Acenaphthylenol-1,2-dihydro acetate: HPLC (Chiralcel® OD-H), tS = 11.88 min; tR = 12.52 min; (hexane/i-PrOH 95:5, flow: 0.5 mL/min). [α]D = −85.9 (c 2.4, CHCL3).
- 6a:
- (S)-(−)-1-(1-Naphthyl)ethyl acetate: HPLC (Chiralcel® OD-H), tS = 9.87 min; tR = 13.50 min; (hexane/i-PrOH 90:10, flow: 0.5 mL/min). [α]D = −49.5 (c 1, CHCl3).
4. Conclusions
In this study, we have displayed the importance of the enantiocomplementarity concept and the efficient routes of access to the R and S enantiomers of the models studied by comparing the chemoenzymatic deracemization via a sequence of hydrolysis/acylation with CAL-B, combined with esterification using Mitsunobu protocol.
We have performed the kinetic resolution of a series of racemic-arylalkylcarbinol acetates 1a–6a via hydrolysis catalysed by CAL-B lipase. The optimization of CAL-B amount is of great importance. We studied this parameter aiming precisely to increase selectivity of our models. For each substrate, the optimum amount of Candida antarctica lipase fraction B (CAL-B) was determined to increase the selectivity in hydrolysis reaction in biphasic media conditions.
The convenient amount of lipase has been used in a deracemization process combining kinetic resolution by hydrolysis and a chemical in situ stereoinversion for substrates 1a–4a. The target (S)-arylalkylcarbinol acetates were obtained in excellent enantiomeric purity ee > 99% for 2a to good, ee = 94% for 1a, ee = 92% for 4a and ee = 71% for 3a and in good yields (76%–89%). Thus, the Racemic acetates 1a–4a have been resolved in an enantiocomplementary way by kinetic acylation and hydrolysis reactions using the Candida antarctica Lipase B. This enantiocomplementarity offers an additional catalysis tool; the strategy (acylation/inversion or hydrolysis/inversion) can be adapted to the desired enantiomer providing it in a quantitative yield.
Acknowledgments
Olivier Riant (IMCN/Louvain Catholic University, Louvain, Belgium) and Claude Rabiller (UFR Sciences, UMR CNRS 6204 U3B, Nantes, France) are acknowledged for welcoming Nassima Bouzemi to carry out chiral chromatographic analysis.
Author Contributions
The ideas in this manuscript arose as the result of many conversations among the Nassima Bouzemi and Louisa Aribi-Zouioueche. Nassima Bouzemi wrote the first draft of the manuscript that was then extensively improved by Louisa Aribi-Zouioueche and as the result of comments from our reviewers. Experimental work was carried out by Ismahane Grib and Zahia Houiene. Each member participated sufficiently in the work to take responsibility of the content.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Itoh, N.; Isotani, K.; Nakamura, M.; Inoue, K.; Isogai, Y.; Makino, Y. Efficient synthesis of optically pure alcohols by asymmetric hydrogen-transfer biocatalysis: Application of engineered enzymes in a 2-propanol-water medium. Appl. Microbiol. Biotechnol. 2012, 93, 1075–1085. [Google Scholar] [CrossRef]
- Ahmed, M.; Kelly, T.; Ghanem, A. Applications of enzymatic and non-enzymatic methods to access enantiomerically pure compounds using kinetic resolution and racemisation. Tetrahedron 2012, 68, 6781–6802. [Google Scholar] [CrossRef]
- Lee, Y.S.; Murphy, J.M.; Ukai, A.; Fu, G.C. Nanoenzymatic dynamic kinetic resolution of Secondary alcohols via acylation: Synthetic and mechanistic studies. J. Am. Chem. Soc. 2012, 134, 15149–15153. [Google Scholar] [CrossRef]
- Adams, T.B.; McGowen, M.M.; Williams, M.C. The FEMA GRAS assessment of aromatic substituted secondary alcohols, ketones, and related esters used as flavor ingredients. Food Chem. Toxicol. 2007, 45, 171–201. [Google Scholar] [CrossRef]
- Pollard, D.J.; Woodley, M.J. Biocatalysis for pharmaceutical intermediates: The future is now. Trends Biotechnol. 2007, 25, 66–73. [Google Scholar] [CrossRef]
- Athawale, V.; Manrekar, N.; Athawale, M. Effect of Reaction Parameters on Synthesis of Citronellyl Methacrylate by Lipase-Catalyzed Transesterification. Biotechnol. Prog. 2003, 19, 298–302. [Google Scholar] [CrossRef]
- Turner, N.J. Deracemisation methods. Curr. Opin. Chem. Biol. 2010, 14, 115–121. [Google Scholar] [CrossRef]
- Bouzemi, N.; Debbeche, H.; Aribi-Zouioueche, L.; Fiaud, J.C. On the use of succinic anhydride as acylating agent for practical resolution of aryl-alkyl alcohols through lipase-catalyzed acylation. Tetrahedron Lett. 2004, 45, 627–630. [Google Scholar] [CrossRef]
- Escorcia, A.M.; Molina, D.; Daza, M.C.; Doerr, M. Acetylation of (R,S)-propranolol catalyzed by Candida antarctica lipase B: An experimental and computational study. J. Mol. Catal. B 2013, 98, 21–29. [Google Scholar] [CrossRef]
- Salezadeh-Asl, R.; Lee-Ruff, E. Enantiomeric resolution of cyclobutanones and related derivatives by enzyme-catalyzed acylation and hydrolysis. Tetrahedron 2005, 16, 3986–3991. [Google Scholar] [CrossRef]
- Strauss, U.T.; Felfer, U.; Faber, K. Biocatalytic transformation of racemates into chiral building blocks in 100% chemical yield and 100% enantiomeric excess. Tetrahedron 1999, 10, 107–117. [Google Scholar] [CrossRef]
- Larissegger-Schnell, B.; Glueck, S.M.; Kroutil, W.; Faber, K. Enantio-complementary deracemization of (±)-2-hydroxy-4-phenylbutanoic acid and (±)-3-phenyllactic acid using lipase-catalyzed kinetic resolution combined with biocatalytic racemization. Tetrahedron 2006, 62, 2912–2916. [Google Scholar]
- Kamaruddin, A.H.; Uzir, M.H.; Aboul-enein, H.Y.; hairul, N.A. Chemoenzymatic and microbial dynamic kinetic resolutions. Chirality 2009, 21, 449–467. [Google Scholar] [CrossRef]
- Traff, A.; Lihammar, R.; Backvall, J.E. A Chemoenzymatic Dynamic Kinetic Resolution Approach to Enantiomerically Pure (R)- and (S)-Duloxetine. J. Org. Chem. 2011, 76, 3917–3921. [Google Scholar] [CrossRef]
- Merabet-Khelassi, M.; Vriamont, N.; Riant, O.; Aribi-Zouioueche, L. Racemization of secondary alcohols catalyzed by ruthenium: Application to chemoenzymatic dynamic resolution. Tetrahedron 2011, 22, 1790–1796. [Google Scholar]
- Hoyos, P.; Pace, V.; Alcántara, A.R. Dynamic Kinetic Resolution via Hydrolase-Metal Combo Catalysis in Stereoselective Synthesis of Bioactive Compounds. Adv. Synth. Catal. 2012, 354, 2585–2611. [Google Scholar] [CrossRef]
- Mantovani, S.M.; Angolini, C.F.; Marsaioli, A.J. Mechanistic investigation of the Candida albicans CCT 0776 stereoinversion system and application to obtain enantiopure secondary alcohols. Tetrahedron 2009, 20, 2635–2638. [Google Scholar] [CrossRef]
- Koszelewski, D.; Pressnitz, D.; Clay, D.; Kroutil, K. Deracemization of mexiletine biocatalyzed by ω-Transaminases. Org. Lett. 2009, 11, 4810–4812. [Google Scholar] [CrossRef]
- Danda, H.; Maehara, A.; Umemura, T. Preparation of (4S)-4-hydroxy-3-methyl-2-(2edirect.yl)-2-cyclopentenone by combination of enzymatic hydrolysis and chemical transformation. Tetrahedron Lett. 1991, 32, 5119–5122. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, I.S. Combined Chemical and Enzymatic Synthesis of (S,S)-2,5-Dimethylpyrrolidine. Synlett 1993, 1993, 767–768. [Google Scholar]
- Turner, N.J. Controlling chirality. Curr. Opin. Biotechnol. 2003, 14, 401–406. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Wallner, A.; Mang, H.; Glueck, S.M.; Steinreiber, A.; Mayer, S.F.; Faber, K. Chemo-enzymatic enantio-convergent asymmetric total synthesis of (S)-(+)-dictyoprolene using a kinetic resolution-stereoinversion protocol. Tetrahedron 2003, 14, 2427–2432. [Google Scholar] [CrossRef]
- Shimada, Y.; Usuda, K.; Okabe, H.; Suzuki, T.; Matsumoto, K. Deracemization of 1,2-diol monotosylate derivatives by a combination of enzymatic hydrolysis with the Mitsunobu inversion using polymer-bound triphenylphosphine. Tetrahedron 2009, 20, 2802–2808. [Google Scholar] [CrossRef]
- Bouzemi, N.; Aribi-Zouioueche, L.; Fiaud, J.C. Combined lipase-catalyzed resolution/Mitsunobu esterification for the production of enantiomerically enriched arylalkyl carbinols. Tetrahedron 2006, 17, 797–800. [Google Scholar] [CrossRef]
- Chênevert, R.; Gravil, S.; Bolte, J. Enzymatic resolution of 1,1-dimethoxybut-3-en-2-ol and 1,1-dimethoxypent-4-en-2-ol, α-hydroxyaldehyde precursors for aldol-type reactions. Tetrahedron 2005, 16, 2081–2086. [Google Scholar] [CrossRef]
- Shen, L.L.; Wang, F.; Mun, H.S.; Suh, M.; Jeong, J.H. Resolution of α-methylbenzylamine via diastereomeric salt formation using the naturally based reagent N-tosyl-(S)-phenylalanine together with a solvent switch technique. Tetrahedron 2008, 19, 1647–1653. [Google Scholar] [CrossRef]
- Steinreiber, A.; Stadler, A.; Mayer, S.F.; Faber, K.; Kappe, C.O. High-speed microwave-promoted Mitsunobu inversions: Application toward the deracemization of sulcatol. Tetrahedron Lett. 2001, 42, 6283–6286. [Google Scholar] [CrossRef]
- Zaks, A.; Tamarez, M.; Li, T. Convergent Synthesis of Both Enantiomers of 4-Hydroxypent-2-ynoic Acid Diphenylamide for a Thrombin Receptor Antagonist Sch 530348 and Himbacin Analogues. Adv. Synth. Catal. 2009, 351, 2351–2357. [Google Scholar] [CrossRef]
- Aribi-Zouioueche, L.; Fiaud, J.C. Kinetic resolution of 1-acenaphthenol and 1-acetoxynaphthene through lipase-catalyzed acylation and hydrolysis. Tetrahedron Lett. 2000, 41, 4085–4088. [Google Scholar] [CrossRef]
- Merabet-khelassi, M.; Bouzemi, N.; Fiaud, J.-C.; Riant, O.; Aribi-Zouioueche, L. Effet de la quantité de lipase sur la sélectivité du dédoublement cinétique par acylation enzymatique des arylalkylcarbinols. Comptes Rendus Chim. 2011, 14, 978–986. (In French) [Google Scholar] [CrossRef]
- Paizs, C.; Tosa, M.; Majdik, C.; Tahtinen, P.; Irimie, F.D.; Kanerva, L.T. Candida antarcticalipase A in the dynamic resolution of novel furylbenzotiazol-based cyanohydrin acetates. Tetrahedron 2003, 14, 619–627. [Google Scholar] [CrossRef]
- Rotticci, D.; Norin, T.; Hult, K. Mass Transport Limitations Reduce the Effective Stereospecificity in Enzyme-Catalyzed Kinetic Resolution. Org. Lett. 2000, 2, 1373–1376. [Google Scholar] [CrossRef]
- Warmerdam, E.; Brussee, J.; Kruse, C.G.; van der Gen, A. Inversion of the configuration of cyanohydrins by a mitsunobu esterification reaction. Tetrahedron 1993, 49, 1063–1070. [Google Scholar] [CrossRef]
- Hillier, M.C.; Desrosiers, J.N.; Marcoux, J.F.; Grabowski, E.J.J. Stereoselective Carbon–Carbon Bond Formation via the Mitsunobu Displacement of Chiral Secondary Benzylic Alcohols. Org. Lett. 2004, 9, 573–576. [Google Scholar]
- Thvedt, T.H.K.; Fuglseth, E.; Sundby, E.; Hoff, B.H. Enantioenriched 1-aryl-2-fluoroethylamines. Efficient lipase-catalysed resolution and limitations to the Mitsunobu inversion protocol. Tetrahedron 2010, 66, 6733–6743. [Google Scholar] [CrossRef]
- Houiene, Z.; Merabet-Khelassi, M.; Bouzemi, N.; Riant, O.; Aribi-Zouioueche, L. A green route to enantioenriched (S)-arylalkyl carbinols by deracemization via combined lipase alkaline-hydrolysis/Mitsunobu esterification. Tetrahedron 2013, 24, 290–296. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).