
Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clustered DNA Lesions. A Challenge to Detect, a Challenge to Repair
2.1. Biological Significance and Detection of Clustered DNA Damage
2.2. The Epigenetic Biomarker γH2AX Detects DNA Double-Strand Breaks
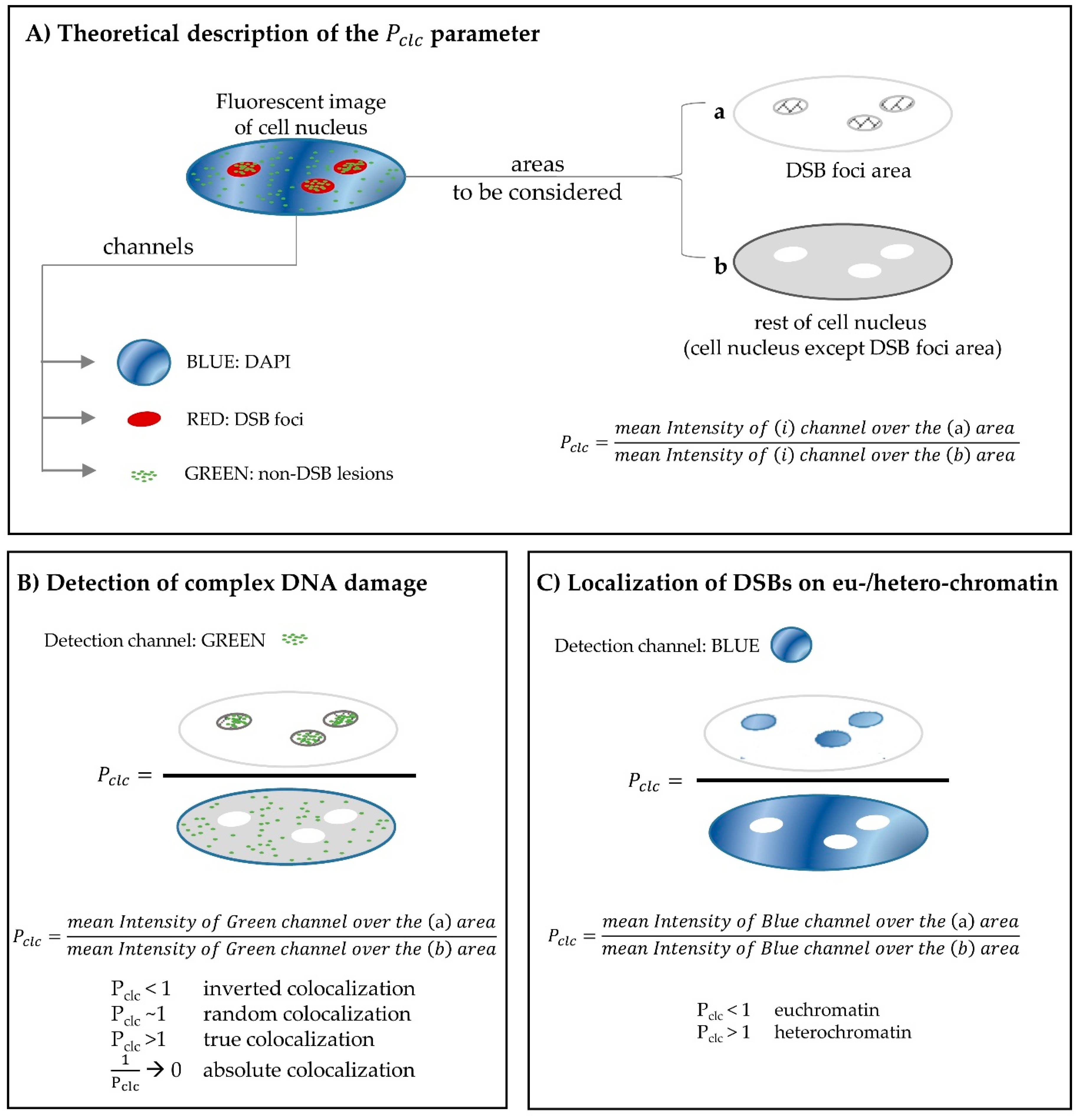
2.3. Using Fluorescence Microscopy for the in situ Detection of Complex DNA Damage. A Useful Tool
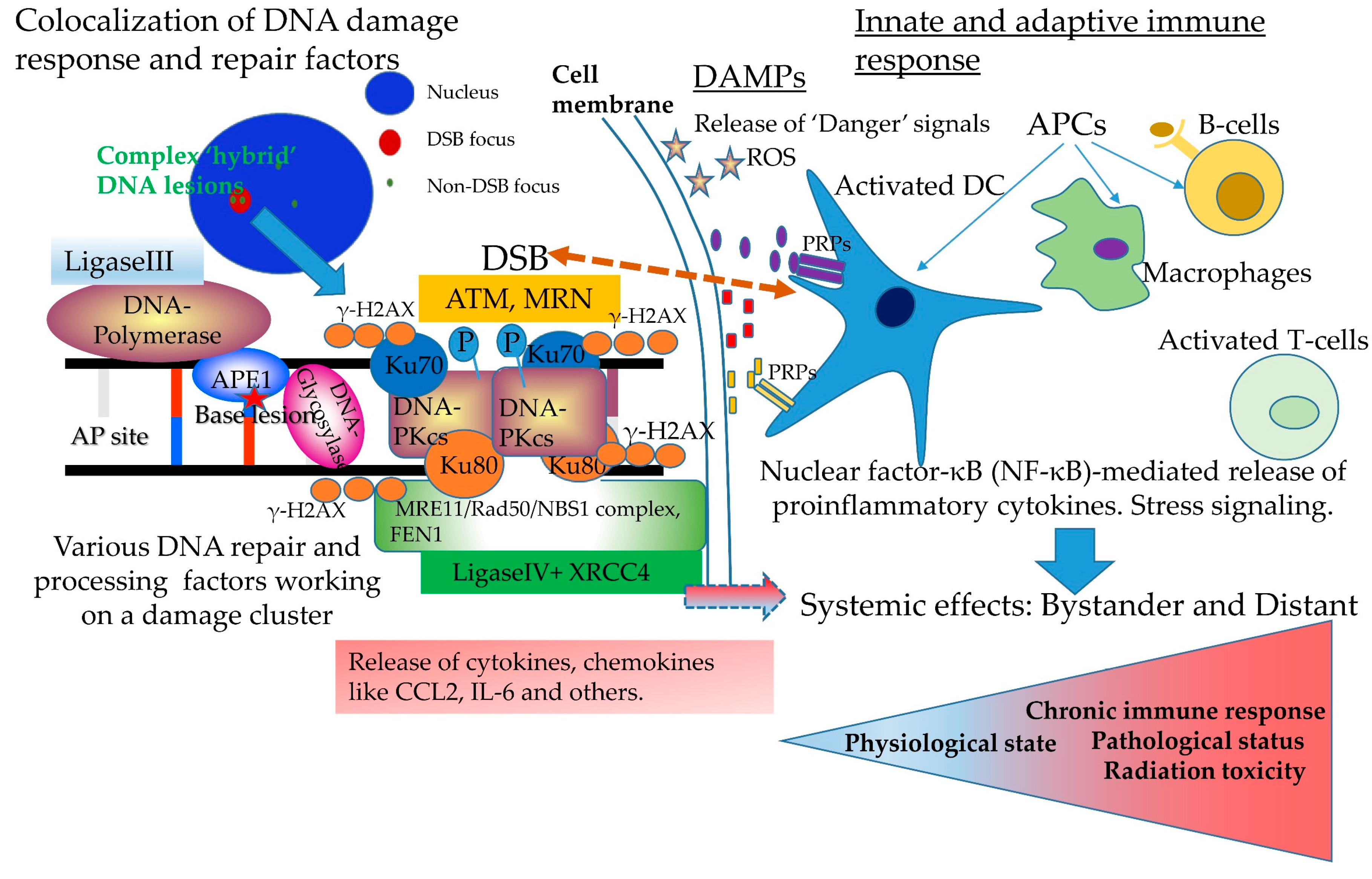
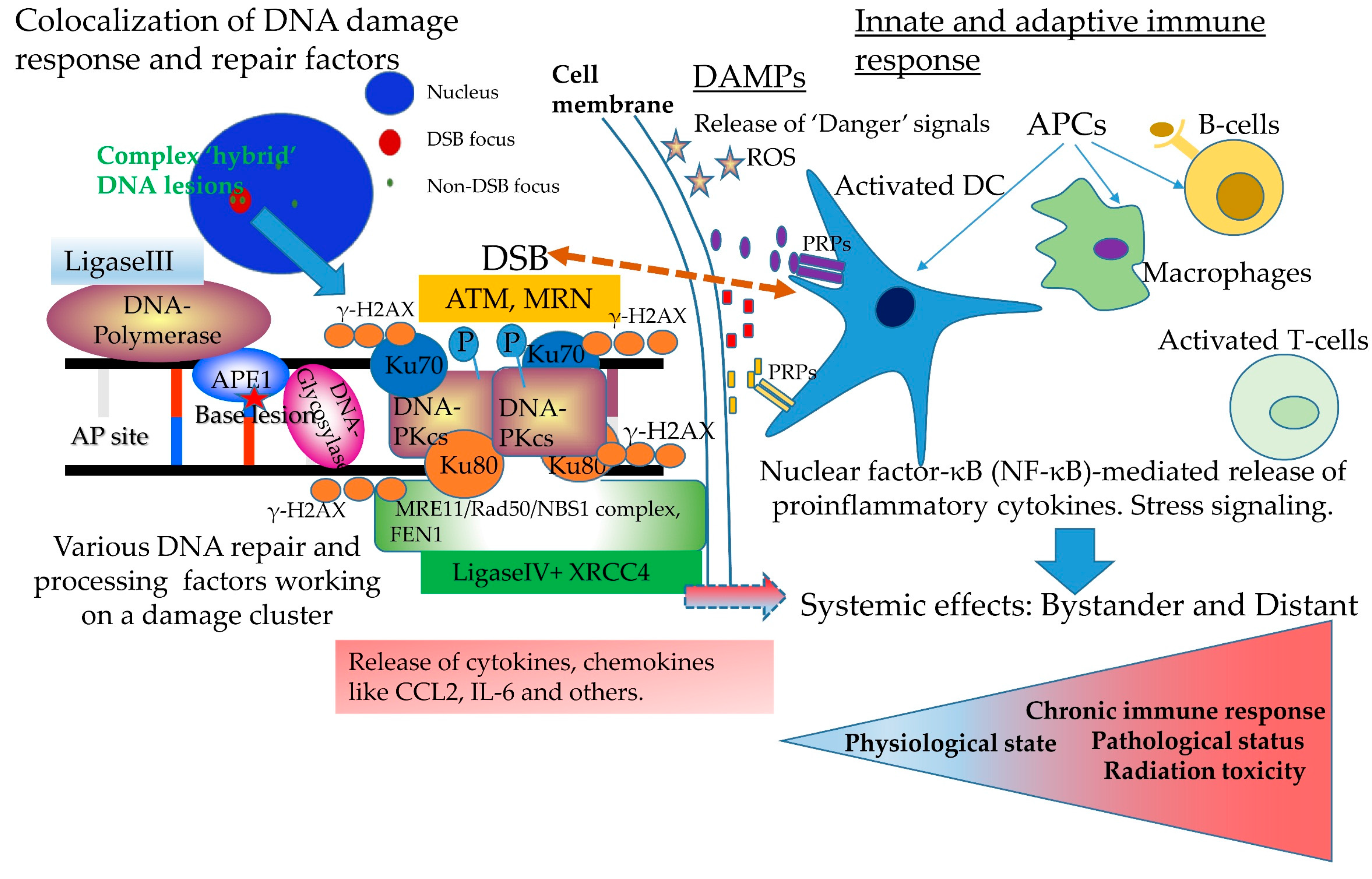
3. Complex DNA Damage, Immune Signaling and Systemic Effects. A Puzzling Case of Triage for the Cell
4. Clinical Implications of Complex DNA Damage
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Apurinic/apyrimidinic |
| BER | Base excision repair |
| bp | Base pairs |
| DAMPs | Damage-associated molecular patterns |
| DNA | Deoxyribonucleic acid |
| DDR/R | DNA damage response and repair |
| DSB | Double strand break |
| GI | Genomic instability |
| HMGB-1 | High-mobility group box 1 |
| IL | Interleukin |
| IR | Ionizing radiation |
| HR | Homologous recombination |
| LET | Linear energy transfer |
| MC | Monte Carlo |
| NHEJ | Non-homologous end joining |
| NTE | Non-targeted effects |
| OCDL | Oxidatively-clustered DNA lesions |
| PBT | Proton beam therapy |
| PRPs | Pattern recognition receptor |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RT | Radiotherapy |
| SPCs | Secondary primary cancers |
| SSB | Single strand break |
| TLRs | Toll-like receptors |
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-R.; Li, K.; Lin, S.-Y.; Hung, W.-C. Connecting the dots: From DNA damage and repair to aging. Int. J. Mol. Sci. 2016, 17, 685. [Google Scholar] [CrossRef] [PubMed]
- Nakad, R.; Schumacher, B. DNA damage response and immune defense: Links and mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Aracely, G.G.; Panayiotidis, M.I.; Franco, R. DNA damage and autophagy. Mutat. Res. 2011, 711, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Eng. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.J.; Sachs, R.K.; Wilson, D.J. Radiation-induced cancer: A modern view. Br. J. Radiol. 2012, 85, e1166–e1173. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, K.; Shimizu, Y.; Suyama, A.; Kasagi, F.; Soda, M.; Grant, E.J.; Sakata, R.; Sugiyama, H.; Kodama, K. Studies of the mortality of atomic bomb survivors, report 14, 1950–2003: An overview of cancer and noncancer diseases. Radiat. Res. 2012, 177, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; Egleston, B.L.; Litwin, S. Comments on “studies of the mortality of atomic bomb survivors, report 14, 1950–2003: An overview of cancer and noncancer diseases” (Radiat. Res., 177, 229–243, 2012). Radiat. Res. 2012, 178, 244–245. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. The threshold vs LNT showdown: Dose rate findings exposed flaws in the LNT model part 2. How a mistake led beir i to adopt LNT. Environ. Res. 2017, 154, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.A.; Pennington, C.W.; Sacks, B. Subjecting radiologic imaging to the linear no-threshold hypothesis: A non sequitur of non-trivial proportion. J. Nucl. Cardiol. 2017, 58, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cox, R. The multi-step nature of carcinogenesis and the implications for risk analysis. Int. J. Radiat. Biol. 1998, 73, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ojima, M.; Kodama, S.; Watanabe, M. Radiation-induced DNA damage and delayed induced genomic instability. Oncogene 2003, 22, 6988–6993. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. 2011, 711, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.B.; Mothersill, C. Radiation-induced bystander effects—Implications for cancer. Nat. Rev. Cancer 2004, 4, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Snyder, A.R.; Morgan, W.F. Radiation-induced genomic instability and its implications for radiation carcinogenesis. Oncogene 2003, 22, 5848–5854. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G. Role of the immune system and inflammation in ionizing radiation effects. Cancer Lett. 2015, 368, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Radons, J. Radiation, inflammation and immune responses in cancer. Front. Oncol. 2012, 2, 58. [Google Scholar] [CrossRef] [PubMed]
- Hada, M.; Georgakilas, A.G. Formation of clustered DNA damage after high-LET irradiation: A review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Lorat, Y.; Timm, S.; Jakob, B.; Taucher-Scholz, G.; Rube, C.E. Clustered double-strand breaks in heterochromatin perturb DNA repair after high linear energy transfer irradiation. Radiother. Oncol. 2016, 121, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G. Induction and Repair of Non-dsb Clustered DNA Lesions: What Do We Know and Do Not Know? In Proceedings of the 41st Annual Meeting of the European Radiation Research Society, Rhodes, Greece, 14–19 September 2014. [Google Scholar]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; Van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational signatures of ionizing radiation in second malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef] [PubMed]
- Detours, V.; Wattel, S.; Venet, D.; Hutsebaut, N.; Bogdanova, T.; Tronko, M.D.; Dumont, J.E.; Franc, B.; Thomas, G.; Maenhaut, C. Absence of a specific radiation signature in post-chernobyl thyroid cancers. Br. J. Cancer 2005, 92, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Plante, I.; Emfietzoglou, D.; Iliakis, G.; Georgakilas, A.G. Non-dsb clustered DNA lesions. Does theory colocalize with the experiment? Radiat. Phys. Chem. 2016, 128, 26–35. [Google Scholar] [CrossRef]
- Okayasu, R. Repair of DNA damage induced by accelerated heavy ions—A mini review. Int. J. Cancer 2011, 130, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Hellweg, C.; Georgakilas, A.G.; Ravanat, J.L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; O’Neill, P.; Goodhead, D.T. Securing genome stability by orchestrating DNA repair: Removal of radiation-induced clustered lesions in DNA. BioEssays 2001, 23, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Davies, K.J.; Medeiros, M.H.; Di Mascio, P.; Wagner, J.R. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic. Biol. Med. 2017, 107, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.-L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively generated base damage to cellular DNA. Free Rad. Biol. Med. 2010, 49, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Galick, H.; Wallace, S.S. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair 2004, 3, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Blaisdell, J.O.; Wallace, S. Abortive base-excision repair of radiation-induced clustered DNA lesions in escherichia coli. Proc. Natl. Acad. Sci. USA 2001, 98, 7426–7430. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, I.; Shikazono, N.; Suzuki, M.; Fujii, K.; Yokoya, A. Efficiency of radiation-induced base lesion excision and the order of enzymatic treatment. Int. J. Radiat. Biol. 2017, 93, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Rahmanian, S.; Nikjoo, H. Spectrum of radiation-induced clustered non-dsb damage - a monte carlo track structure modeling and calculations. Radiat. Res. 2015, 183, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Kozmin, S.G.; Sedletska, Y.; Reynaud-Angelin, A.; Gasparutto, D.; Sage, E. The formation of double-strand breaks at multiply damaged sites is driven by the kinetics of excision/incision at base damage in eukaryotic cells. Nucl. Acids Res. 2009, 37, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Asaithamby, A.; Hu, B.; Chen, D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8293–8298. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Taleei, R.; Liamsuwan, T.; Liljequist, D.; Emfietzoglou, D. Perspectives in radiation biophysics: From radiation track structure simulation to mechanistic models of DNA damage and repair. Radiat. Phys. Chem. 2016, 128, 3–10. [Google Scholar] [CrossRef]
- Nikjoo, H.; Emfietzoglou, D.; Liamsuwan, T.; Taleei, R.; Liljequist, D.; Uehara, S. Radiation track, DNA damage and response-a review. Rep. Prog. Phys. 2016, 79, 116601. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Streitmatter, S.W.; Argento, D.C.; Kirkby, C.; Goorley, J.T.; Moffitt, G.; Jevremovic, T.; Sandison, G.A. Rapid mcnp simulation of DNA double strand break (dsb) relative biological effectiveness (rbe) for photons, neutrons, and light ions. Phys. Med. Biol. 2015, 60, 8249–8274. [Google Scholar] [CrossRef] [PubMed]
- Taleei, R.; Girard, P.M.; Nikjoo, H. Dsb repair model for mammalian cells in early s and g1 phases of the cell cycle: Application to damage induced by ionizing radiation of different quality. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 779, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Rahmanian, S.; Taleei, R.; Nikjoo, H. Radiation induced base excision repair (ber): A mechanistic mathematical approach. DNA Repair 2014, 22, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Taleei, R.; Nikjoo, H. The non-homologous end-joining (nhej) pathway for the repair of DNA double-strand breaks: I. A mathematical model. Radiat. Res. 2013, 179, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Roch-Lefèvre, S.; Mandina, T.; Voisin, P.; Gaëtan, G.; Mesa, J.E.; Valente, M.; Bonnesoeur, P.; García, O.; Voisin, P.; Roy, L. Quantification of γ-H2AX FOCI in human lymphocytes: A method for biological dosimetry after ionizing radiation exposure. Radiat. Res. 2010, 174, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Redon, C.E.; Nakamura, A.J.; Gouliaeva, K.; Rahman, A.; Blakely, W.F.; Bonner, W.M. The use of gamma-h2ax as a biodosimeter for total-body radiation exposure in non-human primates. PLoS ONE 2010, 5, e15544. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.-X. Γ-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. {gamma}-h2ax in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucl. Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. [gamma]H2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Sedelnikova, O.A.; Redon, C.; Pilch, D.R.; Sinogeeva, N.I.; Shroff, R.; Lichten, M.; Bonner, W.M.; Judith, L.C.; et al. Techniques for gamma-H2AX detection. In Methods in Enzymology; Academic Press: San Diego, CA, USA, 2006; pp. 236–250. [Google Scholar]
- Sedelnikova, O.A.; Rogakou, E.P.; Panyutin, I.G.; Bonner, W.M. Quantitative detection of 125idu-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat. Res. 2002, 158, 486–492. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.M.; Bennett, P.V.; Sutherland, J.C.; Laval, J. Clustered DNA damages induced by X-rays in human cells. Radiat. Res. 2002, 157, 611–616. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Sidorkina, O.; Laval, J. Clustered damages and total lesions induced in DNA by ionizing radiation: Oxidized bases and strand breaks. Biochemistry 2000, 39, 8026–8031. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.; Bennett, P.V.; Sidorkina, O.; Laval, J. DNA damage clusters induced by ionizing radiation in isolated DNA and in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Gulston, M.; de Lara, C.; Jenner, T.; Davis, E.; O’Neill, P. Processing of clustered DNA damage generates additional double-strand breaks in mammalian cells post-irradiation. Nucleic Acids Res. 2004, 32, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Bennett, P.V.; Wilson, D.M., III; Sutherland, B.M. Processing of bistranded abasic DNA clusters in gamma-irradiated human hematopoietic cells. Nucleic Acids Res. 2004, 32, 5609–5620. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Mladenov, E.; Mangelis, A.; Laskaratou, D.A.; Fragkoulis, G.I.; Hellweg, C.E.; Martin, O.A.; Emfietzoglou, D.; et al. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET). Free Radic. Res. 2016, 50, S64–S78. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Laskaratou, D.A.; Frey, B.; Candeias, S.M.; Gaipl, U.S.; Lumniczky, K.; Georgakilas, A.G. Key mechanisms involved in ionizing radiation-induced systemic effects. A current review. Toxicol. Res. 2016, 5, 12–33. [Google Scholar] [CrossRef]
- Chen, H.T.; Bhandoola, A.; Difilippantonio, M.J.; Zhu, J.; Brown, M.J.; Tai, X.; Rogakou, E.P.; Brotz, T.M.; Bonner, W.M.; Ried, T.; et al. Response to RAG-mediated VDJ cleavage by nbs1 and gamma-H2AX. Science 2000, 290, 1962–1965. [Google Scholar] [CrossRef] [PubMed]
- Redon, C.E.; Nakamura, A.J.; Zhang, Y.-W.; Ji, J.; Bonner, W.M.; Kinders, R.J.; Parchment, R.E.; Doroshow, J.H.; Pommier, Y. Histone γH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res. 2010, 16, 4532–4542. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.N.; Martin, O.A.; Smith, A.J.; Redon, C.E.; Bonner, W.M.; Martin, R.F.; Lobachevsky, P.N. GammaH2AX foci as a measure of DNA damage: A computational approach to automatic analysis. Mutat. Res. 2011, 711, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Sunter, N.J.; Singh, P.B.; Austin, C.A.; Durkacz, B.W.; Tilby, M.J. Gammah2ax foci form preferentially in euchromatin after ionising-radiation. PLoS ONE 2007, 2, e1057. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone h2ax is phosphorylated in an atr-dependent manner in response to replicational stress. J.Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Derijck, A.A.; van der Heijden, G.W.; Giele, M.; Philippens, M.E.; van Bavel, C.C.; de Boer, P. Gammah2ax signalling during sperm chromatin remodelling in the mouse zygote. DNA Repair 2006, 5, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Suchankova, J.; Kozubek, S.; Legartova, S.; Sehnalova, P.; Kuntziger, T.; Bartova, E. Distinct kinetics of DNA repair protein accumulation at DNA lesions and cell cycle-dependent formation of gammaH2AX- and nbs1-positive repair foci. Biol. Cell 2015, 107, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Djuzenova, C.S.; Zimmermann, M.; Katzer, A.; Fiedler, V.; Distel, L.V.; Gasser, M.; Waaga-Gasser, A.M.; Flentje, M.; Polat, B. A prospective study on histone gamma-H2AX and 53bp1 foci expression in rectal carcinoma patients: Correlation with radiation therapy-induced outcome. BMC Cancer 2015, 15, 856. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, F.; Campa, A.; Esposito, G.; Giardullo, P.; Belli, M.; Dini, V.; Meschini, S.; Simone, G.; Sorrentino, E.; Gerardi, S.; et al. Induction and repair of DNA DSB as revealed by H2AX phosphorylation foci in human fibroblasts exposed to low- and high-let radiation: Relationship with early and delayed reproductive cell death. Radiat. Res. 2015, 183, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Vandevoorde, C.; Franck, C.; Bacher, K.; Breysem, L.; Smet, M.H.; Ernst, C.; De Backer, A.; Van De Moortele, K.; Smeets, P.; Thierens, H. Γ-h2ax foci as in vivo effect biomarker in children emphasize the importance to minimize X-ray doses in paediatric ct imaging. Eur. Radiol. 2014, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, N.I.; Brunton, H.; Watanabe, R.; Shrikhande, A.; Hirayama, R.; Matsufuji, N.; Fujimori, A.; Murakami, T.; Okayasu, R.; Jeggo, P.; et al. Visualisation of gammah2ax foci caused by heavy ion particle traversal; distinction between core track versus non-track damage. PLoS ONE 2013, 8, e70107. [Google Scholar] [CrossRef] [PubMed]
- Staaf, E.; Brehwens, K.; Haghdoost, S.; Czub, J.; Wojcik, A. Gamma-h2ax foci in cells exposed to a mixed beam of X-rays and alpha particles. Genome Integr. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.E.; Zlobinskaya, O.; Multhoff, G. Differences in phosphorylated histone h2ax foci formation and removal of cells exposed to low and high linear energy transfer radiation. Curr. Genom. 2012, 13, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, M.V.; Dickey, J.S.; Bonner, W.M.; Sedelnikova, O.A. Γ-H2AX in bystander cells: Not just a radiation-triggered event, a cellular response to stress mediated by intercellular communication. Cell Cycle 2007, 6, 2210–2212. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Bonner, W.M. Gammah2ax in cancer cells: A potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle 2006, 5, 231–240. [Google Scholar]
- Desai, N.; Davis, E.; O’Neill, P.; Durante, M.; Cucinotta, F.A.; Wu, H. Immunofluorescence detection of clustered gamma-H2AX foci induced by HZE-particle radiation. Radiat. Res. 2005, 164, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; MacPhail, S.H.; Banath, J.P.; Klokov, D.; Olive, P.L. Endogenous expression of phosphorylated histone H2AX in tumors in relation to DNA double-strand breaks and genomic instability. DNA Repair 2006, 5, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Banath, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Neumaier, T.; Swenson, J.; Pham, C.; Polyzos, A.; Lo, A.T.; Yang, P.; Dyball, J.; Asaithamby, A.; Chen, D.J.; Bissell, M.J.; et al. Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. Proc Natl. Acad. Sci. USA 2012, 109, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Schuler, N.; Palm, J.; Kaiser, M.; Betten, D.; Furtwängler, R.; Rübe, C.; Graf, N.; Rübe, C.E. DNA-damage foci to detect and characterize DNA repair alterations in children treated for pediatric malignancies. PLoS ONE 2014, 9, e91319. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Kanagaraj, R.; Mihaljevic, B.; Schwendener, S.; Sartori, A.A.; Gerrits, B.; Shevelev, I.; Janscak, P. Mre11 complex links recq5 helicase to sites of DNA damage. Nucleic Acids Res. 2009, 37, 2645–2657. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, V.B.; Rodier, F.; Choi, Y.; Busuttil, R.A.; Vogel, H.; Vijg, J.; Campisi, J.; Hasty, P. Ku80 deletion suppresses spontaneous tumors and induces a p53-mediated DNA damage response. Cancer Res. 2008, 68, 9497–9502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, C.; Sun, F.; Wei, W.; Hu, B.; Wang, J. Both complexity and location of DNA damage contribute to cellular senescence induced by ionizing radiation. PLoS ONE 2016, 11, e0155725. [Google Scholar] [CrossRef] [PubMed]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 2016, 37–38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA damage and innate immunity: Links and trade-offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. DNA damage: A trigger of innate immunity but a requirement for adaptive immune homeostasis. Nat. Rev. Immunol. 2006, 6, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Ermolaeva, M.A.; Segref, A.; Dakhovnik, A.; Ou, H.-L.; Schneider, J.I.; Utermohlen, O.; Hoppe, T.; Schumacher, B. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature 2013, 501, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppe, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Behrens, A.; van Deursen, J.M.; Rudolph, K.L.; Schumacher, B. Impact of genomic damage and ageing on stem cell function. Nat. Cell Biol. 2014, 16, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Mavragani, I.V.; Laskaratou, D.A.; Gika, V.; Moskvin, V.P.; Theofilatos, K.; Vougas, K.; Stewart, R.D.; Georgakilas, A.G. Systemic mechanisms and effects of ionizing radiation: A new ‘old’ paradigm of how the bystanders and distant can become the players. Semin. Cancer Biol. 2016, 37–38, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.A.; Yin, X.; Forrester, H.B.; Sprung, C.N.; Martin, R.F. Potential strategies to ameliorate risk of radiotherapy-induced second malignant neoplasms. Semin. Cancer Biol. 2016, 37–38, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Siva, S.; MacManus, M.P.; Martin, R.F.; Martin, O.A. Abscopal effects of radiation therapy: A clinical review for the radiobiologist. Cancer Lett. 2015, 356, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, S.; Matzinger, P. Danger signals: Sos to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Kareva, I.G.; Naf, D.; Nowsheen, S.; Kryston, T.B.; Bonner, W.M.; Georgakilas, A.G.; Sedelnikova, O.A. Tumors induce complex DNA damage in distant proliferative tissues in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17992–17997. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Redon, C.E.; Ferguson, N.F.; Kryston, T.B.; Parekh, P.; Dickey, J.S.; Nakamura, A.J.; Mitchell, J.B.; Bonner, W.M.; Martin, O.A. Systemic DNA damage accumulation under in vivo tumor growth can be inhibited by the antioxidant tempol. Cancer Lett. 2014, 353, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.A.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Bonner, W.M. Para-inflammation mediates systemic DNA damage in response to tumor growth. Commun. Integr. Biol. 2011, 4, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, J.; Luo, C.; Pandi, S.P.; Penalva, R.G.; Fitzgerald, D.C.; Xu, H. Para-inflammation-mediated retinal recruitment of bone marrow-derived myeloid cells following whole-body irradiation is CCL2 dependent. Glia 2012, 60, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Siva, S.; Lobachevsky, P.N.; MacManus, M.; Kron, T.; Moller, A.; Lobb, R.; Ventura, J.; Best, N.; Smith, J.; Ball, D.; et al. Radiotherapy for non-small cell lung cancer induces DNA damage response in both irradiated and out-of-field normal tissues. Clin. Cancer Res. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G. Bystander and non-targeted effects: A unifying model from ionizing radiation to cancer. Cancer Lett. 2015, 356, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Kapoor, V.; Buchlis, G.; Cheng, G.; Sun, J.; Wang, L.-C.S.; Singhal, S.; Snyder, L.A.; Albelda, S.M. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; LaRusso, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000, 60, 184–190. [Google Scholar] [PubMed]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakasilioti, I.; Kamileri, I.; Chatzinikolaou, G.; Kosteas, T.; Vergadi, E.; Robinson, A.R.; Tsamardinos, I.; Rozgaja, T.A.; Siakouli, S.; Tsatsanis, C.; et al. DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in ner progeria. Cell Metab. 2013, 18, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Aziz, K.; Nowsheen, S.; Pantelias, G.; Iliakis, G.; Gorgoulis, V.G.; Georgakilas, A.G. Targeting DNA damage and repair: Embracing the pharmacological era for successful cancer therapy. Pharmacol. Ther. 2012, 133, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Regulus, P.; Duroux, B.; Bayle, P.A.; Favier, A.; Cadet, J.; Ravanat, J.L. Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could induce the formation of a cluster DNA lesion. Proc. Natl. Acad. Sci. USA 2007, 104, 14032–14037. [Google Scholar] [CrossRef] [PubMed]
- Bredenfeld, H.; Franklin, J.; Nogova, L.; Josting, A.; Fries, S.; Mailander, V.; Oertel, J.; Diehl, V.; Engert, A. Severe pulmonary toxicity in patients with advanced-stage hodgkin’s disease treated with a modified bleomycin, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone, and gemcitabine (beacopp) regimen is probably related to the combination of gemcitabine and bleomycin: A report of the german hodgkin's lymphoma study group. J. Clin. Oncol. 2004, 22, 2424–2429. [Google Scholar] [PubMed]
- De Lena, M.; Guzzon, A.; Monfardini, S.; Bonadonna, G. Clinical, radiologic, and histopathologic studies on pulmonary toxicity induced by treatment with bleomycin (nsc-125066). Cancer Chemother. Rep. 1972, 56, 343–356. [Google Scholar] [PubMed]
- Cunningham, T.J.; Olson, K.B.; Horton, J.; Wright, A.; Hussain, M.; Davies, J.N.; Harrington, G. A clinical trial of intravenous and intracavitary bleomycin. Cancer 1972, 29, 1413–1419. [Google Scholar] [CrossRef]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to nhej mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bennett, P.V.; Cintron, N.S.; Gros, L.; Laval, J.; Sutherland, B.M. Are endogenous clustered DNA damages induced in human cells? Free Radic. Biol. Med. 2004, 37, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.M.; Bennett, P.V.; Cintron, N.S.; Guida, P.; Laval, J. Low levels of endogenous oxidative damage cluster levels in unirradiated viral and human dnas. Free Rad. Biol. Med. 2003, 35, 495–503. [Google Scholar] [CrossRef]
- Hable, V.; Drexler, G.A.; Bruning, T.; Burgdorf, C.; Greubel, C.; Derer, A.; Seel, J.; Strickfaden, H.; Cremer, T.; Friedl, A.A.; et al. Recruitment kinetics of DNA repair proteins MDC1 and RAD52 but not 53bp1 depend on damage complexity. PLoS ONE 2012, 7, e41943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, A.S.; Hartman, T.; Stenerlow, B. Formation and repair of clustered damaged DNA sites in high let irradiated cells. Int. J. Radiat. Biol. 2015, 91, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Held, K.D.; Kawamura, H.; Kaminuma, T.; Paz, A.E.; Yoshida, Y.; Liu, Q.; Willers, H.; Takahashi, A. Effects of charged particles on human tumor cells. Front. Oncol. 2016, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, O.; Sishc, J.B.; Saha, J.; Pompos, A.; Rahimi, A.; Story, D.M.; Davis, J.A.; Kim, N.D.W. Carbon ion radiotherapy: A review of clinical experiences and preclinical research, with an emphasis on DNA damage/repair. Cancers 2017, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Tsao, D.; Kalogerinis, P.; Tabrizi, I.; Dingfelder, M.; Stewart, R.D.; Georgakilas, A.G. Induction and processing of clustered DNA lesions in human monocytes exposed to low doses of HZE 56Fe particles. Radiat. Res. 2007, 168, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Leloup, C.; Garty, G.; Assaf, G.; Cristovao, A.; Breskin, A.; Chechik, R.; Shchemelinin, S.; Paz-Elizur, T.; Livneh, Z.; Schulte, R.W.; et al. Evaluation of lesion clustering in irradiated plasmid DNA. Int. J. Radiat. Biol. 2005, 81, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Pachnerova Brabcova, K.; Stepan, V.; Karamitros, M.; Karabin, M.; Dostalek, P.; Incerti, S.; Davidkova, M.; Sihver, L. Contribution of indirect effects to clustered damage in DNA irradiated with protons. Radiat. Prot. Dosimetry 2015, 166, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Vysin, L.; Pachnerova Brabcova, K.; Stepan, V.; Moretto-Capelle, P.; Bugler, B.; Legube, G.; Cafarelli, P.; Casta, R.; Champeaux, J.P.; Sence, M.; et al. Proton-induced direct and indirect damage of plasmid DNA. Radiat. Environ. Biophys. 2015, 54, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Marshall, T.I.; Currell, F.J.; Kacperek, A.; Schettino, G.; Prise, K.M. Variations in the processing of DNA double-strand breaks along 60-MEV therapeutic proton beams. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Veiga, L.H.; Lubin, J.H.; Anderson, H.; de Vathaire, F.; Tucker, M.; Bhatti, P.; Schneider, A.; Johansson, R.; Inskip, P.; Kleinerman, R.; et al. A pooled analysis of thyroid cancer incidence following radiotherapy for childhood cancer. Radiat. Res. 2012, 178, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, C.S.; Chou, J.F.; Wolden, S.L.; Bernstein, J.L.; Malhotra, J.; Novetsky Friedman, D.; Mubdi, N.Z.; Leisenring, W.M.; Stovall, M.; Hammond, S.; et al. Breast cancer after chest radiation therapy for childhood cancer. J. Clin. Oncol. 2014, 32, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Grantzau, T.; Thomsen, M.S.; Vaeth, M.; Overgaard, J. Risk of second primary lung cancer in women after radiotherapy for breast cancer. Radiother. Oncol. 2014, 111, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.Y.; Huang, W.Y.; Lin, C.S.; Su, Y.F.; Lo, C.H.; Tsao, C.C.; Liu, M.Y.; Lin, C.L.; Kao, C.H. Risk of second primary malignancies among patients with prostate cancer: A population-based cohort study. PLoS ONE 2017, 12, e0175217. [Google Scholar] [CrossRef] [PubMed]
- Rastgou Talemi, S.; Kollarovic, G.; Lapytsko, A.; Schaber, J. Development of a robust DNA damage model including persistent telomere-associated damage with application to secondary cancer risk assessment. Sci. Rep. 2015, 5, 13540. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, A.J.; Jones, I.M. Second cancers after radiotherapy: Any evidence for radiation-induced genomic instability? Oncogene 2003, 22, 7018–7027. [Google Scholar] [CrossRef] [PubMed]
- Kandula, S.; Zhu, X.; Garden, A.S.; Gillin, M.; Rosenthal, D.I.; Ang, K.K.; Mohan, R.; Amin, M.V.; Garcia, J.A.; Wu, R.; et al. Spot-scanning beam proton therapy vs intensity-modulated radiation therapy for ipsilateral head and neck malignancies: A treatment planning comparison. Med. Dosim. 2013, 38, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, S.; Nakamura, J.L. Radiotherapy-induced malignancies: Review of clinical features, pathobiology, and evolving approaches for mitigating risk. Front. Oncol. 2013, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Makita, C.; Nakamura, T.; Takada, A.; Takayama, K.; Suzuki, M.; Ishikawa, Y.; Azami, Y.; Kato, T.; Tsukiyama, I.; Kikuchi, Y.; et al. Clinical outcomes and toxicity of proton beam therapy for advanced cholangiocarcinoma. Radiat. Oncol. 2014, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Lin, L.; Simone, C.B., 2nd; Berman, A.T. Risk of major cardiac events following adjuvant proton versus photon radiation therapy for patients with thymic malignancies. Acta Oncol. 2017, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, M.; Murayama, S.; Akimoto, T.; Demizu, Y.; Fukushima, T.; Ishida, Y.; Oshiro, Y.; Numajiri, H.; Fuji, H.; Okumura, T.; et al. Long-term follow-up after proton beam therapy for pediatric tumors: A japanese national survey. Cancer Sci. 2017, 108, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Cotter, S.E.; Herrup, D.A.; Friedmann, A.; Macdonald, S.M.; Pieretti, R.V.; Robinson, G.; Adams, J.; Tarbell, N.J.; Yock, T.I. Proton radiotherapy for pediatric bladder/prostate rhabdomyosarcoma: Clinical outcomes and dosimetry compared to intensity-modulated radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Amini, A.; Raben, D.; Crawford, E.D.; Flaig, T.W.; Kessler, E.R.; Lam, E.T.; Maroni, P.; Pugh, T.J. Patient characterization and usage trends of proton beam therapy for localized prostate cancer in the united states: A study of the national cancer database. Urol. Oncol. 2017, 35, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg Robert, A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Julie, J.B.; Patsy, A.T.; Robert, M.L. Non-targeted effects and radiation-induced carcinogenesis: A review. J. Radiol. Prot. 2016, 36, R23. [Google Scholar]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. https://doi.org/10.3390/cancers9070091
Mavragani IV, Nikitaki Z, Souli MP, Aziz A, Nowsheen S, Aziz K, Rogakou E, Georgakilas AG. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers. 2017; 9(7):91. https://doi.org/10.3390/cancers9070091
Chicago/Turabian StyleMavragani, Ifigeneia V., Zacharenia Nikitaki, Maria P. Souli, Asef Aziz, Somaira Nowsheen, Khaled Aziz, Emmy Rogakou, and Alexandros G. Georgakilas. 2017. "Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis" Cancers 9, no. 7: 91. https://doi.org/10.3390/cancers9070091