
Extracellular Molecules Involved in Cancer Cell Invasion

1
Department of Biochemistry, Hellenic Pasteur Institute, Athens 11521, Greece
2
Technological Educational Institute of Athens, Egaleo, Athens 12210, Greece
*
Author to whom correspondence should be addressed.
Cancers 2015, 7(1), 238-265; https://doi.org/10.3390/cancers7010238
Submission received: 12 December 2014
/
Revised: 30 December 2014
/
Accepted: 20 January 2015
/
Published: 26 January 2015
(This article belongs to the Special Issue Cancer Cell Invasion)
Abstract
:Nowadays it is perfectly clear that understanding and eradicating cancer cell invasion and metastasis represent the crucial, definitive points in cancer therapeutics. During the last two decades there has been a great interest in the understanding of the extracellular molecular mechanisms involved in cancer cell invasion. In this review, we highlight the findings concerning these processes, focusing in particular on extracellular molecules, including extracellular matrix proteins and their receptors, growth factors and their receptors, matrix metalloproteinases and extracellular chaperones. We report the molecular mechanisms underlying the important contribution of this pool of molecules to the complex, multi-step phenomenon of cancer cell invasion.
1. Introduction
Metastasis is one of the most important problems concerning mortality in cancer patients [1,2,3]. It is a multistep, complex process composed of a cascade of inter-connected events including: neo-vascularization, the escape of tumor cells from the primary tumor, a process known as local cancer cell invasion, migration through the extracellular matrix (ECM), intravasation, circulation and survival of the tumor cells in the blood or lymphatic circulation, extravasation and invasion of the tumor cells through the endothelium and basement membrane of the target site, and finally growth of the secondary tumor (colonization) [4,5]. During this multistage process, only very small numbers of cancer cells can survive, and give rise to secondary tumors. This type of cancer cell shows resistance under adverse conditions, such as tumor hypoxia and nutrient shortage [6], and chemo- and radio-therapies [7]. Moreover, these die-hard neoplastic cells must have the ability to self-renew and differentiate in order to create new tumor bulks at distant sites from the primary tumor [8]. Cancer Stem Cells (CSC) represent exactly the kind of tumor cells that possess all of these fundamental requisites for cancer cell invasion and metastasis [9].
Cancer cell invasion and secondary tumor outgrowth are regulated by numerous, interconnected molecular networks. In fact, there are many intracellular molecules belonging to the Wnt, Notch, Sonic Hedgehog, NF-κB, Ras/Raf/MEK/MAPK, as well as the AKT/ERK signaling pathways, which control every aspect of each of the stages of cancer cell invasion [4,5,10]. On the other hand, extracellular molecules also contribute in a critical way to the progress of cancer cell invasion. These molecules can be (a) part of the ECM, (b) secreted in the ECM (c) secreted but also attached on the cell surface (d) cell membrane proteins such as receptors. In this review, we focus on the function and regulation of extracellular molecules that take part in cancer cell invasion and metastatic processes (Table 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Molecule Name | Molecule Type | Molecules Co-involved in Cancer Cell Invasion |
|---|---|---|---|
| ECM MOLECULES | Hyaluronan (HA) | glycosaminoglycan | CD44 |
| Fibronectin (FN) | glycoprotein | eHSP90, HSP90, MMP-9, MMP-9, FAK/PI3K/AKT/ERK/NF-κB, PEDF | |
| SIBLING | Small Integrin-Binding Ligand, N-linked Glycoprotein | Pro-MMPs, MMP-2, MMP-9, MMP-3, αvβ3 integrin, FAK/MEK/ERK/NF-Κβ pathway, CD44v6 | |
| ECM RECEPTORS | Integrins | Cell surface receptors | Fibronectin, MMP-9, MMP-2, FAK/ILK/, ERK/, PI3K/NF-κB signaling cascades EGFR, osteopontin |
| CD44 | Cell surface receptors | Hyaluronan (HA), osteopontin | |
| GROWTH FACTORS | TGF-β | Growth factors | TBRI, TBRII, Erk, Ras |
| Heregulin | EGF-like growth and differentiation factor | ErbB3, ErbB4, PAK-1, AMF | |
| GROWTH FACTOR RECEPTORS | EGFR | Cell surface receptor | TGF-α, Grb2, Ras/Raf/MEK/MAPK |
| HER-2 | Cell surface co-receptor | HER-3, eHSP90, MAPK, PI3K/AKT | |
| IGF-R | Cell surface receptor | IGFs, IRS-2, PI3K/AKT, Ras/Raf/MAPK | |
| MATRIXMETALLO-PROTEINASES | Matrix Metalloproteinase (MMP)-9 | Zinc endopeptidase | eHSP90, HSP90, Rab40b, VAMP-4, gelatin type IV collagen, VEGF, bFGF |
| Matrix Metalloproteinase (MMP)-2 | Zinc endopeptidase | gelatine, type IV collagen, eHSP90, HSP90, Rab40b, VAMP-4, VEGF, bFGF | |
| CD10 | Zinc-dependent metalloproteinase | Twist1 | |
| CHAPERONES | eHSP90 | Chaperone | Cdc37, FN, HER-2, EGFR, pro-MMP-2, pro-MMP-9 |
| eCdc37 | Co-chaperone | HSP90, eHSP90, HER2, EGFR, Raf1, CDK4, EGFRvIII, Peuth-Jeghers cancer syndrome-associated kinase | |
| LRP-1 | LRP-1 | Low-density lipoprotein (LDL) receptor | Nexin-1 (PN-1), Erk pathway, MMP-9, eHSP90, EphA2, AKT1, AKT2 |
2. ECM Molecules
2.1. Hyaluronan
Hyaluronan (HA), also known as hyaluronic acid, constitutes the major glycosaminoglycan present in the ECM. HA binds mainly to CD44 receptor (Figure 1A) and promotes tumor growth, survival as well as cancer cell invasion [11]. Moreover, data suggest that HA forms a protecting covering for cancer cells against cytotoxic and chemotherapeutic agents, and that augmented HA synthesis leads to a less dense matrix that facilitates cancer cell motility and invasion. Regarding tumor progression, it has been shown that HA promotes tumor-associated angiogenesis [11,12] and that the expression of this molecule and hyaluronan synthase (HAS), as well as the rate of HA synthesis, are increased in highly metastatic breast carcinoma cells [13,14].
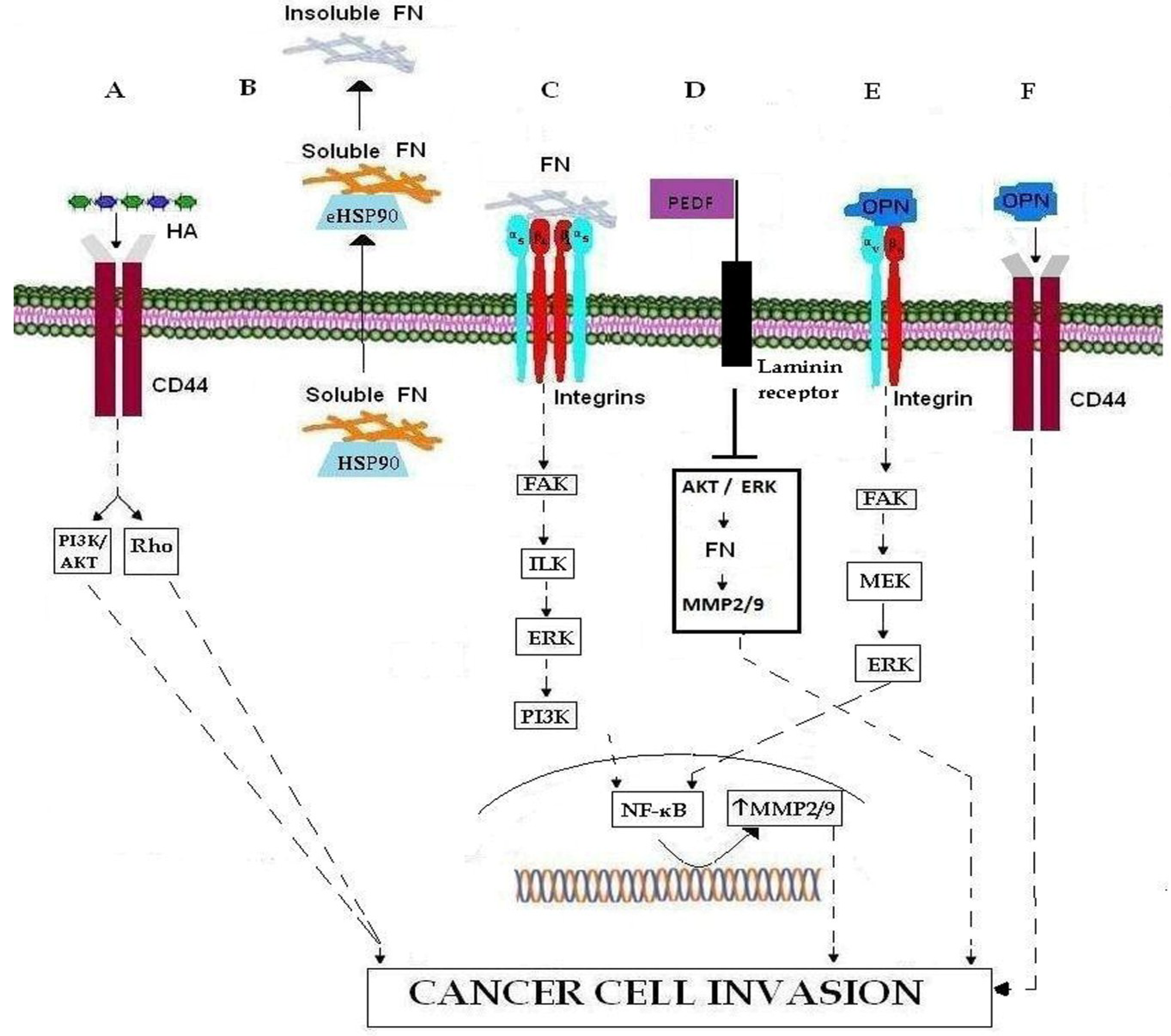
Figure 1.
Important ECM molecules and ECM receptors involved in cancer cell invasion. (A) Binding of HA to CD44 promotes cell invasion. HA-CD44 interaction promotes invasion via the PI3Κ/AKT and Rho signaling pathways. (B) The transport of FN, in its soluble form, to the membrane occurs through the chaperone activity of intracellular HSP90, while eHSP90 regulates the conversion of soluble FN to its insoluble ECM form. (C) Binding of FN to integrin results in MMP-2 and MMP-9 over-expression via the FAK/ILK/ERK/PI3K/NF-κB pathways, and thereby leading to ECM degradation and cancer cell invasion. (D) PEDF binding to integrin results in MMP-2 and MMP-9 down-regulation through inhibition of AKT/ERK signaling pathway. (E) OPN binding to integrin promotes cancer cell invasion by inducing MMP-9 over-expression through the FAK/MEK/ERK/NF-κB pathway. (F) Binding of OPN to CD44 promotes cell invasion.
Figure 1.
Important ECM molecules and ECM receptors involved in cancer cell invasion. (A) Binding of HA to CD44 promotes cell invasion. HA-CD44 interaction promotes invasion via the PI3Κ/AKT and Rho signaling pathways. (B) The transport of FN, in its soluble form, to the membrane occurs through the chaperone activity of intracellular HSP90, while eHSP90 regulates the conversion of soluble FN to its insoluble ECM form. (C) Binding of FN to integrin results in MMP-2 and MMP-9 over-expression via the FAK/ILK/ERK/PI3K/NF-κB pathways, and thereby leading to ECM degradation and cancer cell invasion. (D) PEDF binding to integrin results in MMP-2 and MMP-9 down-regulation through inhibition of AKT/ERK signaling pathway. (E) OPN binding to integrin promotes cancer cell invasion by inducing MMP-9 over-expression through the FAK/MEK/ERK/NF-κB pathway. (F) Binding of OPN to CD44 promotes cell invasion.
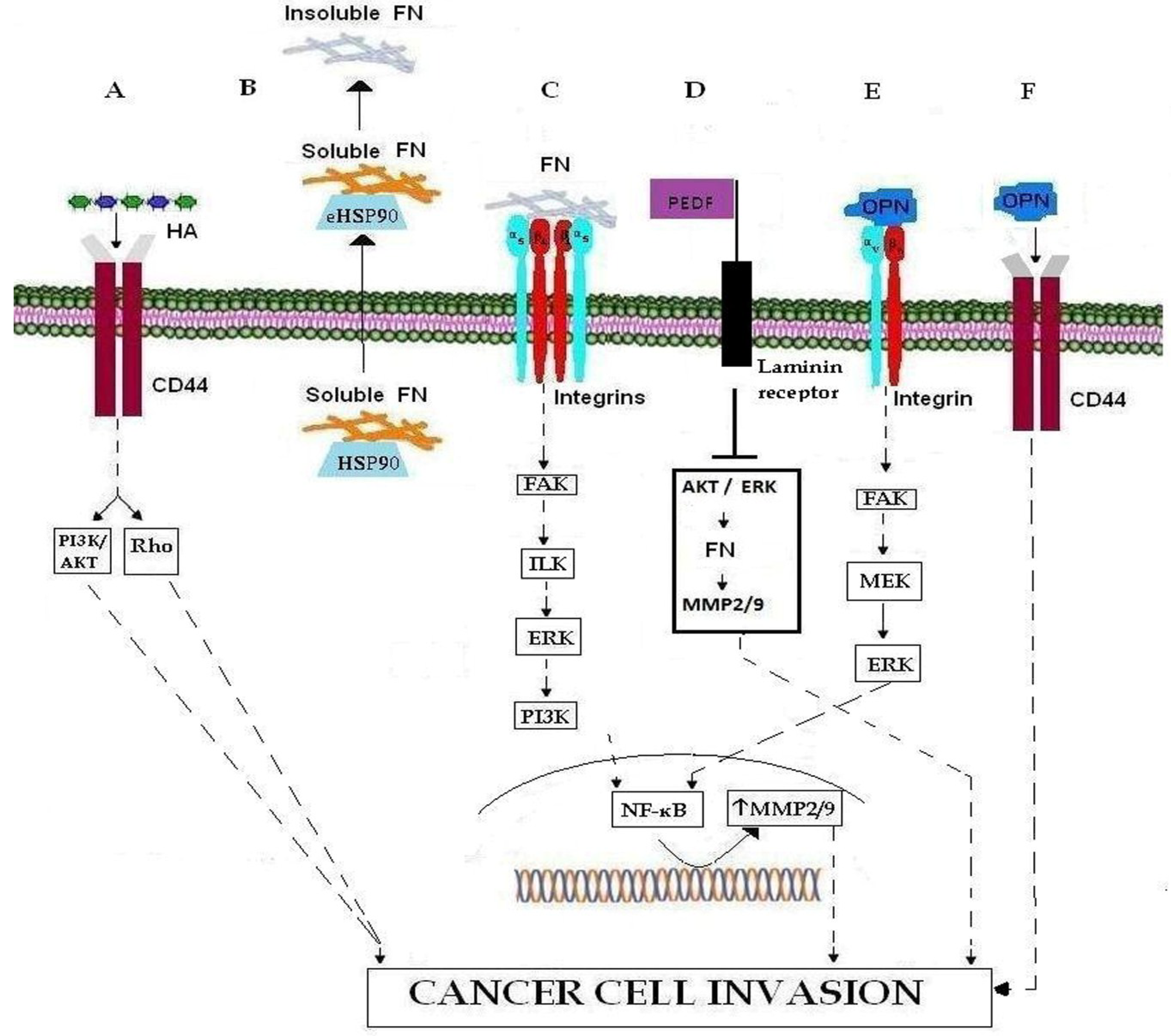
2.2. Fibronectin
ECM glycoprotein fibronectin (FN), a major cell-matrix and cell-cell adhesion mediator, is involved in the regulation of embryogenesis, mesoderm formation, tissue repair, cell migration, differentiation, cell growth as well as certain pathological disorders such as fibrosis, atherosclerosis, tumor invasion and metastasis [15,16,17,18]. FN is secreted by cells as a soluble dimer that is then assembled into an insoluble network of fibers. The change of FN conformation from soluble to insoluble form begins after its secretion, when it binds to cell surface integrins and exposes self-association sites which allow the creation of an insoluble form [19,20,21]. Regarding the regulation of FN function, Hunter et al. suggest that intracellular HSP90 acts as a chaperone for the stabilization and/or transport of soluble FN followed by its export from the cell (Figure 1B). Once secreted, extracellular HSP90 (eHSP90) promotes the conversion of soluble FN to its insoluble form (Figure 1B). The intra- and extracellular roles of HSP90 as an FN chaperone can be considered as another HSP90 mechanism, which promotes cell migration and metastasis through the degradation and remodeling of ECM [17]. FN over-expression has been reported in specimens of various tumor types such as breast, lung, thyroid and esophageal cancer [22]. Additionally, FN over-expression has been correlated with poor clinical outcome in breast cancer patients, whilst its involvement in breast cancer invasion and metastasis has been demonstrated to involve the up-regulation of matrix metalloproteinases (MMPs) MMP-2 and MMP-9 via the FAK/ILK//ERK/PI3K/NF-κB cascade of pathways [23,24] (Figure 1C). Most recently Hong et al. [16] proposed a new model regarding the down-regulation of FN by pigment epithelium-derived factor (PEDF), a molecule well-known for its important anti-cancer role, mainly through the inhibition of angiogenesis and the induction of tumor differentiation and apoptosis in various types of cancer. In particular, they showed that PEDF inhibits MMP-2 and MMP-9 expression by binding to the laminin receptor and consequently inhibiting the AKT/ERK tumorigenic pathway, thereby down-regulating FN expression (Figure 1D).
2.3. SIBLING
The Small Integrin-Binding Ligand, N-linked Glycoprotein (SIBLING) family includes bone sialoprotein (BSP), osteopontin (OPN), dentin matrix protein 1 (DMP1), dentin sialoprotein (DSPP), and matrix extracellular phosphoglycoprotein (MEPE). They comprise a class of non-structural ECM proteins. Expression of SIBLING family members was first characterized in mineralized tissue including bone and teeth. Additionally, SIBLING proteins are localized in neoplastic tissues and induce metastasis [25,26]. Elevated SIBLING expression has been associated with an analogous increased expression of MMPs in breast, stomach, colon, ovarian, rectal and lung cancers [25]. Amongst the SIBLING proteins mentioned above, OPN is a secreted phosphoprotein characterized as a biomarker of tumor metastasis because increased OPN expression was found within tumor cells and in the surrounding stroma of multiple human cancers [27,28,29,30,31,32,33,34]. Nowadays, OPN is considered a serum biomarker in predicting tumor metastasis. Elevated OPN levels can be specific in predicting disease progression in head and neck, gastric, renal, hepatocellular, lung, and pancreatic cancers as well as uveal melanoma. Additionally, it has been established that OPN is a strong prognostic indicator for overall survival as its circulating levels are proportional with tumor stage and metastasis [35,36]. Two main mediators of OPN signaling pathways are ανβ integrins and CD44. OPN binds various types of integrins, such as ανβ3 which participates in the metastatic phenomenon in several ways. ανβ3-OPN interaction promotes cancer cell migration and invasion in prostate and breast cancer as well as in chondrosarcoma where OPN-ανβ3 binding leads to MMP-9 up-regulation through the FAK/MEK/ERK/NF-Κβ pathway [37,38,39,40] (Figure 1E). Moreover, OPN-ανβ3 integrin ligation promotes neo-vascularization by up-regulating endothelial cell migration, survival and lumen formation during angiogenesis [41,42,43,44,45]. Finally, OPN interaction with CD44v6 is observed in metastasis of breast, hepatocellular, pancreatic, lung, colorectal cancers and lymphomas [26,46,47,48,49,50,51] (Figure 1F).
Primary tumor formation and metastatic processes are clearly the result of the co-participation of genetically modified tumor and normal cells. OPN is mainly secreted by tumor cells while in myeloid cells OPN is localized intracellularly. Most recently, Sangaletti et al. clarified an aspect of the dual role of OPN whereby tumor cells secrete OPN in order to support their survival in the blood circulation, whereas both tumor- and host-derived OPN, particularly from myeloid cells, render the metastatic site more immunosuppressive [52].
3. ECM Receptors
3.1. Integrins
Integrins are the major and most characterized cell surface receptors of several ECM proteins such as laminin, fibronectin, collagen IV and vitronectin. Integrins are composed of non-covalent, heterodimeric complexes of an α and β subunit [53]. Many members of the integrin family, such as α5β1, α8β1, αIIbβ3, αVβ3, αVβ5, αVβ6, and αVβ8 recognize an Arg-Gly-Asp (RGD) motif within their ligands, which include FN, fibrinogen, vitronectin, von Willebrand factor, and various other large glycoproteins [54]. Both the α and β subunits are transmembrane glycoproteins. As the cytoplasmic tails of integrins are devoid of enzymatic features, they transduce signals by associating with adaptor proteins that connect the integrin to the cytoskeleton, cytoplasmic kinases, and transmembrane growth factor receptors [55]. Integrins constitute the mediators between ECM and the actin cytoskeleton with focal adhesion sites representing the regions of signal transduction controlling proliferation, differentiation, survival, wound healing, migration, tumorigenesis, etc. [56]. It has been suggested that bone metastasis derived from advanced prostate cancer process is characterized by the integrin-mediated interaction of metastatic cancer cells and bone microenvironment [57]. In fact, it has been shown that in the majority of tumors, ανβ3 integrin is the prime initial receptor to support adhesion and migration to bone matrix. The crucial role of integrins in cancer cell invasion is additionally evidenced by an α5β1 integrin-FN interaction (Figure 1C), which accelerates cell invasion of SiHa cervical cancer cells and promotes the expression and activation of pro-MMP-9, as well as moderate change of pro-MMP-2 activity through the FAK, ILK, ERK, PI3K and NF-κB signaling cascade [58]. Moreover, an α5β1 integrin-FN interaction was found to up-regulate MMP-9 expression and activity in the highly metastatic MDA-MB-231cancer cell line. Additionally, the expression of ανβ3 integrin has been found significantly higher in pancreatic primary tumors with lymph node infiltration, as compared to those without node metastasis, while tumors with high ανβ3 integrin expression showed significantly higher MMP-2 activation ratios than did tumors with low expression of this receptor [59]. Regarding breast cancer-bone metastasis, Takayama et al. reported that breast cancer cells which express αvβ3 integrin acquire the ability to adhere to bone matrix in breast cancer bone metastasis [60]. Finally, a crosstalk between ανβ3 integrin and epidermal growth factor receptor (EGFR) has been shown, through which cancer cell invasion and metastasis are stimulated [61].
3.2. CD44
The CD44 receptors are cell-surface molecules which mediate cell-matrix and cell-cell interactions [26,47]. They constitute a family of transmembrane glycoproteins encoded by a single gene. Alternative splicing and variation in N- and O-glycosylation give rise to the various CD44 isoforms distinguishable by their differential roles in breast cancer CD44s, one of the most expressed CD44 isoforms, is up-regulated in primary tumors but correlates with overall patient survival [62,63]. In fact, it has been shown that CD44s inhibits cancer cell invasion since loss of CD44s in vivo resulted in a marked promotion of cancer metastasis to the lung in a metastatic mouse model of breast carcinoma whereas tumor onset and tumor size were unaffected [63,64]. On the other hand, Rys et al. revealed a strong correlation of CD44ν3 isoform with tumor infiltration by T lymphocytes and cancer metastasis to draining lymph nodes combined with a loss of p53 protein expression [13,64].
Furthermore, as mentioned above, CD44 represents the main receptor of HA [13,65] whose binding with CD44 promotes signaling pathways that induce tumor growth, survival as well as cancer cell invasion [11]. In particular, binding of HA with CD44ν3 triggers downstream Rho and PI3K-AKT signalling pathways, inducing breast cancer cell growth, and invasion (Figure 1A).
Nowadays, CD44 is an established marker of cancer stem populations in breast, prostate, pancreas, ovarian and colorectal cancers [66,67,68]. In particular, the CD44+/CD24low/− combination represents the molecular phenotype of breast CSC sub-population which represents the part of the tumor that can survive during colonization and promote cancer cell invasion. In fact, breast CSC constitute the only cell sub-population of this type of neoplasia that can effectively induce the creation of tumors when injected to immunosuppresed mice [69].
4. Growth Factors
4.1. TGF-β
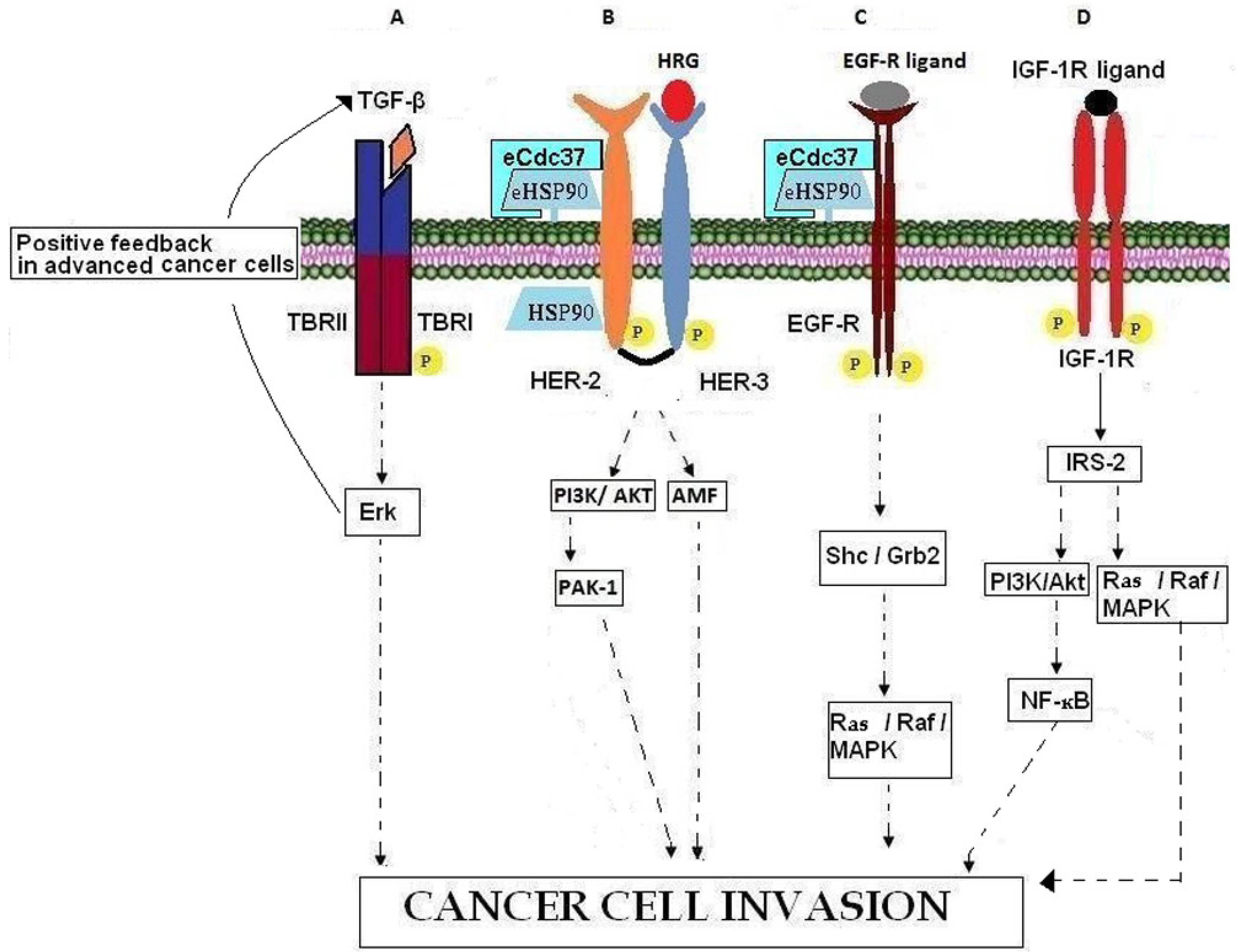
Transforming growth factor β (TGF-β) belongs to a large family of polypeptide growth factors that includes activins, inhibins, and bone morphogenetic proteins (BMPs). There are three known mammalian TGF-β isoforms (TGF-β1, TGF-β2, TGF-β3), which are closely related both structurally and functionally. These isoforms are secreted as latent precursor molecules that are activated by proteolytic cleavage, interaction with integrins, or pH changes in the local environment [70,71,72]. Active TGF-β is implicated in many regulatory activities that influence development, tissue repair, immune defense, inflammation and tumorigenesis [70,73]. The biological effects of TGF-β are mediated through specific receptors (TBRI and TBRII), which are transmembrane serine/threonine kinases (Figure 2A). TGF-β is involved in tumor cell invasion by participating in epithelial mesenchymal transition (EMT) [74,75,76], by enhancing angiogenesis [77] and by mediating immune evasion of tumor cells [78,79]. TGF-β is known to inhibit the cell cycle in benign cells and early stage cancer cells while at the same time it promotes progression of the cell cycle and metastasis in advanced cancer cells [78,79,80,81]. This phenomenon is known as the TGF-β paradox [82]. Recently, Zhang et al. [83] reported that differential activation of Erk in cancer cells is the underlying molecular mechanism of the TGF-β paradox (Figure 2A). More precisely, the inhibition or progression of cell cycle is due to inactivation or activation of the cell proliferation regulator Erk, respectively [83,84].
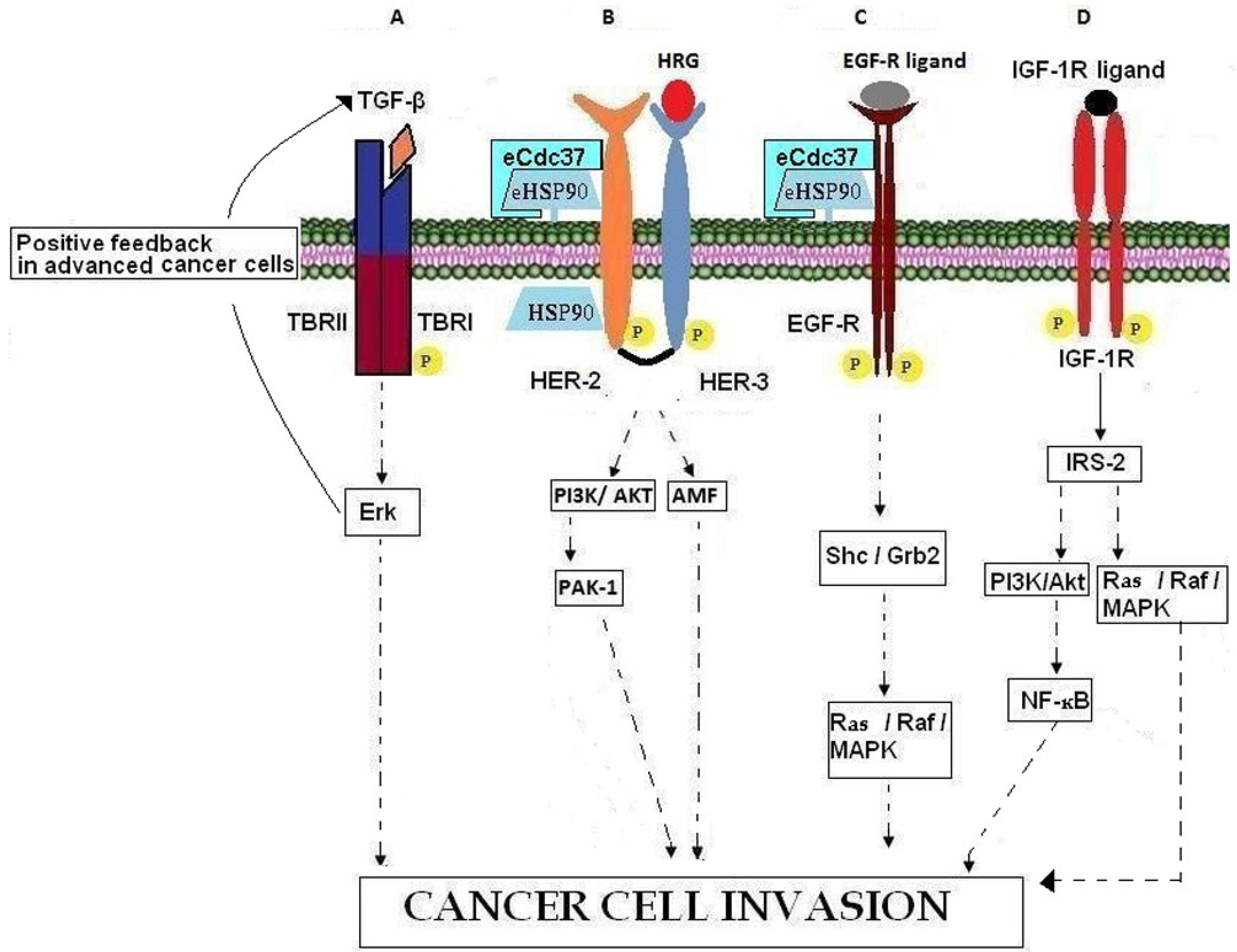
Figure 2.
Involvement of growth factors, growth factor receptors, and chaperones in cancer cell invasion. (A) In advanced cancer cells, TBRII activation promotes cancer cell invasion by binding over-expressed TGF-β and activating Erk which induces a positive feedback loop by further increasing TGF-β. (B) HRG binding to HER-3 leads to the formation of HER-3/HER-2 heterodimers, the activation of downstream kinase signaling pathways and in actin re-arrangement and cell invasion. HSP90 contributes to intracellular HER-2 stabilization while eHSP90-eCdc37-HER-2 heterocomplexes are necessary for HER-2 heterodimerization with HER-3, leading to cell invasion through PI3K/AKT and AMF signaling pathways. (C) EGFR binding to its ligands leads to activation of the Ras/Raf//MAPK pathways resulting in cancer cell invasion. The chaperoning activity of eHSP90, associated with co‑chaperone eCdc37, is necessary for EGFR stabilization. (D) Binding of IGF-1R with its ligand results in IGF-1R auto-phosphorylation, recruitment of adaptor proteins such as IRS-2 and subsequent activation of PI3K/AKT and Ras/Raf/MAPK pathways which promote the invasion processes.
Figure 2.
Involvement of growth factors, growth factor receptors, and chaperones in cancer cell invasion. (A) In advanced cancer cells, TBRII activation promotes cancer cell invasion by binding over-expressed TGF-β and activating Erk which induces a positive feedback loop by further increasing TGF-β. (B) HRG binding to HER-3 leads to the formation of HER-3/HER-2 heterodimers, the activation of downstream kinase signaling pathways and in actin re-arrangement and cell invasion. HSP90 contributes to intracellular HER-2 stabilization while eHSP90-eCdc37-HER-2 heterocomplexes are necessary for HER-2 heterodimerization with HER-3, leading to cell invasion through PI3K/AKT and AMF signaling pathways. (C) EGFR binding to its ligands leads to activation of the Ras/Raf//MAPK pathways resulting in cancer cell invasion. The chaperoning activity of eHSP90, associated with co‑chaperone eCdc37, is necessary for EGFR stabilization. (D) Binding of IGF-1R with its ligand results in IGF-1R auto-phosphorylation, recruitment of adaptor proteins such as IRS-2 and subsequent activation of PI3K/AKT and Ras/Raf/MAPK pathways which promote the invasion processes.
4.2. Heregulin
Heregulin (HRG; also called neregulin (NRG), Neu differentiation factor (NDF), glial growth factor (GGF), and acetylcholine receptor-inducing activity (ARIA)) is a member of the EGF-like growth and differentiation factors and binds with high affinity to the receptors ErbB3 and ErbB4 [85]. The HRG gene family consists of four members, HRG-1, HRG-2, HRG-3, and HRG-4 [86,87,88,89,90] of which a multitude of different isoforms are synthesized by alternative exon splicing [91], showing various tissue distributions, variable potencies, different receptor specificities, and variable biological functions. HRG has been implicated in developmental processes [85,92,93,94], as well as in the patho-physiological processes of psychiatric diseases, cardiac diseases, and various types of cancer. Although increasing data indicate that HRG-2, HRG-3, and HRG-4 may play a role in malignancy, most research interests have focused on the HRG-1 gene. HRG-1 is over-expressed in 30% of human breast cancer patients [95]. On the other hand Raj et al. have reported that low levels of HRG1 in cases of locally advanced breast cancer, are associated with poor prognosis [96]. HRG participates in tumor growth through induction of angiogenesis and invasion. In particular it has been shown that HRG induces EMT process and cell migration in SK-BR-3 and MCF7 breast cancer cells by binding to the HER-3 receptor and signaling through the PI3K/AKT pathway [85,92,93,94,97,98] (Figure 2B). HRG induced cell migration through PI3K takes place also by regulating PAK-1 and enhancing the formation of lamellipodia, membrane ruffles, stress fibers and fidopodia [99]. HRG-induced progression of breast cancer cells into a more aggressive phenotype also involves the regulation of Autocrine Motility Factor (AMF). In fact, it has been shown that HRG stimulates cell motility-associated changes such as cell scattering and actin re-organization by up-regulating the expression of AMF [100]. HRG stimulates the up-regulation of proteins implicated in cell invasion acting both as a ligand which binds its cell surface receptor(s) and activates downstream signaling pathways and as an intracellular mediator of the expression of invasion-related genes. In fact, reported data suggest that HRG may regulate transcription indirectly by recruiting co-factors considered essential for transcriptional control [101,102].
5. Growth Factor Receptors
5.1. ErbB Receptors
The ErbB family of receptor tyrosine kinases (RTK) includes four distinct receptors: the EGFR (also known as ErbB-1/HER-1), ErbB-2 (neu, HER-2), ErbB-3 (HER-3) and ErB-4 (HER-4) [103,104]. With respect to ErbB-receptor binding, ErbB ligands can be classified into three groups: (a) those that bind specifically to EGFR, in particular, transforming growth factor α (TGFα) and amphiregulin (AR); (b) those that show dual specificity by binding EGFR and ErbB-4, specifically betacellulin (BTC), heparin-binding growth factor (HB-EGF) and epiregulin (EPR); (c) the neuregulins (NRGs) which can be divided in two sub-groups based upon their capacity to bind HER-3 and HER-4 (NRG-1 and NGR-2) or only HER-4 (NRG-3 and NGR-4) [86,87,89,105,106]. None of the EGF family of ligands bind HER-2. Ligands binding to the extracellular domain of the respective receptors induce homo- or hetero-dimerization of ErbBs. Dimerization consequently stimulates intrinsic tyrosine kinase activity of the receptors and triggers auto-phosphorylation of specific tyrosine residues within the cytoplasmic regulatory domain. These phosphorylated tyrosines serve as binding sites for various adaptor proteins such as Shc, Grb7, Grb2, Crk, Nck, the phospholipase Cγ (PLCγ), the intracellular kinases Src and PI3K, the protein tyrosine phosphatases SHP1 and SHP2 and Cbl E3 ubiquitin ligase [107,108]. EGFR ligands and receptors induce activation of the Ras/Raf/MEK/MAPK pathway through either Grb2 or Shc adaptor proteins [109,110,111]. These signaling events eventually result in cell proliferation, angiogenesis, resistance to apoptosis, migration and metastasis (Figure 2C). In fact, in normal tissues the availability of EGFR ligands is regulated in order to ensure that the kinetics of cell proliferation precisely match the tissues’ requirements for homeostasis. In the case of neoplasia however, the EGFR is often chronically stimulated, either by EGFR ligands that are over-produced within the tumor microenvironment [106,112] or as a result of EGFR mutation that causes spontaneous receptor activation [113]. EGFR is expressed in various types of neoplasia including those in the lung, head and neck, colon, pancreas, breast, ovary, bladder and kidney as well as in gliomas [114]. The over-expression of EGFR and TGFα by neoplasias confers a more aggressive phenotype by inducing cancer metastasis, resistance to chemotherapy and poor prognosis [115,116,117]. Given its massive presence in several tumors and its key role in metastasis, EGFR is defined as a principal target in anti-cancer therapies [118].
HER-2 is considered a ligandless receptor which shows preferred heterodimerization with HER-1, HER-3, and HER-4 [104,110,119]. HER-2 functions as a co-receptor to mediate signal transduction resulting in cell motility, mitogenesis, apoptosis, angiogenesis and/or cell differentiation. Any alteration of the tightly regulated HER-2 receptor signaling pathways results in major cellular abnormalities and tumorigenesis. HER-2 over-expression is strongly associated with increased progression and metastasis in human breast and prostate cancer [104,110,119,120,121]. Whilst it had been shown already that intracellular HSP90 contributes to the stability of HER-2 via its cytoplasmic kinase domain [122,123,124]. Sidera et al. [125] reported in 2008 that the molecular interaction of cell surface HSP90 with the extracellular domain (ECD) of HER-2 is necessary for breast cancer cell invasion. More specifically, they showed that this interaction is essential for receptor activation and subsequent heterodimerization with HER-3 which in turn mediates signal transduction pathways via MAP kinase and PI3K/AKT, leading to actin re-arrangement necessary for cell motility (Figure 2B). It has been reported previously that the ECD of HER-2 constitutively adopts an extended configuration with its dimerization arm exposed, suggesting that it is always poised to form heterodimers with ligand-activated forms of ErbB-receptors [110,126,127]. Taking this into consideration Sidera et al. speculated that surface HSP90 interacts with the HER-2 ectodomain in order for the receptor to maintain its active conformation. Finally, it should be noted that HER-2–HER-3 heterodimerization is essential for mediating the effects of growth factors such as HRG on cell motility. HRG binds to HER-3 to activate downstream kinase signaling pathways which lead to actin re-arrangement and cell invasion [125].
5.2. IGF-R
The insulin-like growth factor (IGF) machinery comprises (a) the circulating ligands; insulin-like growth factor-1 (IGF-1), IGF-2 and insulin, (b) multiple receptors; IGF-1R, insulin receptor (IR), hybrid receptors containing one chain of IGF-1R and one chain of IR (IGF-1R/IR-A, IGF-1/IR-B) and IGF-2R, (c) multiple adaptor proteins. Under physiological conditions, IGF-1R activation is implicated in fetal growth as well as in linear growth of the skeleton and other organs [128]. In the case of neoplasia, IGF-1R is frequently over-expressed inducing proliferation, cancer cell motility and adhesion, as well as inhibition of apoptosis. It is known to promote metastasis in various cancers, including those of the colon, pancreas, prostate and breast [129,130,131]. IGF-1R is activated by IGFs present in the extracellular environment, in an endocrine, paracrine or autocrine manner. Upon ligand binding, IGF-1R becomes auto-phosphorylated and subsequently recruits specific docking intermediates, including insulin-receptor substrate-2 (IRS-2), that activate PI3K/AKT and Ras/Raf/MAPK pathways in order to promote cell motility and pro-metastatic behaviour in breast cancer cells [129,132,133] (Figure 2D). In models of breast cancer bone metastasis, IGF-1R activation promotes motility of bone-metastatic cells [134]. In this context, it has been reported that bone-derived IGFs, which are released from bone in substantial amounts by osteoclastic bone resorption, activate IGF-IR/AKT/NF-kB signaling pathways in breast cancer cells that are colonizing the bone, thereby increasing their proliferation, decreasing apoptosis and thus promoting the development and progression of bone metastases [135,136].
6. Matrix Metalloproteinases
6.1. MMPs
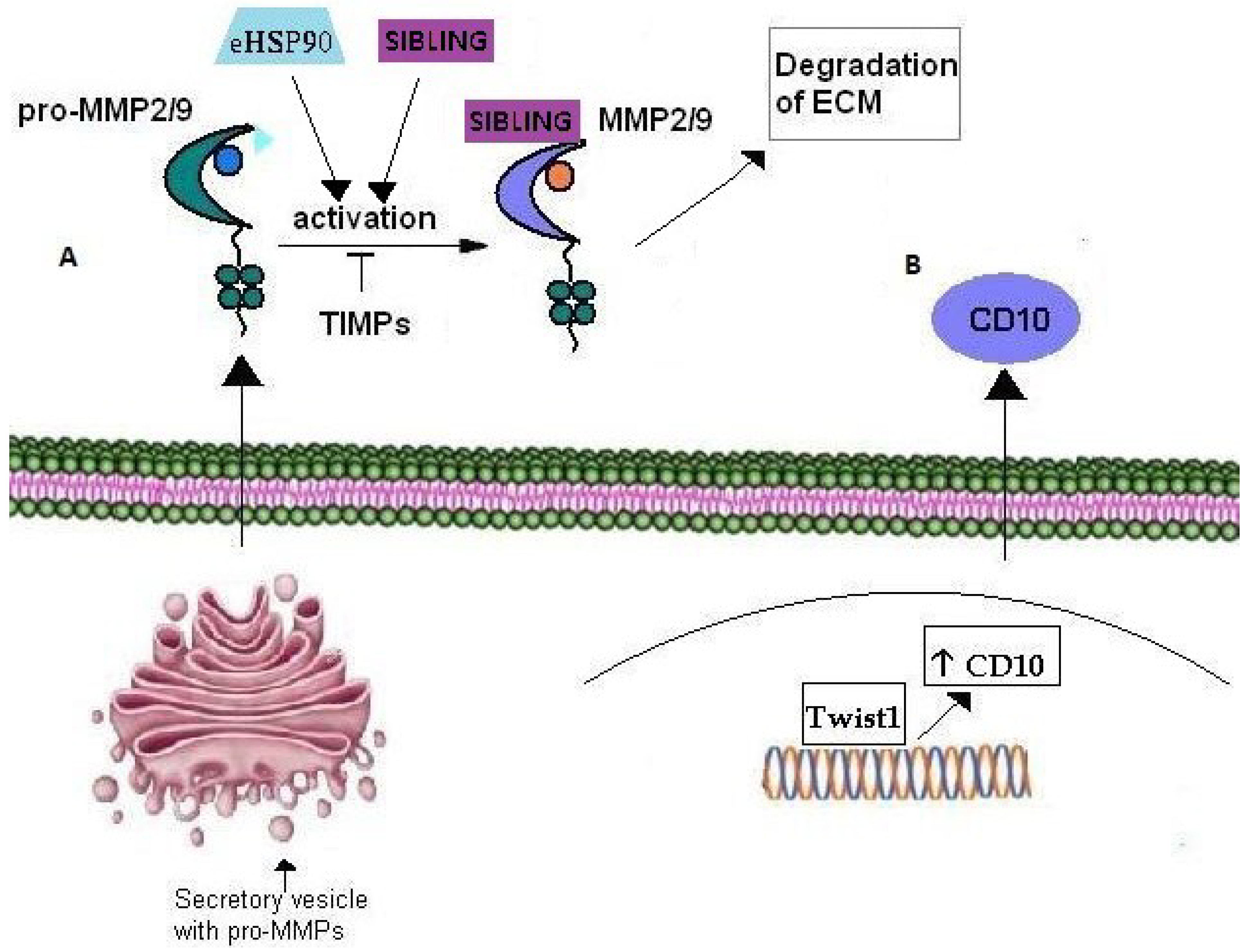
MMPs are a family of zinc-binding endopeptidases that participate in the ECM degradation molecular machinery during tumor invasion [137,138]. In fact, in order for tumor neo-vascularization and cell invasion processes to occur, degradation of the basement membrane as well as matrix remodeling are essential. Amongst the many known MMPs, MMP-2 and MMP-9 degrade gelatin as well as type IV collagen, the central component of the basement membrane. These MMPs are secreted in an inactive form and acquire their active form extracellularly [139,140]. Eustace et al in 2004, demonstrated that eHSP90 activates MMP-2 leading to increased tumor invasiveness in HT-1080 fibrosarcoma cells [141]. The association of eHSP90 with MMPs was further confirmed by Stellas et al. [142] in 2010 with the demonstration that eHSP90 participates in the activation of MMP-2 and MMP-9 in the process of breast cancer cell invasion (Figure 3A). Moreover fluorescence binding and affinity purification studies have shown that three members of the SIBLING family, BSP, DMP1 and OPN, activate pro-MMPs-, MMP-2 MMP-9 and MMP-3, respectively and subsequently bind to the catalytically active MMPs (Figure 3A). Additionally, SIBLING re-activate MMPs that have been de-activated by tissue or exogenous inhibitors [143]. ECM degradation and cancer cell invasion are inter-connected mechanisms that include basement membrane disintegration by actin-rich, finger-like cellular membrane projections located at the ventral side of the cell, named invadopodia [144]. MMP-2 and MMP-9 are enriched in the invadopodia where they contribute to ECM degradation in vitro and in vivo [144]. According to Jacob et al. [145], the intracellular transport and targeting of MMP-2 and MMP-9 to invadopodia during the breast cancer invasion process, is mediated by small monomeric Rab40b GTPase. More specifically, the transport of MMP-2 and MMP-9 from the trans-Golgi network (TGN) is mediated by secretory vesicles containing vesicle-associated membrane protein 4 (VAMP-4) and Rab40b (Figure 3A). Rab GTPases regulate various membrane transport steps including cargo sorting, vesicle budding, transport and targeting to the appropriate target compartment [146]. Jacob et al. showed that Rab40b knockdown, results in mis-targeting of MMP-2 and MMP-9 to lysosomes, where they are usually degraded. Overall, they identified Rab40b GTPase as the key regulator in MMP-2 and MMP-9 transport and targeting to the plasma membrane in the breast cancer invasion process. On the other hand, the down-regulation of MMP activity includes inactivation by extracellular tissue inhibitors of MMPs, named TIMPs [147] (Figure 3A).
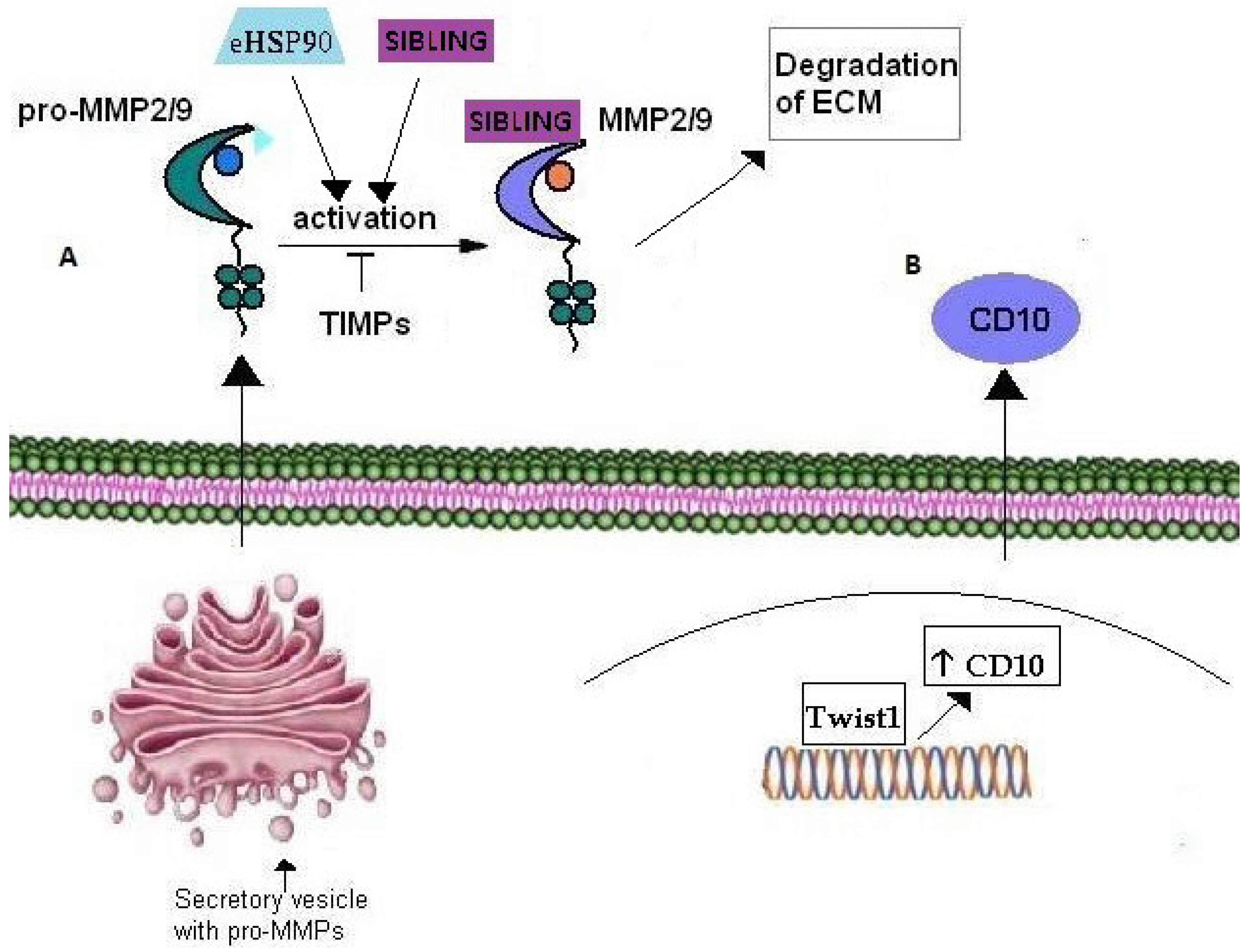
Figure 3.
Role of Matrix Metalloproteinases in cancer cell invasion. (A) Pro-MMP2/9 are transported through secretory vesicles of the Golgi network to the plasma membrane. Their activation occurs extracellularly through their interaction with eHSP90 and SIBLING that consequently bind the active MMP2/9. TIMPs have an inhibitory effect on MMP2/9 activation. (B) CD10 metalloproteinase over-expression is correlated with cancer cell invasion in several tumors. Moreover, CD10 expression is up-regulated by transcription factor Twist1 which is considered a master inductor of EMT and thus, cancer cell invasion.
Figure 3.
Role of Matrix Metalloproteinases in cancer cell invasion. (A) Pro-MMP2/9 are transported through secretory vesicles of the Golgi network to the plasma membrane. Their activation occurs extracellularly through their interaction with eHSP90 and SIBLING that consequently bind the active MMP2/9. TIMPs have an inhibitory effect on MMP2/9 activation. (B) CD10 metalloproteinase over-expression is correlated with cancer cell invasion in several tumors. Moreover, CD10 expression is up-regulated by transcription factor Twist1 which is considered a master inductor of EMT and thus, cancer cell invasion.
Nowadays, it is established that the tumor invasion process is not only based on cancer cell migration but is also a result of the activity of normal cells [148]. In this context, it has been shown that tumor cells induce MMP expression and secretion by stromal cells, including fibroblasts, endothelial cells, and inflammatory cells via cell-cell contact or paracrine mechanisms. MMPs secreted by stromal cells, especiallyMMP-2, MMP-3 and MMP-9, contribute equally or even more to tumor cell invasion than MMPs secreted from cancer cells [137]. More specifically, Min et al. [137] demonstrated that poor overall survival is correlated with the expression of MMP-2 on stromal and tumor cells, as well as the expression of MMP-9 on tumor cells, and suggest that stromal MMP-2 may have a critical role in breast cancer aggressiveness. Finally MMPs promote neovascularization by inducing the secretion of heparin bound growth factors like VEGF and bFGF into their soluble pro-angiogenic forms. In this context, it has been reported that the production of the pro-angiogenic growth factor VEGF is induced by MMP-9 [149,150,151,152].
6.2. CD10
CD10, also called neprilysin and Common Acute Lymphoblastic Leukemia/Lymphoma Antigen (CALLA), belongs to the family of membrane bound, zinc-dependent metalloproteinases, members of which regulate the physiological action of various proteins by lowering their extracellular concentration available for receptor binding [153]. In fact, CD10 is involved in numerous biological activities through regulation of signal transduction of bioactive neuropeptides and vasoactive peptides [154,155]. In particular, it is expressed in the central nervous system, regulating various substrates such as encephalin, an opioid peptide liberated by neurons in response to pain, whilst polymorphisms in the CD10 gene augment the risk for Alzheimer’s disease. Moreover, CD10 is involved in regulating mechanisms of the immune system by controlling, through degradation, the activation of inflammatory peptides [156]. Additionally, it is considered as a stem cell regulator in breast, [157,158], lung [159], bone marrow [159] and adipose tissues [160]. The role of CD10 in tumor growth is still quite controversial. CD10 can be a good prognostic marker with various carcinomas such as cervical and non-small cell lung carcinomas [161,162] but on the other hand indicates poor prognosis with solid tumors such as gastric, pancreatic, and colorectal tumors where it is associated with disease progression and metastatic potential. Ikenaga et al. [163] reported that CD10+ pancreatic stellate cells promoted the invasiveness of pancreatic cancer cells in vitro. In particular, they suggested that CD10+ pancreatic stellate cells promoted invasiveness of tumor cells by secretion of MMP-3, and thus, ECM degradation. Moreover, increased expression of CD10 in tumor and stromal cells of bladder carcinoma is strongly correlated with tumor progression, invasion and metastasis in human bladder cancer [164,165]. Additionally, in invasive duct breast cancer, CD10 expression by stromal cells was positively correlated with a large tumor size, high tumor grade, presence of lymph node metastasis and low overall survival [166,167]. The molecular mechanisms underlying the role of CD10 in cancer invasion remain largely unclear. Nevertheless, very recently, Lee et al. associated CD10 over-expression in esophageal squamous cell carcinoma (ESCC) cells with activity of the transcriptional factor Twist 1. It had been already shown that Twist1 induces EMT in esophageal squamous cell carcinoma (ESCC) cell lines by up-regulating several genes [168]. Lee et al. showed that CD10 over-expression in ESCC cells is directly induced by Twist1, which binds to a specific site on the CD10 gene [169] (Figure 3B).
7. Chaperones
7.1. eHSP90
HSP90 is considered one of the most abundant cytoplasmic chaperones in unstressed normal cells, where it performs housekeeping functions, controlling the stability, activity, intracellular disposition and proteolytic turnover of a variety of client proteins. Moreover, HSP90 interacts with a great number of molecules that are involved in the development and/or survival of cancer cells, allowing mutant proteins to retain or gain function. Additionally, HSP90 allows tumor cells to tolerate genetic alterations, including mutations of critical signaling molecules that would otherwise be lethal. It actually functions as a biochemical buffer for the genetic instability found in cancers by stabilizing and permitting the accumulation of mutant proteins. As a result of this buffering capacity, phenotypic diversity within the tumor population increases and the evolution of invasive metastatic and drug resistant phenotype accelerates while permitting cancer cells to tolerate the imbalanced signaling that such oncoproteins create. HSP90 has also been identified in the extracellular milieu and has been shown to chaperone a finite number of extracellular proteins involved in cell migration and invasion [125].
While the existence of the cytoplasmic pool of HSP90 has been demonstrated and studied for the past three decades, it was not until 2004 that Sidera et al. identified an extracellular pool of HSP90 on the cell surface of developing neuronal cells that plays a critical role in cell motility. Through the development and use of a novel cell-impermeable, HSP90 function-blocking monoclonal antibody, namely mAb 4C5, they showed that eHSP90 is necessary for cell migration and is associated with actin-reorganization of migrating cells [170]. In the same year Eustace et al. revealed the presence of HSP90 on the cell surface and in the conditioned media of fibrosarcoma cells and associated the increased tumor invasiveness of these cells in vitro with the activation of MMP-2 by eHSP90 (Figure 3A) [141]. Furthermore, Becker et al. [171] showed the presence of eHSP90 on the surface of melanoma cells and correlated its over-expression with melanoma malignancy. In this context, Stellas et al. showed that anti-HSP90 mAb 4C5 inhibited cell invasion and metastasis in melanoma cells [172]. Additionally, an involvement of eHSP90 in the re-organization of the actin cytoskeleton and cancer cell invasion in prostate and bladder cancer was reported by Tsutsumi et al. [173], whilst Yang et al. demonstrated that hyperacetylation of eHSP90 promoted its extracellular location and caused increased breast cancer cell invasion [174]. Participation of eHSP90 in breast cancer cell invasion was also shown by Sidera et al. (2008) who revealed interaction of eHSP90 with the extracellular domain of HER-2 during the invasion process [125] (Figure 2B). It has been shown that several hostile environmental conditions such as serum starvation, hypoxia and oxidative stress trigger the extracellular localization of HSP90. In particular, according to Li et al., hypoxic conditions trigger the accumulation of HIF-1, with the consequent secretion of HSP90 and the involvement of the latter in enhanced skin cell migration and wound healing. Hypoxia is a typical condition both in wound healing and in the tumor invasion processes. And, whilst normal cells release eHSP90 in response to tissue injury, this chaperone is constitutively secreted by tumors and promotes metastatic phenomena [175]. Finally, and given the previously mentioned capacity of CSC to survive under severe hypoxia conditions and nutrient shortage as well as to give rise to secondary tumors, our preliminary data suggest that eHSP90 is over-expressed on CSC [176], indicating a further involvement of this protein in cancer cell invasion and metastasis. Overall, increasing evidence indicates that eHSP90, either secreted in the ECM or loosely attached on the cell surface, acts as a chaperone for the activation of proteins involved in the processes of cancer cell invasion and metastasis and therefore might be of critical importance for the these processes.
7.2. eCdc37
Cdc37 was initially identified as part of a protein complex involving HSP90 and the Rous sarcoma virus-encoded oncogene pp60v-src. It interacts with various oncogenic protein kinases such as Raf1 and CDK4, the oncogenic mutant epidermal growth factor receptor tyrosine kinase EGFRvIII and the Peuth-Jeghers cancer syndrome-associated kinase [177]. Intracellularly, Cdc37 acts as a crucial co-chaperone in the HSP90 chaperone machinery, playing a decisive role in the maturation and/or stabilization of a large subset of protein kinases, implicated in signal transduction, proliferation and survival [178]. Cdc37 acts as an adaptor or scaffold, facilitating client kinase interaction with HSP90 [179] and subsequently by recruiting these client kinases into the HSP90 complex, it stabilizes and/or maintains them in a folding-competent conformation [180]. Many client proteins interact directly with both Cdc37 and HSP90 and their folding, maturation and stability depend on the activity of both chaperones. Vice versa, the complex relationship between Cdc37 and HSP90 is illustrated by the finding that their interaction is stabilized by the client protein [181]. In 2012, El Hamidieh et al. identified a cell surface pool of Cdc37 (eCdc37) that participated in breast cancer cell invasion. More specifically the authors showed that eCdc37 is localized on the surface of MDA-MB-453 and MDA-MB-231 breast cancer cells, where it is necessary for the motility of these cells and similarly to its intracellular counterpart it specifically interacts with eHSP90. Moreover, immunoprecipitation experiments using MDA-MB-453 and MDA-MB-231 cell lysates showed that eCdc37 also interacts with HER-2 and EGFR respectively. Thus, the authors concluded that eCdc37 possibly acts in a similar way to its intracellular counterpart, by functioning as a co-chaperone molecule for eHSP90 [182] (Figure 2B,C).
8. LRP-1
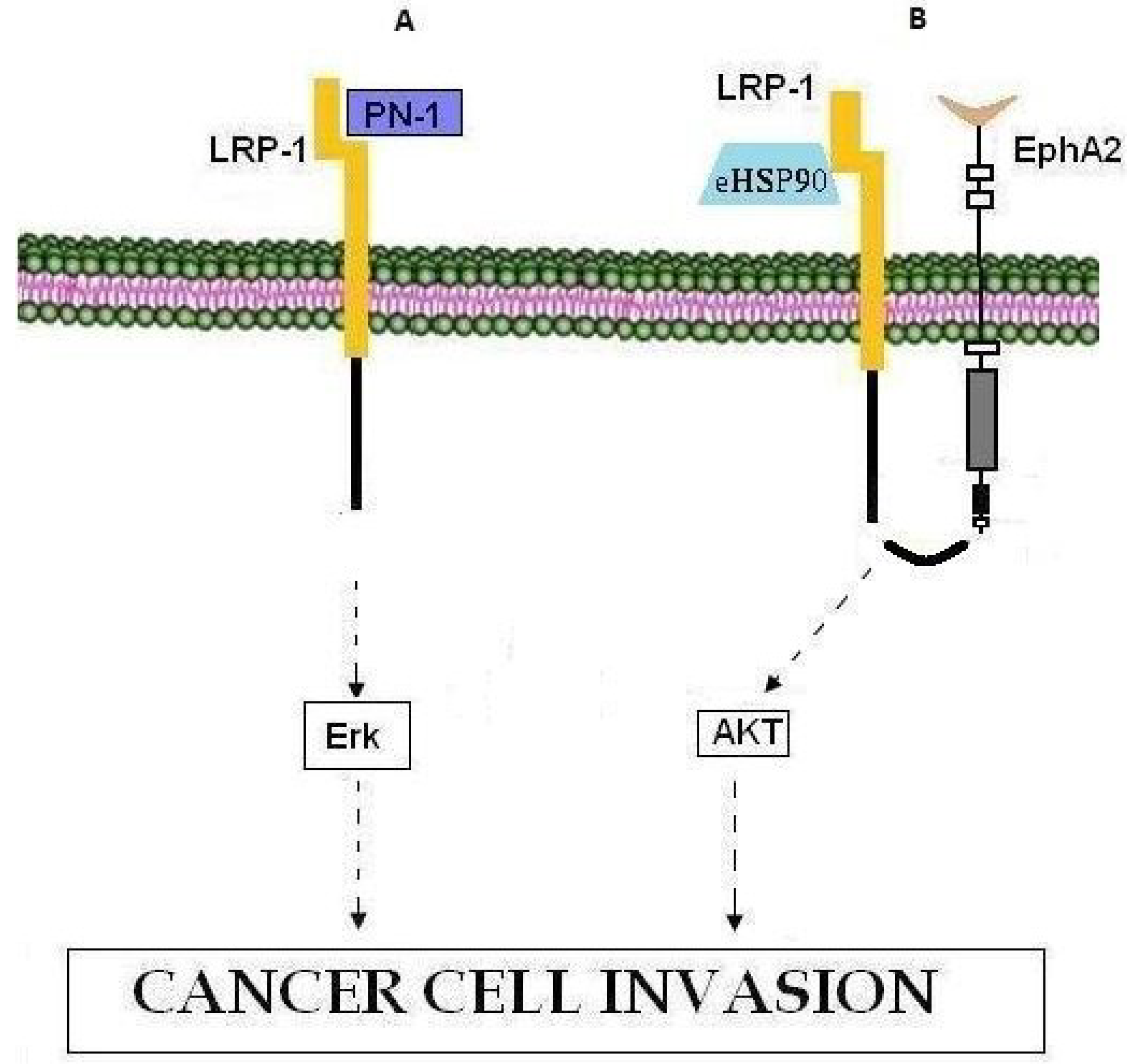
LDL receptor-related protein-1 (LRP-1, also called CD91) is a member of the low-density lipoprotein (LDL) receptor superfamily, which includes at least 11 structurally related members. LRP-1 is a multifunctional molecule as it binds more than 40 ligands. Previous reports [175,183,184,185,186] have shown that nexin-1 (PN-1) binds LRP-1 and thus activates the ERK signaling pathway, controlling MMP-9 expression and inducing metastatic spread (Figure 4A) [183]. In this context, according to Montel et al. [187] silencing of LRP-1 in breast cancer has a negative effect on metastatic spread rather than on primary tumor growth. Moreover, it has been established that eHSP90 binds the extracellular sub-domain II of LRP-1 and activates downstream AKT1 and AKT2 kinase signaling pathways through the cytoplasmic NPVY motif of LRP-1, thus promoting cell motility and being essential for wound healing as well as tumor invasion [186]. In this context, during glioblastoma cell invasion, eHSP90-LRP1-mediated AKT signaling, necessitates the interaction of LRP-1 with pro-motility receptor tyrosine kinase EphA2 whose over-expression is common in cancers and is associated with oncogenic activity, cell invasiveness, metastatic potential and poor prognosis [188,189] (Figure 4B). Moreover, LRP-1 has been proposed to mediate the invasive properties of breast and thyroid cancer cells [184,187,190]. All in all, given its role in cancer cell invasiveness, LRP-1 represents a therapeutic target against metastasis whose inhibition will most probably constitute a selective pharmacological approach.
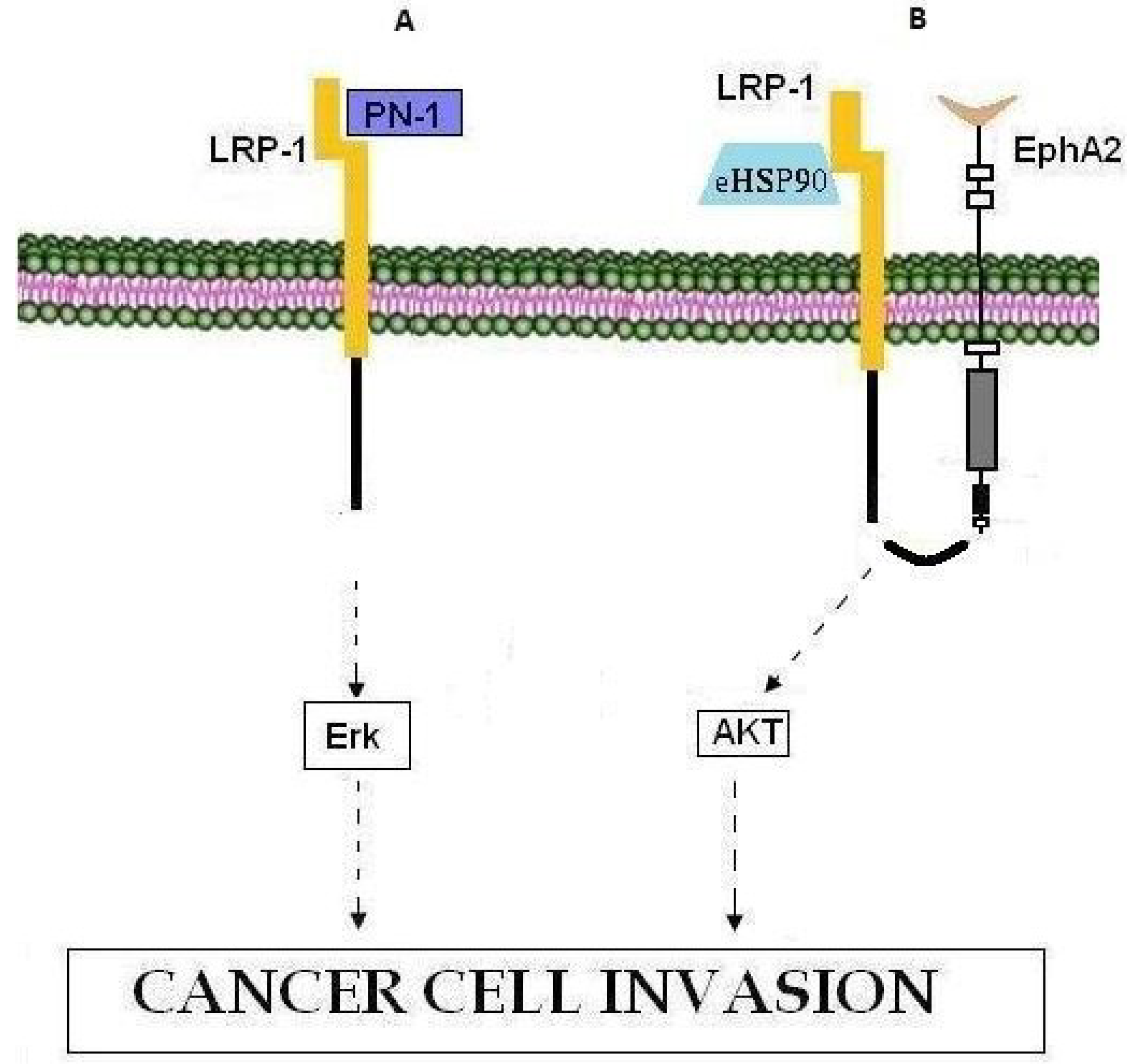
Figure 4.
LRP1 involvement in cancer cell invasion. (A) PN-1 binding to LRP-1 leads to activation of the Erk signaling pathway and cell invasion. (B) The eHSP90-LRP1-EphA2 complex promotes tumor invasion through activation of the AKT signaling pathway.
Figure 4.
LRP1 involvement in cancer cell invasion. (A) PN-1 binding to LRP-1 leads to activation of the Erk signaling pathway and cell invasion. (B) The eHSP90-LRP1-EphA2 complex promotes tumor invasion through activation of the AKT signaling pathway.
9. Conclusions
Cancer cell invasion represents the core of the complex phenomenon of metastasis and is the result of a dense and multifaceted network of molecular interactions taking place inside the cancer cell or within the cancer cell microenvironment. In the present review we have focused on the contribution of extracellular molecules in cancer cell invasion processes. Major components of the extracellular milieu include ECM proteins and their receptors, growth factors and their receptors as well as metalloproteinases. More recently, increasing evidence shows the participation of extracellular chaperone molecules such as eHSP90 and eCdc37 in cancer cell invasion. These chaperone proteins act either after secretion or as cell surface molecules loosely tethered to the cell membrane. All the above mentioned molecules participate in various aspects of the metastatic cascade such as ECM degradation, the stabilization and activation of extracellular oncogenic proteins, neo-vascularization and in cell-cell and cell-matrix interactions. In fact, they constitute the complex extracellular network of molecules that, in concert with their downstream intracellular signaling cascades, promote cancer cell invasion. Thus, one of the major tasks of oncology research is the development of inhibitors selectively targeting extracellular molecules that constitute a crucial inter-connection of multiple metastatic pathways.
Acknowledgments
The authors would like to thank Georgia Vartzi for her valuable assistance in illustration of the figures and Lesley Probert for reviewing and editing the manuscript. This research has been co-financed by the European Union (European Social Fund—ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)—Research Funding Program: ARCHIMEDES III. Investing in knowledge society through the European Social Fund.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.; Fodde, R. Cancer stemness and metastasis: Therapeutic consequences and perspectives. Eur J. Cancer 2010, 46, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.T.; Ye, Y.P.; Deng, Y.J.; Bian, X.W.; Ding, Y.Q. Metastatic cancer stem cells: From the concept to therapeutics. Am. J. Stem. Cells 2014, 3, 46–62. [Google Scholar] [PubMed]
- Spano, D.; Heck, C.; de Antonellis, P.; Christofori, G.; Zollo, M. Molecular networks that regulate cancer metastasis. Semin. Cancer Biol. 2012, 22, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, M.; Dello Sbarba, P. Environmental restrictions within tumor ecosystems select for a convergent, hypoxia-resistant phenotype of cancer stem cells. Cell Cycle 2008, 7, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Fabian, A.; Barok, M.; Vereb, G.; Szollosi, J. Die hard: Are cancer stem cells the bruce willises of tumor biology? Cytometry A 2009, 75, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tiede, B.; Massague, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Brivanlou, A.H. Signaling pathways in cancer and embryonic stem cells. Stem. Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Jaracz, S.; Chen, J.; Kuznetsova, L.V.; Ojima, I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M.; Yip, G.W. Heparanase, hyaluronan, and CD44 in cancers: A breast carcinoma perspective. Cancer Res. 2006, 66, 10233–10237. [Google Scholar] [CrossRef] [PubMed]
- Udabage, L.; Brownlee, G.R.; Nilsson, S.K.; Brown, T.J. The over-expression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp. Cell Res. 2005, 310, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Gould, V.E.; Koukoulis, G.K.; Virtanen, I. Extracellular matrix proteins and their receptors in the normal, hyperplastic and neoplastic breast. Cell Differ. Dev. 1990, 32, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Zhou, T.; Fang, S.; Jia, M.; Xu, Z.; Dai, Z.; Li, C.; Li, S.; Li, L.; Zhang, T.; et al. Pigment epithelium-derived factor (PEDF) inhibits breast cancer metastasis by down-regulating fibronectin. Breast Cancer Res. Treat. 2014, 148, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.C.; O’Hagan, K.L.; Kenyon, A.; Dhanani, K.C.; Prinsloo, E.; Edkins, A.L. Hsp90 binds directly to fibronectin (FN) and inhibition reduces the extracellular fibronectin matrix in breast cancer cells. PLOS ONE 2014, 9, e86842. [Google Scholar] [CrossRef] [PubMed]
- Ioachim, E.; Charchanti, A.; Briasoulis, E.; Karavasilis, V.; Tsanou, H.; Arvanitis, D.L.; Agnantis, N.J.; Pavlidis, N. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: Their prognostic value and role in tumour invasion and progression. Eur. J. Cancer 2002, 38, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Schwarzbauer, J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.E.; Foty, R.A.; Corbett, S.A. Fibronectin matrix assembly regulates α5β1-mediated cell cohesion. Mol. Biol. Cell 2004, 15, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicka-Patynowski, I.; Schwarzbauer, J.E. The ins and outs of fibronectin matrix assembly. J. Cell Sci. 2003, 116, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Kim, A.; Kim, M.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum. Pathol. 2013, 44, 2028–2037. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Banerji, A.; Frei, E.; Chatterjee, A. Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci. 2008, 82, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Maity, G.; Choudhury, P.R.; Sen, T.; Ganguly, K.K.; Sil, H.; Chatterjee, A. Culture of human breast cancer cell line (MDA-MB-231) on fibronectin-coated surface induces pro-matrix metalloproteinase-9 expression and activity. Tumour Biol. 2011, 32, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.W.; Jain, A.; Tayback, M.; Fedarko, N.S. Small integrin binding ligand n-linked glycoprotein gene family expression in different cancers. Clin. Cancer Res. 2004, 10, 8501–8511. [Google Scholar] [CrossRef] [PubMed]
- Wai, P.Y.; Kuo, P.C. Osteopontin: Regulation in tumor metastasis. Cancer Metastasis Rev. 2008, 27, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Bellahcene, A.; Castronovo, V. Increased expression of osteonectin and osteopontin, two bone matrix proteins, in human breast cancer. Am. J. Pathol. 1995, 146, 95–100. [Google Scholar] [PubMed]
- Brown, L.F.; Papadopoulos-Sergiou, A.; Berse, B.; Manseau, E.J.; Tognazzi, K.; Perruzzi, C.A.; Dvorak, H.F.; Senger, D.R. Osteopontin expression and distribution in human carcinomas. Am. J. Pathol. 1994, 145, 610–623. [Google Scholar] [PubMed]
- Casson, A.G.; Wilson, S.M.; McCart, J.A.; O’Malley, F.P.; Ozcelik, H.; Tsao, M.S.; Chambers, A.F. Ras mutation and expression of the ras-regulated genes osteopontin and cathepsin l in human esophageal cancer. Int. J. Cancer 1997, 72, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, M.; Sakamoto, M.; Kanetaka, K.; Chuuma, M.; Hirohashi, S. Overexpression of osteopontin in hepatocellular carcinoma. Pathol. Int. 2002, 52, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Ito, A.; Nagoshi, J.; Takeda, M.; Kurata, A.; Takatsuka, Y.; Kohri, K.; Nomura, S.; Kitamura, Y. Expression of bone matrix protein messenger ribonucleic acids in human breast cancers. Possible involvement of osteopontin in development of calcifying foci. Lab. Invest. 1995, 72, 64–69. [Google Scholar] [PubMed]
- Senger, D.R.; Perruzzi, C.A.; Gracey, C.F.; Papadopoulos, A.; Tenen, D.G. Secreted phosphoproteins associated with neoplastic transformation: Close homology with plasma proteins cleaved during blood coagulation. Cancer Res. 1988, 48, 5770–5774. [Google Scholar] [PubMed]
- Singhal, H.; Bautista, D.S.; Tonkin, K.S.; O’Malley, F.P.; Tuck, A.B.; Chambers, A.F.; Harris, J.F. Elevated plasma osteopontin in metastatic breast cancer associated with increased tumor burden and decreased survival. Clin. Cancer Res. 1997, 3, 605–611. [Google Scholar] [PubMed]
- Tuck, A.B.; O’Malley, F.P.; Singhal, H.; Harris, J.F.; Tonkin, K.S.; Kerkvliet, N.; Saad, Z.; Doig, G.S.; Chambers, A.F. Osteopontin expression in a group of lymph node negative breast cancer patients. Int. J. Cancer 1998, 79, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Bramwell, V.H.; Doig, G.S.; Tuck, A.B.; Wilson, S.M.; Tonkin, K.S.; Tomiak, A.; Perera, F.; Vandenberg, T.A.; Chambers, A.F. Serial plasma osteopontin levels have prognostic value in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 3337–3343. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Kim, H.J.; Chang, J.; Ahn, C.M.; Kim, S.K. Elevated circulating level of osteopontin is associated with advanced disease state of non-small cell lung cancer. Lung Cancer 2007, 57, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, A.; Festuccia, C.; D’Andrea, G.; Teti, A.; Bologna, M. Osteopontin modulates prostate carcinoma invasive capacity through rgd-dependent upregulation of plasminogen activators. Biol Chem. 2002, 383, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Furger, K.A.; Allan, A.L.; Wilson, S.M.; Hota, C.; Vantyghem, S.A.; Postenka, C.O.; Al-Katib, W.; Chambers, A.F.; Tuck, A.B. Beta(3) integrin expression increases breast carcinoma cell responsiveness to the malignancy-enhancing effects of osteopontin. Mol. Cancer Res. 2003, 1, 810–819. [Google Scholar] [PubMed]
- Senger, D.R.; Perruzzi, C.A. Cell migration promoted by a potent grgds-containing thrombin-cleavage fragment of osteopontin. Biochim. Biophys. Acta 1996, 1314, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wei, Y.Y.; Chen, H.T.; Fong, Y.C.; Hsu, C.J.; Tsai, C.H.; Hsu, H.C.; Liu, S.H.; Tang, C.H. Osteopontin increases migration and MMP-9 up-regulation via alphavbeta3 integrin, FAK, ERK, and NF-kappaB-dependent pathway in human chondrosarcoma cells. J. Cell Physiol. 2009, 221, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Bayless, K.J.; Salazar, R.; Davis, G.E. RGD-dependent vacuolation and lumen formation observed during endothelial cell morphogenesis in three-dimensional fibrin matrices involves the αvβ3 and α5β1 integrins. Am. J. Pathol. 2000, 156, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Montgomery, A.M.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Scatena, M.; Almeida, M.; Chaisson, M.L.; Fausto, N.; Nicosia, R.F.; Giachelli, C.M. NF-kappaB mediates αvβ3 integrin-induced endothelial cell survival. J. Cell Biol. 1998, 141, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Ledbetter, S.R.; Claffey, K.P.; Papadopoulos-Sergiou, A.; Peruzzi, C.A.; Detmar, M. Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the αvβ3 integrin, osteopontin, and thrombin. Am. J. Pathol. 1996, 149, 293–305. [Google Scholar] [PubMed]
- Asosingh, K.; Gunthert, U.; Bakkus, M.H.; de Raeve, H.; Goes, E.; van Riet, I.; van Camp, B.; Vanderkerken, K. In vivo induction of insulin-like growth factor-I receptor and CD44V6 confers homing and adhesion to murine multiple myeloma cells. Cancer Res. 2000, 60, 3096–3104. [Google Scholar] [PubMed]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef]
- Gunthert, U.; Hofmann, M.; Rudy, W.; Reber, S.; Zoller, M.; Haussmann, I.; Matzku, S.; Wenzel, A.; Ponta, H.; Herrlich, P. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 1991, 65, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Rudzki, Z.; Jothy, S. CD44 and the adhesion of neoplastic cells. Mol. Pathol. 1997, 50, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Takahashi, F.; Hirama, M.; Tanabe, K.K.; Fukuchi, Y. Restoration of CD44S in non-small cell lung cancer cells enhanced their susceptibility to the macrophage cytotoxicity. Lung Cancer 2003, 41, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Strokovich, K.; Springer, T.A.; Walz, T. Structure of integrin alpha5beta1 in complex with fibronectin. Embo J. 2003, 22, 4607–4615. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Berrier, A.L.; Yamada, K.M. Cell-matrix adhesion. J. Cell Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Keller, E.T.; Brown, J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J. Cell Biochem. 2004, 91, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adh. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Hosotani, R.; Kawaguchi, M.; Masui, T.; Koshiba, T.; Ida, J.; Fujimoto, K.; Wada, M.; Doi, R.; Imamura, M. Expression of integrin alphavbeta3 in pancreatic carcinoma: Relation to MMP-2 activation and lymph node metastasis. Pancreas 2002, 25, e30–e35. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The relationship between bone metastasis from human breast cancer and integrin alpha(v)beta3 expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar] [PubMed]
- Ricono, J.M.; Huang, M.; Barnes, L.A.; Lau, S.K.; Weis, S.M.; Schlaepfer, D.D.; Hanks, S.K.; Cheresh, D.A. Specific cross-talk between epidermal growth factor receptor and integrin alphavbeta5 promotes carcinoma cell invasion and metastasis. Cancer Res. 2009, 69, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Berner, H.S.; Suo, Z.; Risberg, B.; Villman, K.; Karlsson, M.G.; Nesland, J.M. Clinicopathological associations of CD44 mrna and protein expression in primary breast carcinomas. Histopathology 2003, 42, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.K.; Zhou, X.; Wright, E.T.; Cristofanilli, M.; Smith, T.; Yang, Y.; Sneige, N.; Sahin, A.; Gilcrease, M.Z. CD44 expression is associated with increased survival in node-negative invasive breast carcinoma. Clin. Cancer Res. 2005, 11, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- Rys, J.; Kruczak, A.; Lackowska, B.; Jaszcz-Gruchala, A.; Brandys, A.; Stelmach, A.; Reinfuss, M. The role of CD44V3 expression in female breast carcinomas. Pol. J. Pathol. 2003, 54, 243–247. [Google Scholar] [PubMed]
- Hiscox, S.; Baruha, B.; Smith, C.; Bellerby, R.; Goddard, L.; Jordan, N.; Poghosyan, Z.; Nicholson, R.I.; Barrett-Lee, P.; Gee, J. Overexpression of CD44 accompanies acquired tamoxifen resistance in MCF7 cells and augments their sensitivity to the stromal factors, heregulin and hyaluronan. BMC Cancer 2012. [Google Scholar] [CrossRef]
- Bapat, S.A. Human ovarian cancer stem cells. Reproduction 2010, 140, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Wang, H.; He, L.; Zhang, J.; Ni, B.; Wang, X.; Jin, H.; Cahuzac, N.; Mehrpour, M.; Lu, Y.; Chen, Q. CD44 is of functional importance for colorectal cancer stem cells. Clin. Cancer Res. 2008, 14, 6751–6760. [Google Scholar] [CrossRef] [PubMed]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as cancer stem cell markers: An enduring ambiguity. Clin. Dev. Immunol. 2012. [Google Scholar] [CrossRef]
- Wang, R.; Lv, Q.; Meng, W.; Tan, Q.; Zhang, S.; Mo, X.; Yang, X. Comparison of mammosphere formation from breast cancer cell lines and primary breast tumors. J. Thorac. Dis. 2014, 6, 829–837. [Google Scholar] [PubMed]
- Buck, M.B.; Knabbe, C. TGF-beta signaling in breast cancer. Ann. N. Y. Acad. Sci. 2006, 1089, 119–126. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Vogelmann, R.; Nguyen-Tat, M.D.; Giehl, K.; Adler, G.; Wedlich, D.; Menke, A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and pten. J. Cell Sci. 2005, 118, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B. Molecular and cell biology of TGF-beta. Miner. Electrolyte MeTable. 1998, 24, 111–119. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of Cancer. Proc. Jpn. Acad. B Phys. Biol. Sci. 2009, 85, 314–323. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- ten Dijke, P.; Goumans, M.J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008, 11, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; ten Dijke, P. TGF-beta signaling in breast cancer cell invasion and bone metastasis. J. Mammary Gland Biol. Neoplasia 2011, 16, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J. Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr. Opin. Genet. Dev. 2011, 21, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-beta: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014. [Google Scholar] [CrossRef]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-beta paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Mysteries of TGF-beta paradox in benign and malignant cells. Front. Oncol. 2014. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of TGF-beta signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar] [PubMed]
- Garratt, A.N. “To erb-b or not to erb-b...” Neuregulin-1/erbb signaling in heart development and function. J. Mol. Cell Cardiol. 2006, 41, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Riese, D.J., 2nd; Gilbert, W.; Stern, D.F.; McMahan, U.J. Ligands for erbb-family receptors encoded by a neuregulin-like gene. Nature 1997, 387, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Harari, D.; Tzahar, E.; Romano, J.; Shelly, M.; Pierce, J.H.; Andrews, G.C.; Yarden, Y. Neuregulin-4: A novel growth factor that acts through the ErbB-4 receptor tyrosine kinase. Oncogene 1999, 18, 2681–2689. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.E.; Sliwkowski, M.X.; Akita, R.W.; Henzel, W.J.; Lee, J.; Park, J.W.; Yansura, D.; Abadi, N.; Raab, H.; Lewis, G.D.; et al. Identification of heregulin, a specific activator of p185erbb2. Science 1992, 256, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L., 3rd; Sliwkowski, M.X.; Akita, R.; Platko, J.V.; Guy, P.M.; Nuijens, A.; Diamonti, A.J.; Vandlen, R.L.; Cantley, L.C.; Cerione, R.A. The ErbB3 gene product is a receptor for heregulin. J. Biol. Chem. 1994, 269, 14303–14306. [Google Scholar] [PubMed]
- Stove, C.; Bracke, M. Roles for neuregulins in human Cancer. Clin. Exp. Metastasis. 2004, 21, 665–684. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.M.; Pankonin, M.S.; Loeb, J.A. Neuregulins: Versatile growth and differentiation factors in nervous system development and human disease. Brain Res. Rev. 2006, 51, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Falls, D.L. Neuregulins and the neuromuscular system: 10 years of answers and questions. J. Neurocytol. 2003, 32, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cleary, S.; Mandarano, M.A.; Long, W.; Birchmeier, C.; Jones, F.E. The breast proto-oncogene, hrgalpha regulates epithelial proliferation and lobuloalveolar development in the mouse mammary gland. Oncogene 2002, 21, 4900–4907. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Mehmi, I.; Lupu, R. Trastuzumab in combination with heregulin-activated Her-2 (ErbB-2) triggers a receptor-enhanced chemosensitivity effect in the absence of Her-2 overexpression. J. Clin. Oncol. 2006, 24, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Raj, E.H.; Skinner, A.; Mahji, U.; Nirmala, K.N.; Ravichandran, K.; Shanta, V.; Hurst, H.C.; Gullick, W.J.; Rajkumar, T. Neuregulin 1-alpha expression in locally advanced breast cancer. Breast (Edinburgh, Scotland) 2001, 10, 41–45. [Google Scholar] [CrossRef]
- Cheng, L.; Zha, Z.; Lang, B.; Liu, J.; Yao, X. Heregulin-beta1 promotes metastasis of breast cancer cell line SKBR3 through upregulation of snail and induction of epithelial-mesenchymal transition. Cancer Lett. 2009, 280, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jeong, H.; Lee, Y.; Kim, C.; Kim, H.; Kim, A. HRG-beta1-driven ErbB3 signaling induces epithelial-mesenchymal transition in breast cancer cells. BMC Cancer 2013, 13, 383. [Google Scholar] [CrossRef] [PubMed]
- Adam, L.; Vadlamudi, R.; Kondapaka, S.B.; Chernoff, J.; Mendelsohn, J.; Kumar, R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J. Biol. Chem. 1998, 273, 28238–28246. [Google Scholar] [CrossRef] [PubMed]
- Talukder, A.H.; Adam, L.; Raz, A.; Kumar, R. Heregulin regulation of autocrine motility factor expression in human tumor cells. Cancer Res. 2000, 60, 474–480. [Google Scholar] [PubMed]
- Cress, W.D.; Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell Physiol. 2000, 184, 1–16. [Google Scholar] [CrossRef]
- Loeb, J.A.; Fischbach, G.D. Aria can be released from extracellular matrix through cleavage of a heparin-binding domain. J. Cell Biol. 1995, 130, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L., 3rd; Weber, J.L.; Unger, M.J.; Ledesma, J.; Yu, N.; Gassmann, M.; Lai, C. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature 1997, 387, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ErbB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.D.; Yarden, Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 2004, 23, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B. Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 2002, 3, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G. ErbB-4: Mechanism of action and biology. Exp. Cell Res. 2003, 284, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Skaria, K.B.; Yarden, Y. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Exp. Cell Res. 2003, 284, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- De Jong, K.P.; Stellema, R.; Karrenbeld, A.; Koudstaal, J.; Gouw, A.S.; Sluiter, W.J.; Peeters, P.M.; Slooff, M.J.; de Vries, E.G. Clinical relevance of transforming growth factor alpha, epidermal growth factor receptor, p53, and ki67 in colorectal liver metastases and corresponding primary tumors. Hepatology 1998, 28, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Douziech, N.; Calvo, E.; Laine, J.; Morisset, J. Activation of map kinases in growth responsive pancreatic cancer cells. Cell Signal. 1999, 11, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J. The epidermal growth factor receptor as a target for cancer therapy. Endocr. Relat. Cancer 2001, 8, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell Biol. 1996, 16, 5276–5287. [Google Scholar] [PubMed]
- Muss, H.B.; Thor, A.D.; Berry, D.A.; Kute, T.; Liu, E.T.; Koerner, F.; Cirrincione, C.T.; Budman, D.R.; Wood, W.C.; Barcos, M.; et al. C-ErbB-2 expression and response to adjuvant therapy in women with node-positive early breast Cancer. N. Engl. J. Med. 1994, 330, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the Her-2/NEU proto-oncogene in human breast and ovarian Cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Mimnaugh, E.G.; Chavany, C.; Neckers, L. Polyubiquitination and proteasomal degradation of the p185c-ErbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J. Biol. Chem. 1996, 271, 22796–22801. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Mimnaugh, E.; Rosser, M.F.; Nicchitta, C.; Marcu, M.; Yarden, Y.; Neckers, L. Sensitivity of mature ErbB2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein hsp90. J. Biol. Chem. 2001, 276, 3702–3708. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Kochupurakkal, B.S.; Yarden, Y. The achilles heel of ErbB-2/Her2: Regulation by the hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle 2004, 3, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Gaitanou, M.; Stellas, D.; Matsas, R.; Patsavoudi, E. A critical role for hsp90 in cancer cell invasion involves interaction with the extracellular domain of Her-2. J. Biol. Chem. 2008, 283, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other erbb receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The ErbB/Her receptor protein-tyrosine kinases and cancer. Biochem. Biophys. Res. Commun. 2004, 319, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D. Regulation of breast cancer metastasis by IGF signaling. J. Mammary Gland Biol. Neoplasia 2008, 13, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Hanahan, D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002, 1, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.; Zhang, X.; Matise, I.; Gaillard-Kelly, M.; Yee, D. The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene 2010, 29, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Zhang, X.; Yoneda, T.; Yee, D. Regulation of breast cancer cell motility by insulin receptor substrate-2 (IRS-2) in metastatic variants of human breast cancer cell lines. Oncogene 2001, 20, 7318–7325. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Faria, T.N.; Stannard, B.; Roberts, C.T., Jr.; LeRoith, D. Essential role of tyrosine residues 1131, 1135, and 1136 of the insulin-like growth factor-I (IGF-I) receptor in IGF-I action. Mol. Endocrinol. 1994, 8, 40–50. [Google Scholar] [PubMed]
- Yoneda, T.; Williams, P.J.; Hiraga, T.; Niewolna, M.; Nishimura, R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res. 2001, 16, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Myoui, A.; Hashimoto, N.; Sasaki, A.; Hata, K.; Morita, Y.; Yoshikawa, H.; Rosen, C.J.; Mundy, G.R.; Yoneda, T. Bone-derived IGF mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res. 2012, 72, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Saldana, S.M.; Lee, H.H.; Lowery, F.J.; Khotskaya, Y.B.; Xia, W.; Zhang, C.; Chang, S.S.; Chou, C.K.; Steeg, P.S.; Yu, D.; et al. Inhibition of type I insulin-like growth factor receptor signaling attenuates the development of breast cancer brain metastasis. PLOS ONE 2013, 8, e73406. [Google Scholar] [CrossRef] [PubMed]
- Min, K.W.; Kim, D.H.; Do, S.I.; Kim, K.; Lee, H.J.; Chae, S.W.; Sohn, J.H.; Pyo, J.S.; Oh, Y.H.; Kim, W.S.; et al. Expression patterns of stromal MMP-2 and tumoural MMP-2 and -9 are significant prognostic factors in invasive ductal carcinoma of the breast. Apmis 2014, 122, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; Patsavoudi, E. Inhibiting matrix metalloproteinases, an old story with new potentials for cancer treatment. Anticancer Agents Med. Chem. 2012, 12, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [PubMed]
- Zuo, J.H.; Zhu, W.; Li, M.Y.; Li, X.H.; Yi, H.; Zeng, G.Q.; Wan, X.X.; He, Q.Y.; Li, J.H.; Qu, J.Q.; et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing emt-like phenotype change and MMP-9-mediated degradation of E-cadherin. J. Cell Biochem. 2011, 112, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- Eustace, B.K.; Jay, D.G. Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 2004, 3, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; El Hamidieh, A.; Patsavoudi, E. Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 2010. [Google Scholar] [CrossRef]
- Fedarko, N.S.; Jain, A.; Karadag, A.; Fisher, L.W. Three small integrin binding ligand n-linked glycoproteins (siblings) bind and activate specific matrix metalloproteinases. FASEB. J. 2004, 18, 734–736. [Google Scholar] [PubMed]
- Murphy, D.A.; Courtneidge, S.A. The “ins” and “outs” of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Jing, J.; Lee, J.; Schedin, P.; Gilbert, S.M.; Peden, A.A.; Junutula, J.R.; Prekeris, R. Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. J. Cell Sci. 2013, 126, 4647–4658. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Rab gtpases: Specifying and deciphering organelle identity and function. Trends Cell Biol. 2001, 11, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef]
- Campbell, N.E.; Kellenberger, L.; Greenaway, J.; Moorehead, R.A.; Linnerth-Petrik, N.M.; Petrik, J. Extracellular matrix proteins and tumor angiogenesis. J. Oncol. 2010. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Zuidwijk, K.; Verspaget, H.W.; de Jonge-Muller, E.S.; van Duijn, W.; Ferreira, V.; Fontijn, R.D.; David, G.; Hommes, D.W.; Lamers, C.B.; et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer 2008, 44, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Stathopoulos, C.; Steffen, A.; Wiedemann, P.; Kohen, L.; Bringmann, A. Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest. Ophthalmol. Vis. Sci. 2007, 48, 4360–4367. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jilani, S.M.; Nikolova, G.V.; Carpizo, D.; Iruela-Arispe, M.L. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J. Cell Biol. 2005, 169, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Malfroy, B.; Kuang, W.J.; Seeburg, P.H.; Mason, A.J.; Schofield, P.R. Molecular cloning and amino acid sequence of human enkephalinase (neutral endopeptidase). FEBS Lett. 1988, 229, 206–210. [Google Scholar] [CrossRef]
- Ishimaru, F.; Mari, B.; Shipp, M.A. The type 2 CD10/neutral endopeptidase 24.11 promoter: Functional characterization and tissue-specific regulation by CBF/NF-Y isoforms. Blood 1997, 89, 4136–4145. [Google Scholar] [PubMed]
- Sumitomo, M.; Shen, R.; Walburg, M.; Dai, J.; Geng, Y.; Navarro, D.; Boileau, G.; Papandreou, C.N.; Giancotti, F.G.; Knudsen, B.; et al. Neutral endopeptidase inhibits prostate cancer cell migration by blocking focal adhesion kinase signaling. J. Clin. Invest. 2000, 106, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Shipp, M.A.; Stefano, G.B.; Switzer, S.N.; Griffin, J.D.; Reinherz, E.L. CD10 (CALLA)/neutral endopeptidase 24.11 modulates inflammatory peptide-induced changes in neutrophil morphology, migration, and adhesion proteins and is itself regulated by neutrophil activation. Blood 1991, 78, 1834–1841. [Google Scholar] [PubMed]
- Bachelard-Cascales, E.; Chapellier, M.; Delay, E.; Pochon, G.; Voeltzel, T.; Puisieux, A.; Caron de Fromentel, C.; Maguer-Satta, V. The CD10 enzyme is a key player to identify and regulate human mammary stem cells. Stem Cells 2010, 28, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Raouf, A.; Emerman, J.T.; Eaves, C.J. Epithelial progenitors in the normal human mammary gland. J. Mammary Gland Biol. Neoplasia 2005, 10, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Sunday, M.E.; Hua, J.; Torday, J.S.; Reyes, B.; Shipp, M.A. CD10/neutral endopeptidase 24.11 in developing human fetal lung. Patterns of expression and modulation of peptide-mediated proliferation. J. Clin. Invest. 1992, 90, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Buhring, H.J.; Battula, V.L.; Treml, S.; Schewe, B.; Kanz, L.; Vogel, W. Novel markers for the prospective isolation of human msc. Ann. N. Y. Acad. Sci. 2007, 1106, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Terauchi, M.; Kajiyama, H.; Shibata, K.; Ino, K.; Mizutani, S.; Kikkawa, F. Anti-progressive effect of neutral endopeptidase 24.11 (NEP/CD10) on cervical carcinoma in vitro and in vivo. Oncology 2005, 69, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Tokuhara, T.; Adachi, M.; Hashida, H.; Ishida, H.; Taki, T.; Higashiyama, M.; Kodama, K.; Tachibana, S.; Sasaki, S.; Miyake, M. Neutral endopeptidase/CD10 and aminopeptidase N/CD13 gene expression as a prognostic factor in non-small cell lung Cancer. Jpn. J. Thorac. Cardiovasc. Surg. 2001, 49, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology 2010, 139, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Maguer-Satta, V.; Besancon, R.; Bachelard-Cascales, E. Concise review: Neutral endopeptidase (CD10): A multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 2011, 29, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Omran, O.M. CD10 and E-cad expression in urinary bladder urothelial and squamous cell carcinoma. J. Environ. Pathol. Toxicol. Oncol. 2012, 31, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, G.Y.; Kim, Y.W.; Park, Y.K.; Song, J.Y.; Lim, S.J. Stromal CD10 expression and relationship to the E-cadherin/beta-catenin complex in breast carcinoma. Histopathology 2010, 56, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Makretsov, N.A.; Hayes, M.; Carter, B.A.; Dabiri, S.; Gilks, C.B.; Huntsman, D.G. Stromal CD10 expression in invasive breast carcinoma correlates with poor prognosis, estrogen receptor negativity, and high grade. Mod. Pathol. 2007, 20, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Kim, J.H.; Han, S.; Sung, C.O.; Do, I.G.; Ko, Y.H.; Um, S.H.; Kim, S.H. Twist1 is an independent prognostic factor of esophageal squamous cell carcinoma and associated with its epithelial-mesenchymal transition. Ann. Surg. Oncol. 2012, 19, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Sung, C.O.; Kim, J.H.; Kang, M.; Yoo, H.Y.; Kim, H.H.; Um, S.H.; Kim, S.H. CD10 expression is enhanced by twist1 and associated with poor prognosis in esophageal squamous cell carcinoma with facilitating tumorigenicity in vitro and in vivo. Int. J. Cancer 2015, 136, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Samiotaki, M.; Yfanti, E.; Panayotou, G.; Patsavoudi, E. Involvement of cell surface hsp90 in cell migration reveals a novel role in the developing nervous system. J. Biol. Chem. 2004, 279, 45379–45388. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Multhoff, G.; Farkas, B.; Wild, P.J.; Landthaler, M.; Stolz, W.; Vogt, T. Induction of hsp90 protein expression in malignant melanomas and melanoma metastases. Exp. Dermatol. 2004, 13, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; Karameris, A.; Patsavoudi, E. Monoclonal antibody 4C5 immunostains human melanomas and inhibits melanoma cell invasion and metastasis. Clin. Cancer Res. 2007, 13, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Beebe, K.; Neckers, L. Impact of heat-shock protein 90 on cancer metastasis. Future Oncol. 2009, 5, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rao, R.; Shen, J.; Tang, Y.; Fiskus, W.; Nechtman, J.; Atadja, P.; Bhalla, K. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008, 68, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sahu, D.; Tsen, F. Secreted heat shock protein-90 (hsp90) in wound healing and Cancer. Biochim. Biophys. Acta 2012, 1823, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Stivarou, T.; Vartzi, G.; Thomaidou, D.; Patsavoudi, E.; Hellenic Pasteur Institute, Athens, Greece. 2015; Personal observations.
- Pearl, L.H. Hsp90 and cdc37—A chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 2005, 15, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Workman, P. Targeting cdc37: An alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle 2009, 8, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M.; Grammatikakis, N.; Cochran, B.H.; Chinkers, M.; Pratt, W.B. P50(cdc37) binds directly to the catalytic domain of raf as well as to a site on hsp90 that is topologically adjacent to the tetratricopeptide repeat binding site. J. Biol. Chem. 1998, 273, 20090–20095. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Rutherford, S.L.; Miyata, Y.; Yahara, I.; Freeman, B.C.; Yue, L.; Morimoto, R.I.; Lindquist, S. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997, 11, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Hartson, S.D.; Irwin, A.D.; Shao, J.; Scroggins, B.T.; Volk, L.; Huang, W.; Matts, R.L. P50(cdc37) is a nonexclusive hsp90 cohort which participates intimately in hsp90-mediated folding of immature kinase molecules. Biochemistry 2000, 39, 7631–7644. [Google Scholar] [CrossRef] [PubMed]
- El Hamidieh, A.; Grammatikakis, N.; Patsavoudi, E. Cell surface cdc37 participates in extracellular hsp90 mediated cancer cell invasion. PLOS ONE 2012, 7, e42722. [Google Scholar] [CrossRef] [PubMed]
- Fayard, B.; Bianchi, F.; Dey, J.; Moreno, E.; Djaffer, S.; Hynes, N.E.; Monard, D. The serine protease inhibitor protease nexin-1 controls mammary cancer metastasis through LRP-1-mediated MMP-9 expression. Cancer Res. 2009, 69, 5690–5698. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Li, Y.; Lee, J.; Schwartz, A.L.; Bu, G. Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res. 2009, 69, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Staudt, N.D.; Jo, M.; Hu, J.; Bristow, J.M.; Pizzo, D.P.; Gaultier, A.; VandenBerg, S.R.; Gonias, S.L. Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res. 2013, 73, 3902–3912. [Google Scholar] [CrossRef] [PubMed]
- Tsen, F.; Bhatia, A.; O’Brien, K.; Cheng, C.F.; Chen, M.; Hay, N.; Stiles, B.; Woodley, D.T.; Li, W. Extracellular heat shock protein 90 signals through subdomain II and the npvy motif of LRP-1 receptor to AKT1 and AKT2: A circuit essential for promoting skin cell migration in vitro and wound healing in vivo. Mol. Cell Biol. 2013, 33, 4947–4959. [Google Scholar] [CrossRef] [PubMed]
- Montel, V.; Gaultier, A.; Lester, R.D.; Campana, W.M.; Gonias, S.L. The low-density lipoprotein receptor-related protein regulates cancer cell survival and metastasis development. Cancer Res. 2007, 67, 9817–9824. [Google Scholar] [CrossRef] [PubMed]
- Gopal, U.; Bohonowych, J.E.; Lema-Tome, C.; Liu, A.; Garrett-Mayer, E.; Wang, B.; Isaacs, J.S. A novel extracellular hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLOS ONE 2011, 6, e17649. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Debinski, W. The EphA2 receptor and ephrina1 ligand in solid tumors: Function and therapeutic targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Dedieu, S.; Langlois, B.; Devy, J.; Sid, B.; Henriet, P.; Sartelet, H.; Bellon, G.; Emonard, H.; Martiny, L. LRP-1 silencing prevents malignant cell invasion despite increased pericellular proteolytic activities. Mol. Cell Biol. 2008, 28, 2980–2995. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stivarou, T.; Patsavoudi, E. Extracellular Molecules Involved in Cancer Cell Invasion. Cancers 2015, 7, 238-265. https://doi.org/10.3390/cancers7010238
AMA Style
Stivarou T, Patsavoudi E. Extracellular Molecules Involved in Cancer Cell Invasion. Cancers. 2015; 7(1):238-265. https://doi.org/10.3390/cancers7010238
Chicago/Turabian StyleStivarou, Theodora, and Evangelia Patsavoudi. 2015. "Extracellular Molecules Involved in Cancer Cell Invasion" Cancers 7, no. 1: 238-265. https://doi.org/10.3390/cancers7010238