HiJAK’d Signaling; the STAT3 Paradox in Senescence and Cancer Progression

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Inflammatory Microenvironment; from Cancer Suppression to Cancer Progression
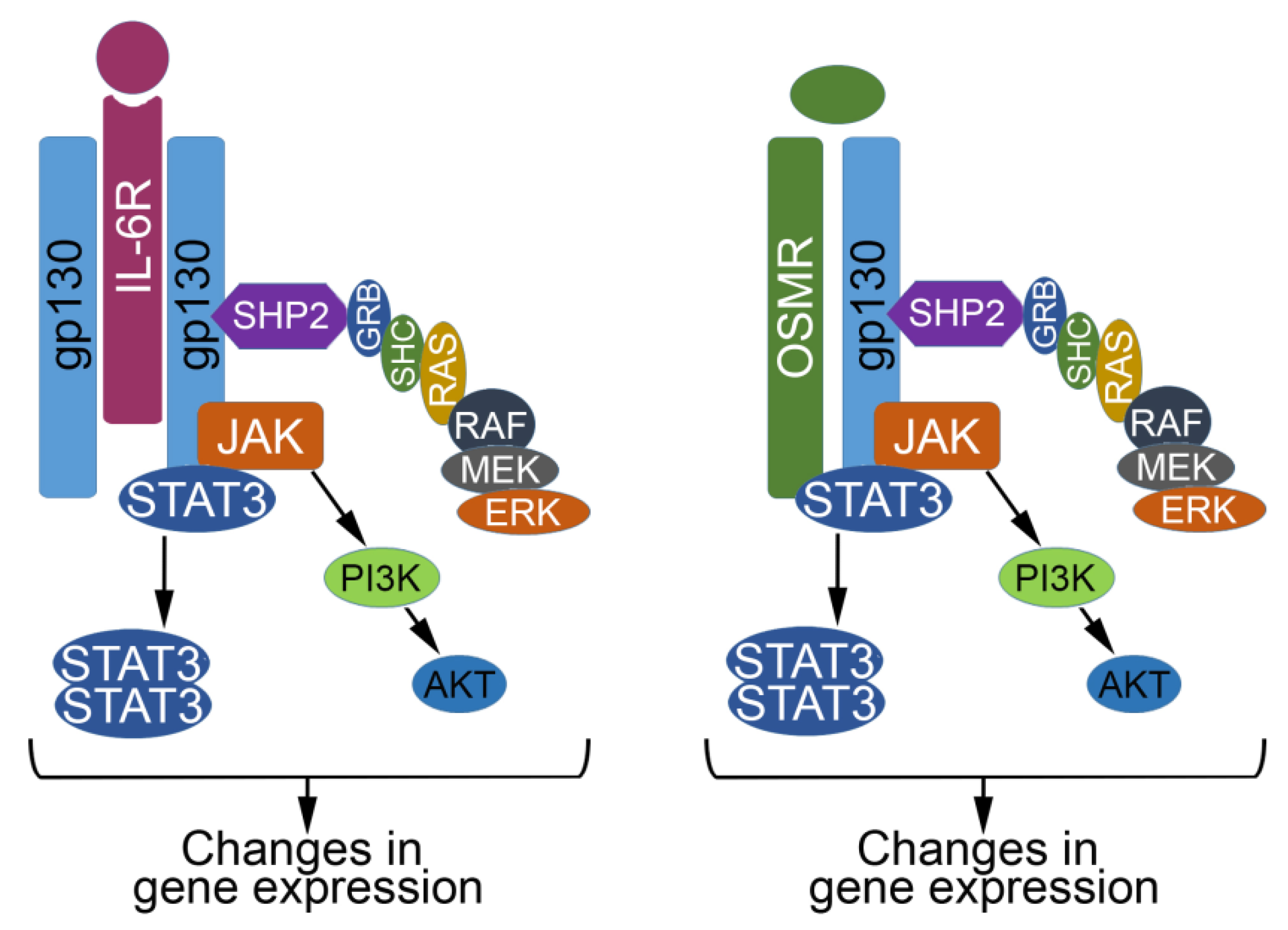
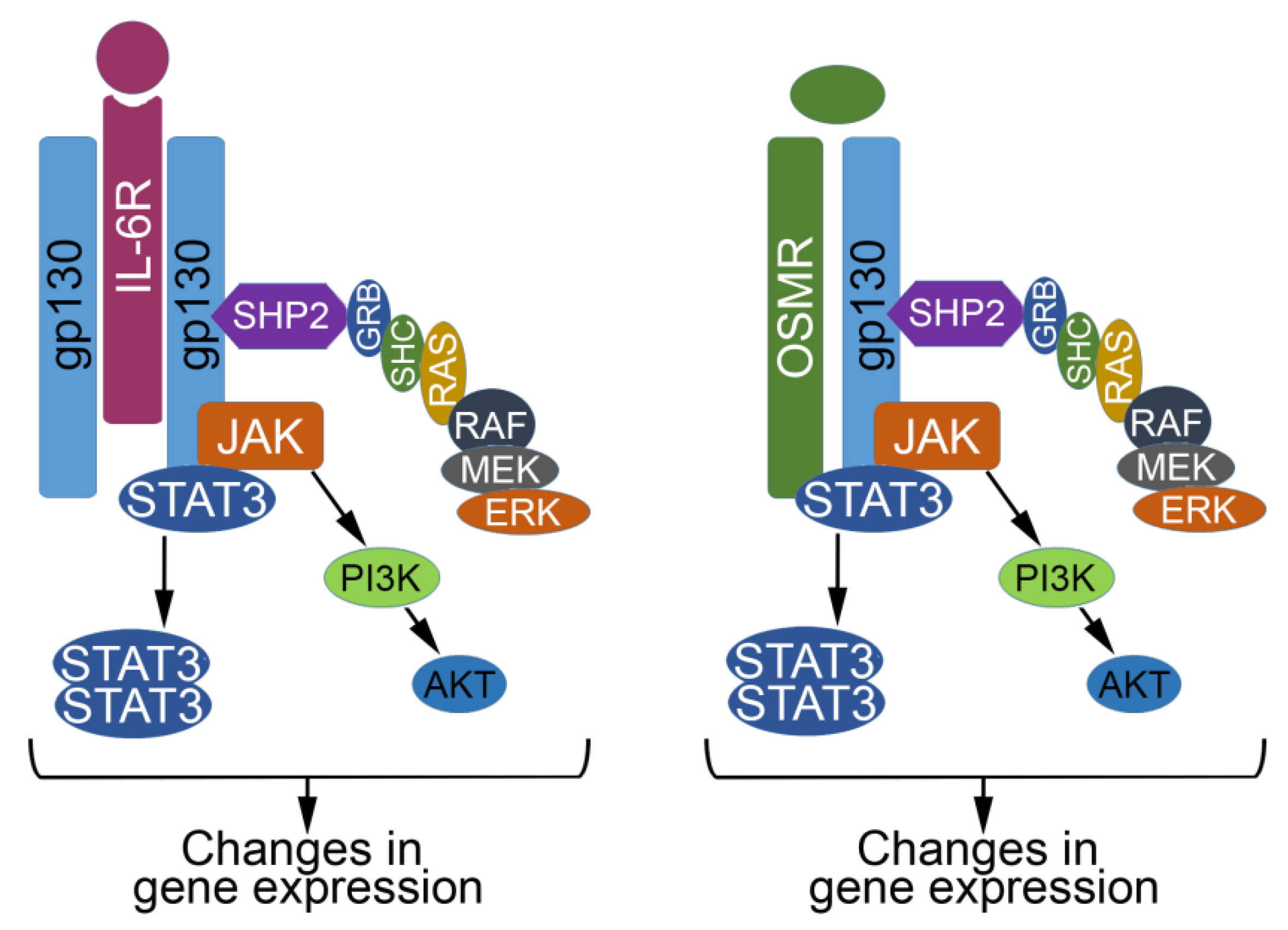
2. The IL6 Family of Cytokines
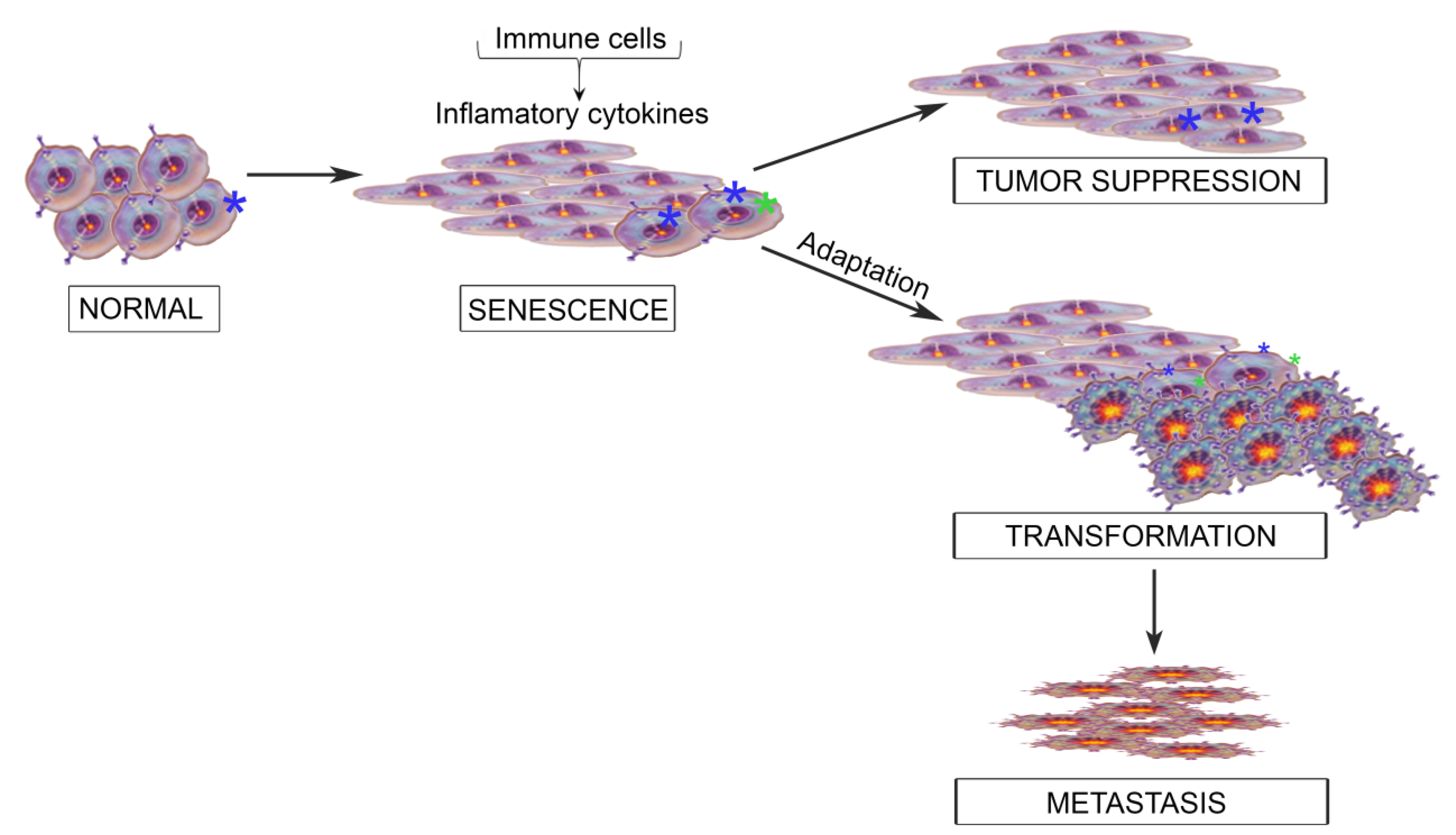
3. When OSM and JAK/STAT Signaling Behave; Acting as a Tumor Suppressor
4. OSM Turns to the Dark Side; Acting as a Tumor Promoter
5. Beyond Transformation: OSM and JAK/STAT3 Signaling in Metastasis and Cancer Stem Cell Phenotypes
6. Integrating the Paradoxical Roles for OSM into a Model of Tumor Progression
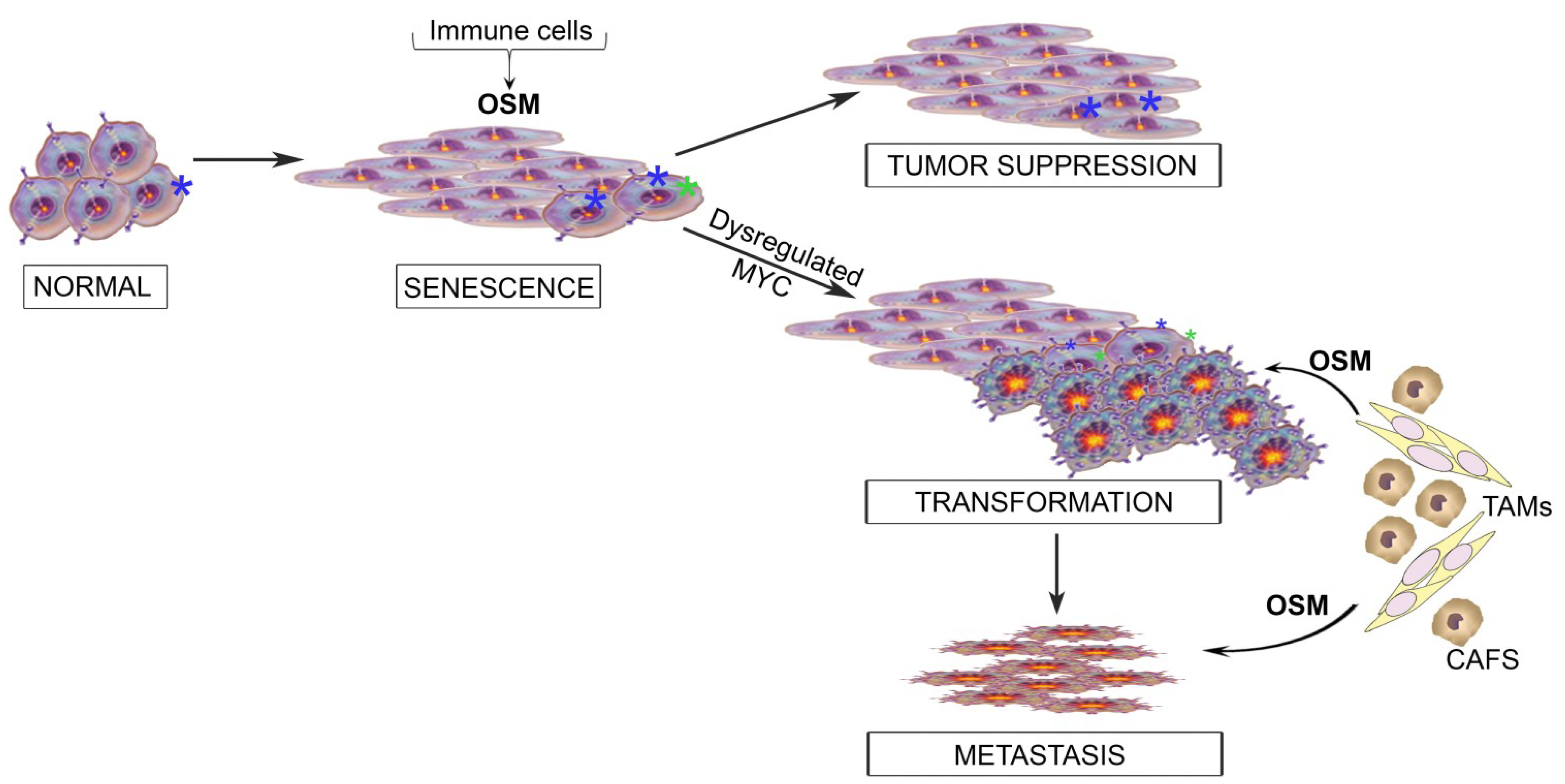
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef]
- Lu, H.; Ouyang, W.; Huang, C. Inflammation, a key event in cancer development. Mol. Cancer Res. 2006, 4, 221–233. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. “Cancer associated fibroblasts”—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Rajaram, M.; Li, J.; Egeblad, M.; Powers, R.S. System-wide analysis reveals a complex network of tumor-fibroblast interactions involved in tumorigenicity. PLoS Genet. 2013, 9, e1003789. [Google Scholar] [CrossRef]
- Kojima, H.; Inoue, T.; Kunimoto, H.; Nakajima, K. Il-6-stat3 signaling and premature senescence. JAKSTAT 2013, 2, e25763. [Google Scholar]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and stat3 shape the cancer microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef]
- Tanaka, M.; Miyajima, A. Oncostatin M, a multifunctional cytokine. Rev. Physiol. Biochem. Pharmacol. 2003, 149, 39–52. [Google Scholar] [CrossRef]
- Silver, J.S.; Hunter, C.A. Gp130 at the nexus of inflammation, autoimmunity, and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of stat3 in mediating the cell growth, differentiation and survival signals relayed through the il-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef]
- Hsieh, F.C.; Cheng, G.; Lin, J. Evaluation of potential stat3-regulated genes in human breast cancer. Biochem. Biophys. Res. Commun. 2005, 335, 292–299. [Google Scholar] [CrossRef]
- Zarling, J.M.; Shoyab, M.; Marquardt, H.; Hanson, M.B.; Lioubin, M.N.; Todaro, G.J. Oncostatin M: A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9739–9743. [Google Scholar] [CrossRef]
- Kan, C.E.; Cipriano, R.; Jackson, M.W. C-myc functions as a molecular switch to alter the response of human mammary epithelial cells to Oncostatin M. Cancer Res. 2011, 71, 6930–6939. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16ink4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Mooi, W.J.; Peeper, D.S. Braf(e600) in benign and malignant human tumours. Oncogene 2008, 27, 877–895. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. Brafe600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Mallette, F.A.; Gaumont-Leclerc, M.F.; Ferbeyre, G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007, 21, 43–48. [Google Scholar] [CrossRef]
- Larsson, L.G. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin. Cancer Biol. 2011, 21, 367–376. [Google Scholar] [CrossRef]
- Reddy, J.P.; Li, Y. Oncogene-induced senescence and its role in tumor suppression. J. Mammary Gland Biol. Neoplasia 2011, 16, 247–256. [Google Scholar] [CrossRef]
- Yaswen, P.; Campisi, J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell 2007, 128, 233–234. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Halazonetis, T.D. Oncogene-induced senescence: The bright and dark side of the response. Curr. Opin. Cell Biol. 2010, 22, 816–827. [Google Scholar] [CrossRef]
- Clark, C.J.; Whang, S.; Paige, K.T. Incidence of precancerous lesions in breast reduction tissue: A pathologic review of 562 consecutive patients. Plast. Reconstr. Surg. 2009, 124, 1033–1039. [Google Scholar] [CrossRef]
- Desantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef]
- Nielsen, M.; Thomsen, J.L.; Primdahl, S.; Dyreborg, U.; Andersen, J.A. Breast cancer and atypia among young and middle-aged women: A study of 110 medicolegal autopsies. Br. J. Cancer 1987, 56, 814–819. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef]
- Garcia-Tunon, I.; Ricote, M.; Ruiz, A.; Fraile, B.; Paniagua, R.; Royuela, M. Osm, lif, its receptors, and its relationship with the malignance in human breast carcinoma (in situ and in infiltrative). Cancer Investig. 2008, 26, 222–229. [Google Scholar] [CrossRef]
- Royuela, M.; Ricote, M.; Parsons, M.S.; Garcia-Tunon, I.; Paniagua, R.; de Miguel, M.P. Immunohistochemical analysis of the il-6 family of cytokines and their receptors in benign, hyperplasic, and malignant human prostate. J. Pathol. 2004, 202, 41–49. [Google Scholar] [CrossRef]
- Savarese, T.M.; Campbell, C.L.; McQuain, C.; Mitchell, K.; Guardiani, R.; Quesenberry, P.J.; Nelson, B.E. Coexpression of Oncostatin M and its receptors and evidence for stat3 activation in human ovarian carcinomas. Cytokine 2002, 17, 324–334. [Google Scholar] [CrossRef]
- Levano, K.S.; Jung, E.H.; Kenny, P.A. Breast cancer subtypes express distinct receptor repertoires for tumor-associated macrophage derived cytokines. Biochem. Biophys. Res. Commun. 2011, 411, 107–110. [Google Scholar] [CrossRef]
- Bolin, C.; Tawara, K.; Sutherland, C.; Redshaw, J.; Aranda, P.; Moselhy, J.; Anderson, R.; Jorcyk, C.L. Oncostatin M promotes mammary tumor metastasis to bone and osteolytic bone degradation. Genes Cancer 2012, 3, 117–130. [Google Scholar] [CrossRef]
- Holzer, R.G.; Ryan, R.E.; Tommack, M.; Schlekeway, E.; Jorcyk, C.L. Oncostatin M stimulates the detachment of a reservoir of invasive mammary carcinoma cells: Role of cyclooxygenase-2. Clin. Exp. Metastasis 2004, 21, 167–176. [Google Scholar] [CrossRef]
- Jorcyk, C.L.; Holzer, R.G.; Ryan, R.E. Oncostatin M induces cell detachment and enhances the metastatic capacity of t-47d human breast carcinoma cells. Cytokine 2006, 33, 323–336. [Google Scholar] [CrossRef]
- Singh, R.A.; Sodhi, A. Cisplatin-treated macrophages produce Oncostatin M: Regulation by serine/threonine and protein tyrosine kinases/phosphatases and Ca2+/calmodulin. Immunol. Lett. 1998, 62, 159–164. [Google Scholar] [CrossRef]
- Sodhi, A.; Shishodia, S.; Shrivastava, A. Cisplatin-stimulated murine bone marrow-derived macrophages secrete Oncostatin M. Immunol. Cell Biol. 1997, 75, 492–496. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef]
- Bastid, J. Emt in carcinoma progression and dissemination: Facts, unanswered questions, and clinical considerations. Cancer Metastasis Rev. 2012, 31, 277–283. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 2012, 12, 23–38. [Google Scholar]
- Balanis, N.; Wendt, M.K.; Schiemann, B.J.; Wang, Z.; Schiemann, W.P.; Carlin, C.R. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent stat3 signaling pathway. J. Biol. Chem. 2013, 288, 17954–17967. [Google Scholar]
- Foroni, C.; Broggini, M.; Generali, D.; Damia, G. Epithelial-mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat. Rev. 2012, 38, 689–697. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef]
- Angelucci, C.; Maulucci, G.; Lama, G.; Proietti, G.; Colabianchi, A.; Papi, M.; Maiorana, A.; de Spirito, M.; Micera, A.; Balzamino, O.B.; et al. Epithelial-stromal interactions in human breast cancer: Effects on adhesion, plasma membrane fluidity and migration speed and directness. PLoS One 2012, 7, e50804. [Google Scholar] [CrossRef]
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429. [Google Scholar] [CrossRef]
- Sleeman, J.P. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012, 31, 429–440. [Google Scholar] [CrossRef]
- Cichon, M.A.; Degnim, A.C.; Visscher, D.W.; Radisky, D.C. Microenvironmental influences that drive progression from benign breast disease to invasive breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 389–397. [Google Scholar] [CrossRef]
- Gao, D.; Vahdat, L.T.; Wong, S.; Chang, J.C.; Mittal, V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012, 72, 4883–4889. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Vlaicu, P.; Mertins, P.; Mayr, T.; Widschwendter, P.; Ataseven, B.; Hogel, B.; Eiermann, W.; Knyazev, P.; Ullrich, A. Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific egfr ligands and a stat3 activator. BMC Cancer 2013, 13, 197. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated lin-28-let-7-hmga2 and mir-200-zeb1 circuits initiate and maintain Oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast cancer cells stimulate neutrophils to produce Oncostatin M: Potential implications for tumor progression. Cancer Res. 2005, 65, 8896–8904. [Google Scholar] [CrossRef]
- Grant, S.L.; Hammacher, A.; Douglas, A.M.; Goss, G.A.; Mansfield, R.K.; Heath, J.K.; Begley, C.G. An unexpected biochemical and functional interaction between gp130 and the egf receptor family in breast cancer cells. Oncogene 2002, 21, 460–474. [Google Scholar] [CrossRef]
- He, S.; Nakada, D.; Morrison, S.J. Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol. 2009, 25, 377–406. [Google Scholar] [CrossRef]
- Spradling, A.; Drummond-Barbosa, D.; Kai, T. Stem cells find their niche. Nature 2001, 414, 98–104. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef]
- Yamashita, T.; Honda, M.; Nio, K.; Nakamoto, Y.; Yamashita, T.; Takamura, H.; Tani, T.; Zen, Y.; Kaneko, S. Oncostatin M renders epithelial cell adhesion molecule-positive liver cancer stem cells sensitive to 5-fluorouracil by inducing hepatocytic differentiation. Cancer Res. 2010, 70, 4687–4697. [Google Scholar] [CrossRef]
- Douglas, A.M.; Grant, S.L.; Goss, G.A.; Clouston, D.R.; Sutherland, R.L.; Begley, C.G. Oncostatin M induces the differentiation of breast cancer cells. Int. J. Cancer 1998, 75, 64–73. [Google Scholar] [CrossRef]
- David, E.; Tirode, F.; Baud'huin, M.; Guihard, P.; Laud, K.; Delattre, O.; Heymann, M.F.; Heymann, D.; Redini, F.; Blanchard, F. Oncostatin M is a growth factor for ewing sarcoma. Am. J. Pathol. 2012, 181, 1782–1795. [Google Scholar] [CrossRef]
- Nightingale, J.; Patel, S.; Suzuki, N.; Buxton, R.; Takagi, K.I.; Suzuki, J.; Sumi, Y.; Imaizumi, A.; Mason, R.M.; Zhang, Z. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via jak/stat pathway activation. J. Am. Soc. Nephrol. 2004, 15, 21–32. [Google Scholar] [CrossRef]
- West, N.R.; Murray, J.I.; Watson, P.H. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2013. [Google Scholar] [CrossRef]
- Koo, K.H.; Kim, H.; Bae, Y.K.; Kim, K.; Park, B.K.; Lee, C.H.; Kim, Y.N. Salinomycin induces cell death via inactivation of stat3 and downregulation of skp2. Cell Death Dis. 2013, 4, e693. [Google Scholar] [CrossRef]
- Chung, S.S.; Aroh, C.; Vadgama, J.V. Constitutive activation of stat3 signaling regulates htert and promotes stem cell-like traits in human breast cancer cells. PLoS One 2013, 8, e83971. [Google Scholar] [CrossRef]
- Garner, J.M.; Fan, M.; Yang, C.H.; Du, Z.; Sims, M.; Davidoff, A.M.; Pfeffer, L.M. Constitutive activation of signal transducer and activator of transcription 3 (stat3) and nuclear factor kappab signaling in glioblastoma cancer stem cells regulates the notch pathway. J. Biol. Chem. 2013, 288, 26167–26176. [Google Scholar]
- Liu, A.Y.; Cai, Y.; Mao, Y.; Lin, Y.; Zheng, H.; Wu, T.; Huang, Y.; Fang, X.; Lin, S.; Feng, Q.; et al. Twist2 promotes self-renewal of liver cancer stem-like cells by regulating cd24. Carcinogenesis 2013, 35, 537–545. [Google Scholar]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant tgf-beta and il-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450. [Google Scholar] [CrossRef]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. Ccl2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef]
- Di, X.; Bright, A.T.; Bellott, R.; Gaskins, E.; Robert, J.; Holt, S.; Gewirtz, D.; Elmore, L. A chemotherapy-associated senescence bystander effect in breast cancer cells. Cancer Biol. Ther. 2008, 7, 864–872. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and tgfbeta-nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine “bystander senescence”. Aging 2012, 4, 932–951. [Google Scholar]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Dimauro, T.; David, G. Ras-induced senescence and its physiological relevance in cancer. Curr. Cancer Drug Targets 2010, 10, 869–876. [Google Scholar] [CrossRef]
- Jamerson, M.H.; Johnson, M.D.; Dickson, R.B. Of mice and myc: C-myc and mammary tumorigenesis. J. Mammary Gland Biol. Neoplasia 2004, 9, 27–37. [Google Scholar] [CrossRef]
- Dang, C.V. C-myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of tgf-beta-induced emt during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-smad tgf-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Loewen, G.M.; Tracy, E.; Blanchard, F.; Tan, D.; Yu, J.; Raza, S.; Matsui, S.; Baumann, H. Transformation of human bronchial epithelial cells alters responsiveness to inflammatory cytokines. BMC Cancer 2005, 5, 145. [Google Scholar] [CrossRef]
- Lacreusette, A.; Lartigue, A.; Nguyen, J.M.; Barbieux, I.; Pandolfino, M.C.; Paris, F.; Khammari, A.; Dreno, B.; Jacques, Y.; Blanchard, F.; et al. Relationship between responsiveness of cancer cells to Oncostatin M and/or il-6 and survival of stage iii melanoma patients treated with tumour-infiltrating lymphocytes. J. Pathol. 2008, 216, 451–459. [Google Scholar] [CrossRef]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Acosta, J.C.; Gil, J. Senescence: A new weapon for cancer therapy. Trends Cell Biol. 2012, 22, 211–219. [Google Scholar] [CrossRef]
- Pazolli, E.; Stewart, S.A. Senescence: The good the bad and the dysfunctional. Curr. Opin. Genet. Dev. 2008, 18, 42–47. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Junk, D.J.; Bryson, B.L.; Jackson, M.W. HiJAK’d Signaling; the STAT3 Paradox in Senescence and Cancer Progression. Cancers 2014, 6, 741-755. https://doi.org/10.3390/cancers6020741
Junk DJ, Bryson BL, Jackson MW. HiJAK’d Signaling; the STAT3 Paradox in Senescence and Cancer Progression. Cancers. 2014; 6(2):741-755. https://doi.org/10.3390/cancers6020741
Chicago/Turabian StyleJunk, Damian J., Benjamin L. Bryson, and Mark W. Jackson. 2014. "HiJAK’d Signaling; the STAT3 Paradox in Senescence and Cancer Progression" Cancers 6, no. 2: 741-755. https://doi.org/10.3390/cancers6020741