The Role of STAT3 in Non-Small Cell Lung Cancer


Abstract
:1. Introduction
2. Characteristics of STATs
3. STAT3 in Tumor Cell Proliferation
4. Prognostic Impact of STAT3 Activation in NSCLC
5. STAT3 in Primary Resistance to Treatment
6. Activation of STAT3 Independent of EGFR Downstream Pathways
7. STAT3 in EGFR Signaling Pathways
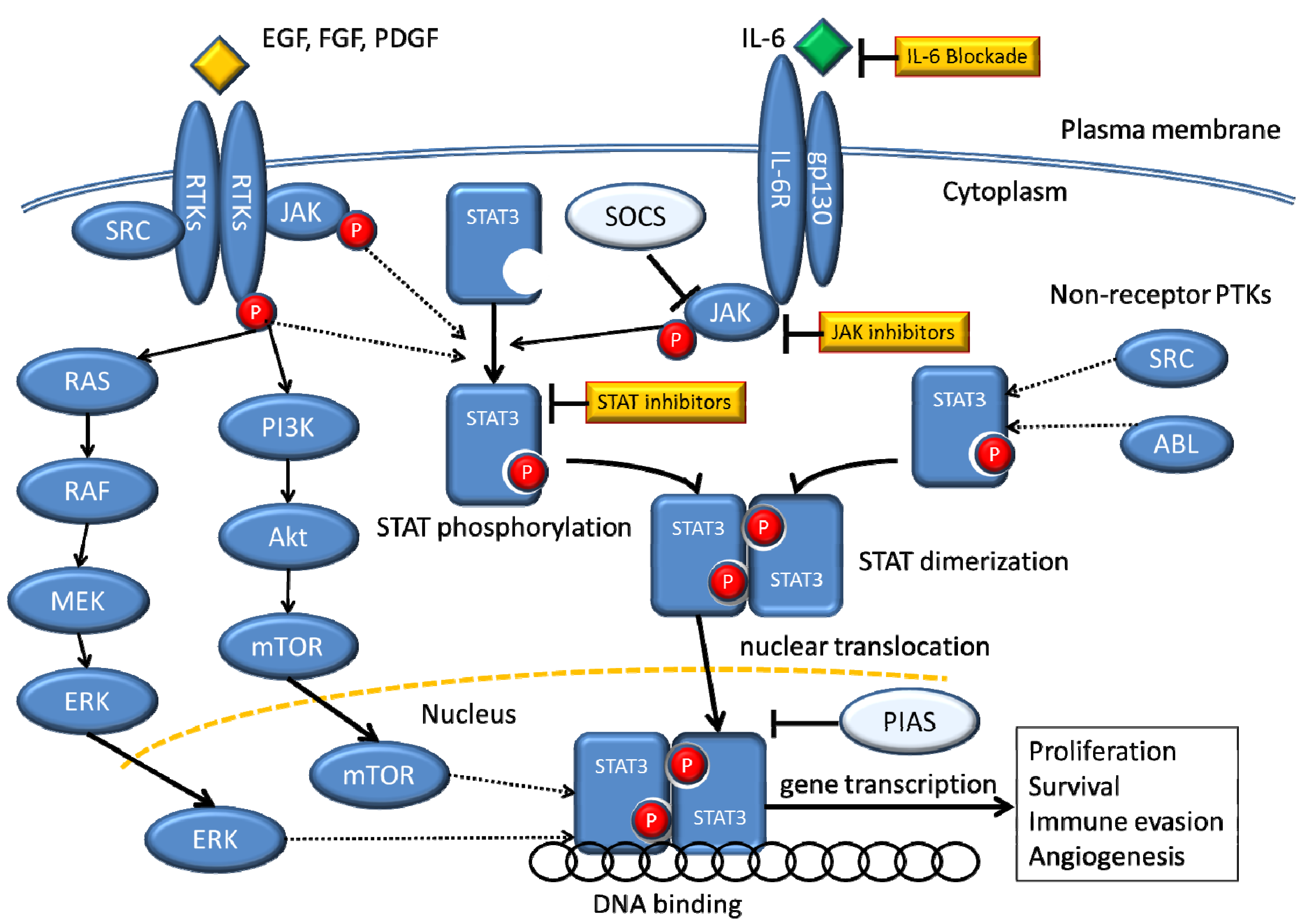
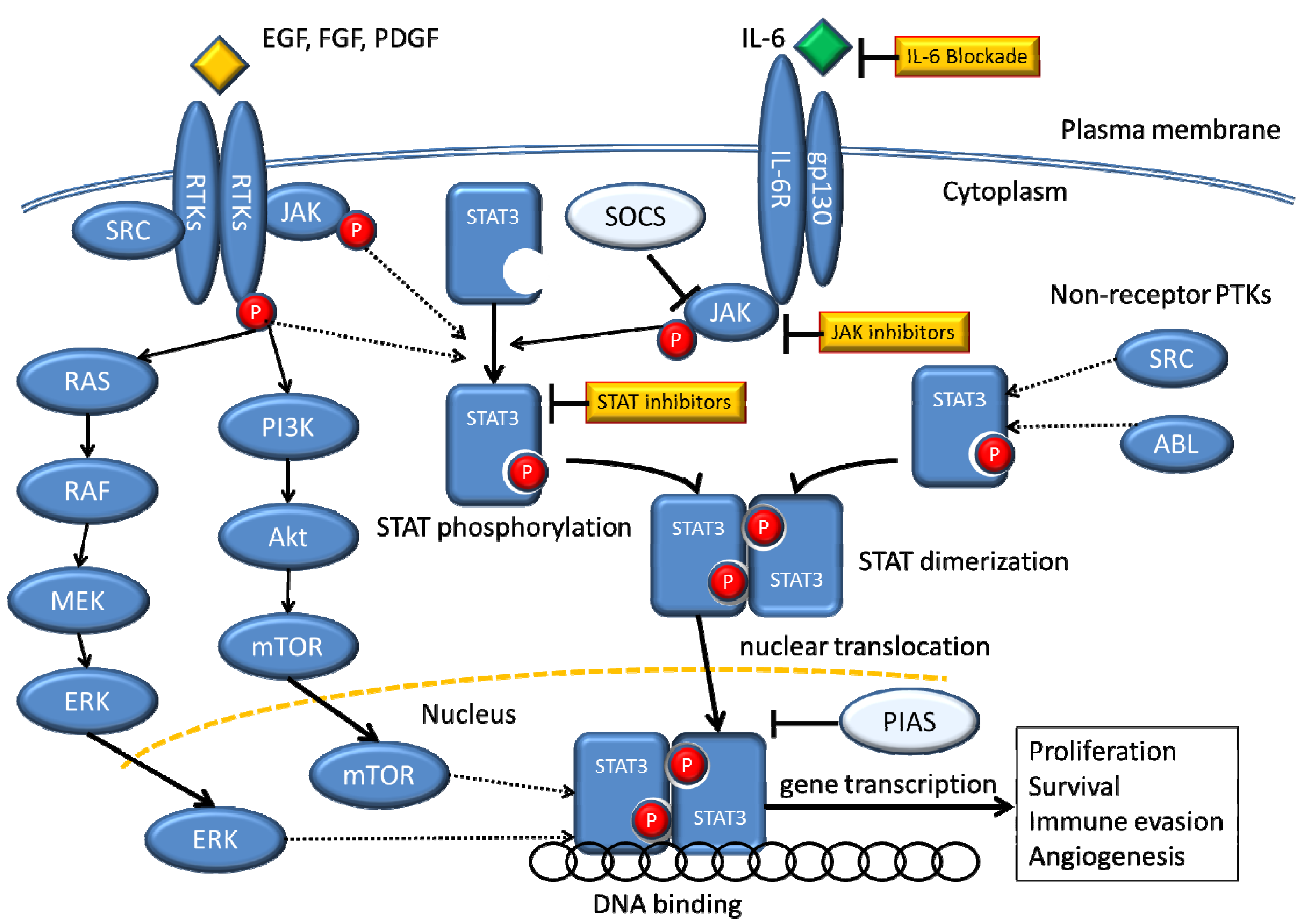
8. Relationship between STAT3 and Acquired Resistance to EGFR-TKI
9. STAT3 as a Target for Cancer Therapy

{kind=link}
| Drug | Target | Indication | Status |
|---|---|---|---|
| Siltuximab (CNTO-328) | IL-6 (monoclonal antibody) | Ovarian, pancreatic, colorectal, head and neck, and lung cancer | Phase I Phase II |
| Ruxolitinib (INCB018424) | JAK1/2 | Chronic myeloproliferative disorders, leukemia, myelodysplastic syndrome, myeloproliferative neoplasms, unspecified childhood solid tumor | Phase I |
| AZD1480 | JAK1/2 | Advanced solid tumor, hepatocellular carcinomaEGFR and/or ROS mutant lung cancer, metastatic lung cancer, gastric cancer | Phase I |
| INCB047986 | JAK | Advanced solid tumor, Hodgkin's lymphoma, non-Hodgkin's lymphoma | Phase I |
| OPB-31121 | JAK2 and gp130 | Advanced solid tumor | Phase I |
| WP1066 | JAK2 and gp130 | Glioblastoma multiforme, melanoma | Phase I |
| OPB-51602 | STAT3 phosphorylation | Advanced cancer | Phase I |
| SAR302503 | JAK | Solid malignancy | Phase I |
10. Conclusions
Acknowledgments
Conflicts of Interest
References
- Zimmer, S.; Kahl, P.; Buhl, T.M.; Steiner, S.; Wardelmann, E.; Merkelbach-Bruse, S.; Buettner, R.; Heukamp, L.C. Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J. Cancer Res. Clin. Oncol. 2009, 135, 723–730. [Google Scholar] [CrossRef]
- Looyenga, B.D.; Hutchings, D.; Cherni, I.; Kingsley, C.; Weiss, G.J.; Mackeigan, J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One 2012, 7, e30820. [Google Scholar]
- Jiang, R.; Jin, Z.; Liu, Z.; Sun, L.; Wang, L.; Li, K. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol. Diagn. Ther. 2011, 15, 347–352. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Verstovsek, S. Therapeutic potential of JAK2 inhibitors. Hematol. Am. Soc. Hematol. Educ. Program. 2009. [Google Scholar] [CrossRef]
- Murakami, T.; Takigawa, N.; Ninomiya, T.; Ochi, N.; Yasugi, M.; Honda, Y.; Kubo, T.; Ichihara, E.; Hotta, K.; Tanimoto, M.; et al. Effect of AZD1480 in an epidermal growth factor receptor-driven lung cancer model. Lung Cancer 2014, 83, 30–36. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Egan, S.E.; Weinberg, R.A. The pathway to signal achievement. Nature 1993, 365, 781–783. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Barre, B.; Avril, S.; Coqueret, O. Opposite regulation of myc and p21waf1 transcription by STAT3 proteins. J. Biol. Chem. 2003, 278, 2990–2996. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Blaskovich, M.A.; Sun, J.; Cantor, A.; Turkson, J.; Jove, R.; Sebti, S.M. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003, 63, 1270–1279. [Google Scholar]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. Stats in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Haura, E.B.; Zheng, Z.; Song, L.; Cantor, A.; Bepler, G. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 8288–8294. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Invest. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. Il-17 can promote tumor growth through an IL-6-STAT3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Shain, K.H.; Yarde, D.N.; Meads, M.B.; Huang, M.; Jove, R.; Hazlehurst, L.A.; Dalton, W.S. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: Implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009, 69, 1009–1015. [Google Scholar] [CrossRef]
- Ara, T.; Song, L.; Shimada, H.; Keshelava, N.; Russell, H.V.; Metelitsa, L.S.; Groshen, S.G.; Seeger, R.C.; DeClerck, Y.A. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009, 69, 329–337. [Google Scholar] [CrossRef]
- Shen, Y.; Devgan, G.; Darnell, J.E., Jr.; Bromberg, J.F. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc. Natl. Acad. Sci. USA 2001, 98, 1543–1548. [Google Scholar] [CrossRef]
- Cai, L.; Zhang, G.; Tong, X.; You, Q.; An, Y.; Wang, Y.; Guo, L.; Wang, T.; Zhu, D.; Zheng, J. Growth inhibition of human ovarian cancer cells by blocking STAT3 activation with small interfering RNA. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 148, 73–80. [Google Scholar] [CrossRef]
- Burke, W.M.; Jin, X.; Lin, H.J.; Huang, M.; Liu, R.; Reynolds, R.K.; Lin, J. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 2001, 20, 7925–7934. [Google Scholar] [CrossRef]
- Horiguchi, A.; Asano, T.; Kuroda, K.; Sato, A.; Asakuma, J.; Ito, K.; Hayakawa, M.; Sumitomo, M.; Asano, T. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br. J. Cancer 2010, 102, 1592–1599. [Google Scholar] [CrossRef]
- Byers, L.A.; Sen, B.; Saigal, B.; Diao, L.; Wang, J.; Nanjundan, M.; Cascone, T.; Mills, G.B.; Heymach, J.V.; Johnson, F.M. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin. Cancer Res. 2009, 15, 6852–6861. [Google Scholar] [CrossRef]
- Chiu, H.C.; Chou, D.L.; Huang, C.T.; Lin, W.H.; Lien, T.W.; Yen, K.J.; Hsu, J.T. Suppression of STAT3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem. Pharmacol. 2011, 81, 1263–1270. [Google Scholar] [CrossRef]
- Song, L.; Rawal, B.; Nemeth, J.A.; Haura, E.B. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol. Cancer Ther. 2011, 10, 481–494. [Google Scholar] [CrossRef]
- Sen, B.; Saigal, B.; Parikh, N.; Gallick, G.; Johnson, F.M. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009, 69, 1958–1965. [Google Scholar] [CrossRef]
- Xu, Y.H.; Lu, S. A meta-analysis of STAT3 and phospho-STAT3 expression and survival of patients with non-small-cell lung cancer. Eur. J. Surg. Oncol. 2014, 40, 311–317. [Google Scholar] [CrossRef]
- Zhao, M.; Gao, F.H.; Wang, J.Y.; Liu, F.; Yuan, H.H.; Zhang, W.Y.; Jiang, B. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer 2011, 73, 366–374. [Google Scholar] [CrossRef]
- Barre, B.; Vigneron, A.; Perkins, N.; Roninson, I.B.; Gamelin, E.; Coqueret, O. The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol. Med. 2007, 13, 4–11. [Google Scholar] [CrossRef]
- Ikuta, K.; Takemura, K.; Kihara, M.; Nishimura, M.; Ueda, N.; Naito, S.; Lee, E.; Shimizu, E.; Yamauchi, A. Overexpression of constitutive signal transducer and activator of transcription 3 mRNA in cisplatin-resistant human non-small cell lung cancer cells. Oncol. Rep. 2005, 13, 217–222. [Google Scholar]
- Su, W.P.; Cheng, F.Y.; Shieh, D.B.; Yeh, C.S.; Su, W.C. PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int. J. Nanomed. 2012, 7, 4269–4283. [Google Scholar]
- Kulesza, D.W.; Carre, T.; Chouaib, S.; Kaminska, B. Silencing of the transcription factor STAT3 sensitizes lung cancer cells to DNA damaging drugs, but not to TNFalpha- and NK cytotoxicity. Exp. Cell Res. 2013, 319, 506–516. [Google Scholar] [CrossRef]
- Zhang, W.; Wan, M.; Ma, L.; Liu, X.; He, J. Protective effects of ADAM8 against cisplatin-mediated apoptosis in non-small-cell lung cancer. Cell Biol. Int. 2013, 37, 47–53. [Google Scholar] [CrossRef]
- Sano, S.; Chan, K.S.; Kira, M.; Kataoka, K.; Takagi, S.; Tarutani, M.; Itami, S.; Kiguchi, K.; Yokoi, M.; Sugasawa, K.; et al. Signal transducer and activator of transcription 3 is a key regulator of keratinocyte survival and proliferation following UV irradiation. Cancer Res. 2005, 65, 5720–5729. [Google Scholar] [CrossRef]
- You, S.; Li, R.; Park, D.; Xie, M.; Sica, G.L.; Cao, Y.; Xiao, Z.Q.; Deng, X. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol. Cancer Ther. 2014, 13, 606–616. [Google Scholar] [CrossRef]
- Sun, Y.; Moretti, L.; Giacalone, N.J.; Schleicher, S.; Speirs, C.K.; Carbone, D.P.; Lu, B. Inhibition of JAK2 signaling by TG101209 enhances radiotherapy in lung cancer models. J. Thorac. Oncol. 2011, 6, 699–706. [Google Scholar] [CrossRef]
- Jin, H.O.; Lee, Y.H.; Park, J.A.; Kim, J.H.; Hong, S.E.; Kim, H.A.; Kim, E.K.; Noh, W.C.; Kim, B.H.; Ye, S.K.; et al. Blockage of STAT3 enhances the sensitivity of NSCLC cells to PI3K/mTOR inhibition. Biochem. Biophys. Res. Commun. 2014, 444, 502–508. [Google Scholar] [CrossRef]
- Frank, D.A. STAT signaling in the pathogenesis and treatment of cancer. Mol. Med. 1999, 5, 432–456. [Google Scholar]
- Lou, W.; Ni, Z.; Dyer, K.; Tweardy, D.J.; Gao, A.C. Interleukin-6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate 2000, 42, 239–242. [Google Scholar] [CrossRef]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: Direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar]
- Hung, W.; Elliott, B. Co-operative effect of c-Src tyrosine kinase and Stat3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J. Biol. Chem. 2001, 276, 12395–12403. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef]
- Niu, G.; Bowman, T.; Huang, M.; Shivers, S.; Reintgen, D.; Daud, A.; Chang, A.; Kraker, A.; Jove, R.; Yu, H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 2002, 21, 7001–7010. [Google Scholar] [CrossRef]
- Yan, C.; Naltner, A.; Martin, M.; Naltner, M.; Fangman, J.M.; Gurel, O. Transcriptional stimulation of the surfactant protein B gene by STAT3 in respiratory epithelial cells. J. Biol. Chem. 2002, 277, 10967–10972. [Google Scholar]
- Li, Y.; Du, H.; Qin, Y.; Roberts, J.; Cummings, O.W.; Yan, C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007, 67, 8494–8503. [Google Scholar]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Sordella, R. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Tsao, M.S.; Sakurada, A.; Cutz, J.C.; Zhu, C.Q.; Kamel-Reid, S.; Squire, J.; Lorimer, I.; Zhang, T.; Liu, N.; Daneshmand, M.; et al. Erlotinib in lung cancer—Molecular and clinical predictors of outcome. N. Engl. J. Med. 2005, 353, 133–144. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Yatabe, Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007, 98, 1817–1824. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Greulich, H.; Sellers, W.R.; Meyerson, M.; Frank, D.A. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006, 66, 3162–3168. [Google Scholar] [CrossRef]
- Greulich, H.; Chen, T.H.; Feng, W.; Janne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Invest. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Mendelsohn, J.; Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003, 21, 2787–2799. [Google Scholar] [CrossRef]
- Inoue, A.; Suzuki, T.; Fukuhara, T.; Maemondo, M.; Kimura, Y.; Morikawa, N.; Watanabe, H.; Saijo, Y.; Nukiwa, T. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J. Clin. Oncol. 2006, 24, 3340–3346. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 2012, 11, 2254–2264. [Google Scholar] [CrossRef]
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Soderberg-Naucler, C.; Smit, M.J. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci. Signal. 2010, 3, ra58. [Google Scholar]
- Blackwell, T.S.; Christman, J.W. The role of nuclear factor-kappa B in cytokine gene regulation. Am. J. Respir. Cell Mol. Biol. 1997, 17, 3–9. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Pu, Y.S.; Hour, T.C.; Chuang, S.E.; Cheng, A.L.; Lai, M.K.; Kuo, M.L. Interleukin-6 is responsible for drug resistance and anti-apoptotic effects in prostatic cancer cells. Prostate 2004, 60, 120–129. [Google Scholar] [CrossRef]
- Conze, D.; Weiss, L.; Regen, P.S.; Bhushan, A.; Weaver, D.; Johnson, P.; Rincon, M. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001, 61, 8851–8858. [Google Scholar]
- Akira, S.; Taga, T.; Kishimoto, T. Interleukin-6 in biology and medicine. Adv. Immunol. 1993, 54, 1–78. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yamamoto, Y.; Yokota, S.; Nakagawa, M.; Ito, M.; Ogura, T. Involvement of interleukin-6 in the elevation of plasma fibrinogen levels in lung cancer patients. Jpn. J. Clin. Oncol. 1998, 28, 740–744. [Google Scholar] [CrossRef]
- Yanagawa, H.; Sone, S.; Takahashi, Y.; Haku, T.; Yano, S.; Shinohara, T.; Ogura, T. Serum levels of interleukin 6 in patients with lung cancer. Br. J. Cancer 1995, 71, 1095–1098. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Ochi, N.; Ninomiya, T.; Yasugi, M.; Kubo, T.; Takeda, H.; Ichihara, E.; Ohashi, K.; Takata, S.; et al. JAK2-related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor-activating mutation. Cancer Sci. 2012, 103, 1795–1802. [Google Scholar] [CrossRef]
- Li, R.; Hu, Z.; Sun, S.Y.; Chen, Z.G.; Owonikoko, T.K.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Khuri, F.R.; Deng, X. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol. Cancer Ther. 2013, 12, 2200–2212. [Google Scholar] [CrossRef]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef]
- Huang, M.; Dorsey, J.F.; Epling-Burnette, P.K.; Nimmanapalli, R.; Landowski, T.H.; Mora, L.B.; Niu, G.; Sinibaldi, D.; Bai, F.; Kraker, A.; et al. Inhibition of Bcr-Abl kinase activity by PD180970 blocks constitutive activation of Stat5 and growth of CML cells. Oncogene 2002, 21, 8804–8816. [Google Scholar] [CrossRef]
- Levis, M.; Allebach, J.; Tse, K.F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99, 3885–3891. [Google Scholar] [CrossRef]
- Morishita, R.; Tomita, N.; Kaneda, Y.; Ogihara, T. Molecular therapy to inhibit NFkappaB activation by transcription factor decoy oligonucleotides. Curr. Opin. Pharmacol. 2004, 4, 139–146. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Wang, L.; Wei, H.; Tian, Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC Cancer 2007, 7, 149. [Google Scholar] [CrossRef]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef]
- Puchalski, T.; Prabhakar, U.; Jiao, Q.; Berns, B.; Davis, H.M. Pharmacokinetic and pharmacodynamic modeling of an anti-interleukin-6 chimeric monoclonal antibody (siltuximab) in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2010, 16, 1652–1661. [Google Scholar]
- Kim, M.J.; Nam, H.J.; Kim, H.P.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013, 335, 145–152. [Google Scholar] [CrossRef]
- Ioannidis, S.; Lamb, M.L.; Wang, T.; Almeida, L.; Block, M.H.; Davies, A.M.; Peng, B.; Su, M.; Zhang, H.J.; Hoffmann, E.; et al. Discovery of 5-chloro-N2-[(1s)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1h-pyrazol-3-yl)p yrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J. Med. Chem. 2011, 54, 262–276. [Google Scholar] [CrossRef]
- Haghikia, A.; Hoch, M.; Stapel, B.; Hilfiker-Kleiner, D. STAT3 regulation of and by microRNAs in development and disease. JAKSTAT 2012, 1, 143–150. [Google Scholar]
- Lin, H.Y.; Chiang, C.H.; Hung, W.C. STAT3 upregulates miR-92a to inhibit RECK expression and to promote invasiveness of lung cancer cells. Br. J. Cancer 2013, 109, 731–738. [Google Scholar] [CrossRef]
- Dai, B.; Meng, J.; Peyton, M.; Girard, L.; Bornmann, W.G.; Ji, L.; Minna, J.D.; Fang, B.; Roth, J.A. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011, 71, 3658–3668. [Google Scholar] [CrossRef]
- Vishwamitra, D.; Li, Y.; Wilson, D.; Manshouri, R.; Curry, C.V.; Shi, B.; Tang, X.M.; Sheehan, A.M.; Wistuba, I.I.; Shi, P.; et al. MicroRNA 96 is a post-transcriptional suppressor of anaplastic lymphoma kinase expression. Am. J. Pathol. 2012, 180, 1772–1780. [Google Scholar] [CrossRef]
- Du, L.; Subauste, M.C.; DeSevo, C.; Zhao, Z.; Baker, M.; Borkowski, R.; Schageman, J.J.; Greer, R.; Yang, C.R.; Suraokar, M.; et al. miR-337–3p and its targets STAT3 and RAP1A modulate taxane sensitivity in non-small cell lung cancers. PLoS One 2012, 7, e39167. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers 2014, 6, 708-722. https://doi.org/10.3390/cancers6020708
Harada D, Takigawa N, Kiura K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers. 2014; 6(2):708-722. https://doi.org/10.3390/cancers6020708
Chicago/Turabian StyleHarada, Daijiro, Nagio Takigawa, and Katsuyuki Kiura. 2014. "The Role of STAT3 in Non-Small Cell Lung Cancer" Cancers 6, no. 2: 708-722. https://doi.org/10.3390/cancers6020708
APA StyleHarada, D., Takigawa, N., & Kiura, K. (2014). The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers, 6(2), 708-722. https://doi.org/10.3390/cancers6020708