Structural Pathways of Cytokines May Illuminate Their Roles in Regulation of Cancer Development and Immunotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
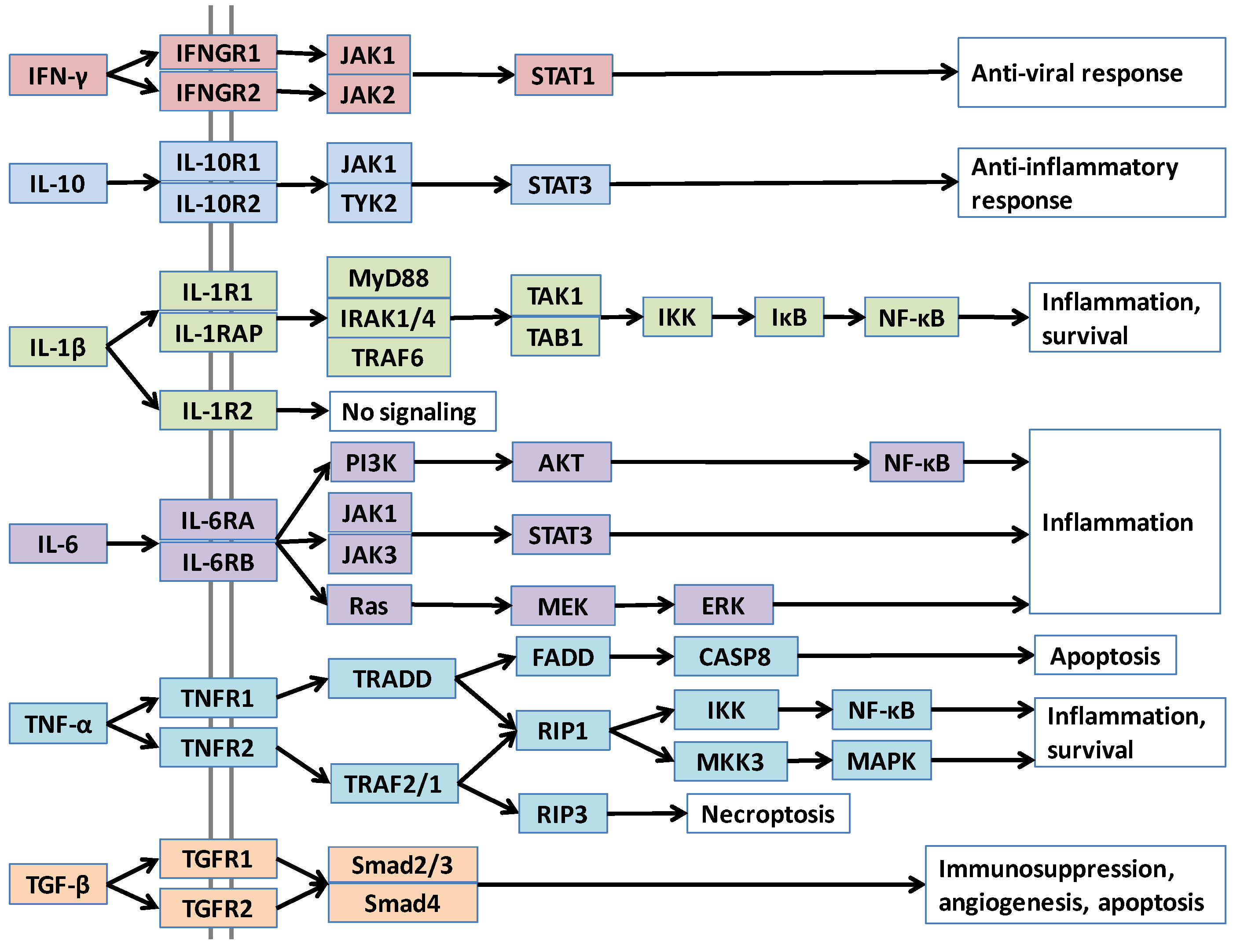
2. Cytokines and Their Roles in Cancer
3. Approaches in Construction of Structural Pathways
4. Case Studies
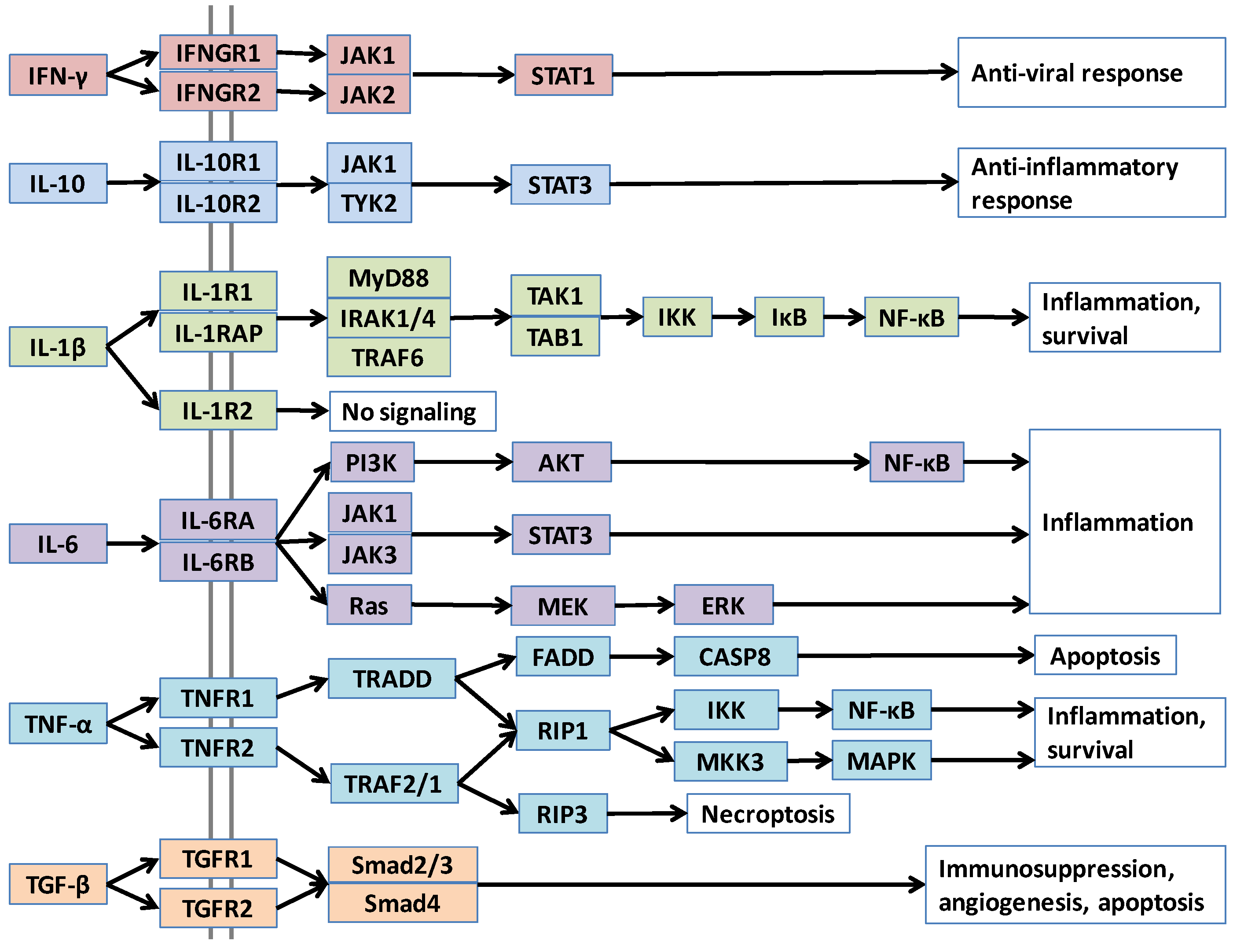
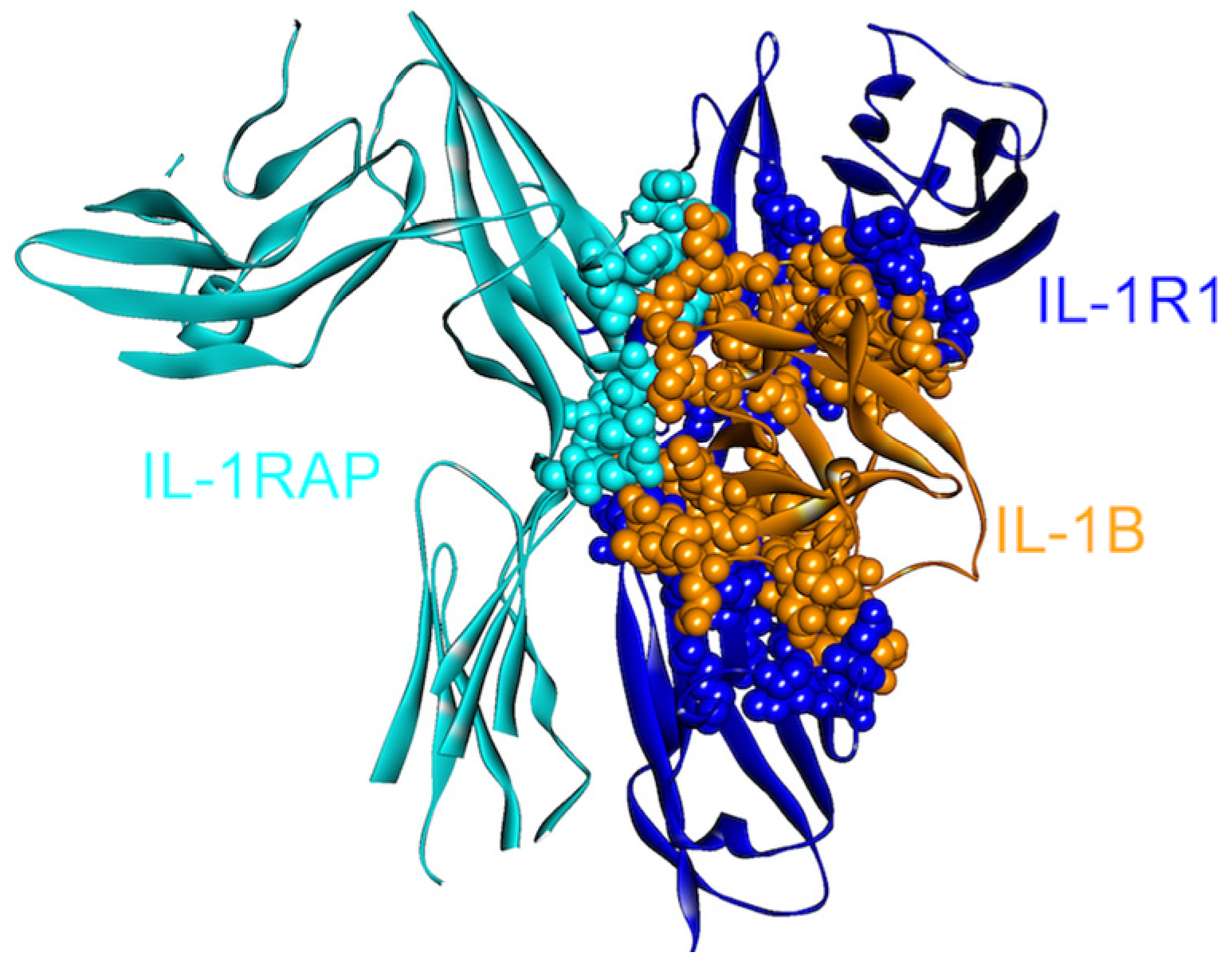
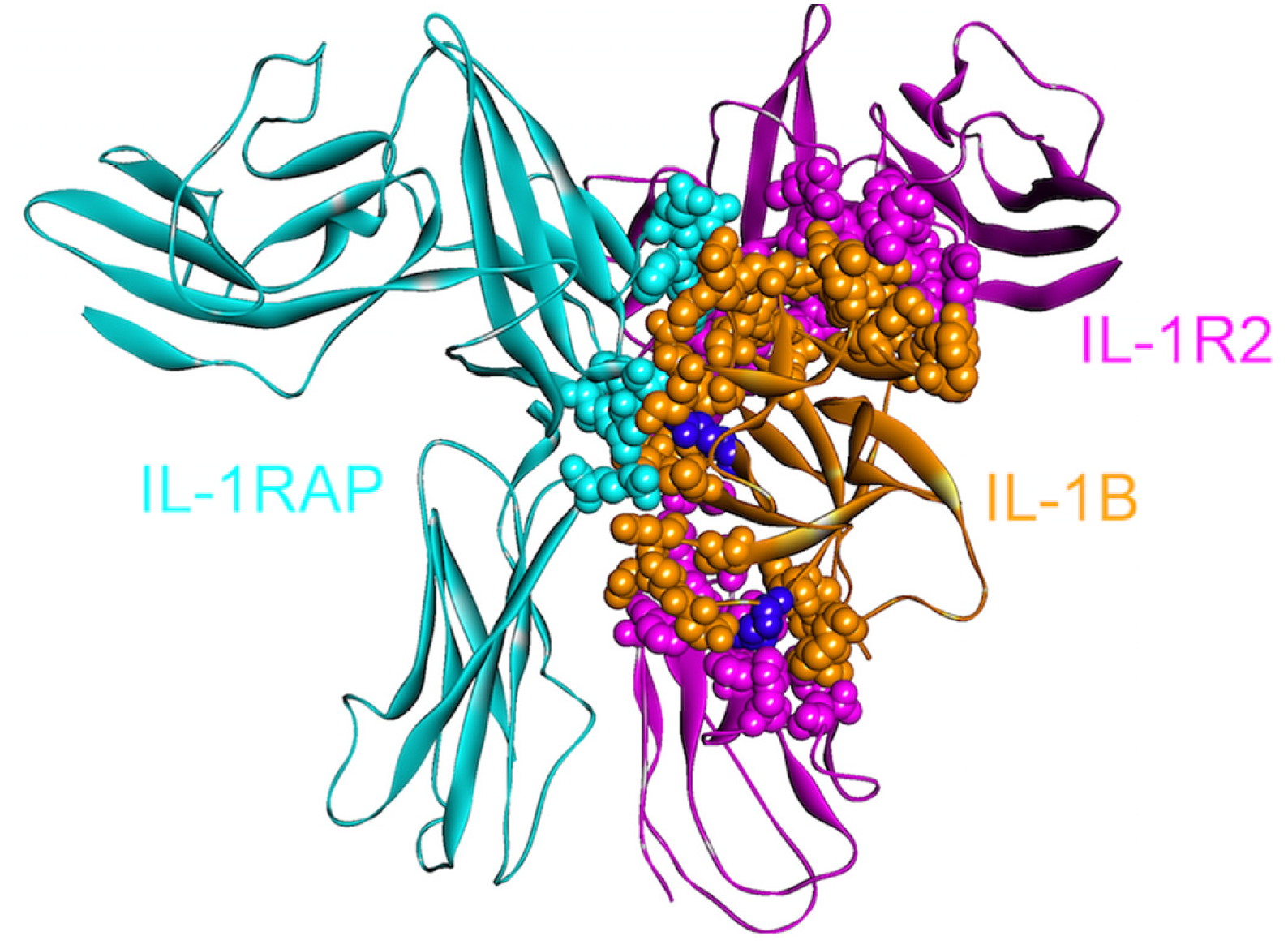
4.1. IL-1β


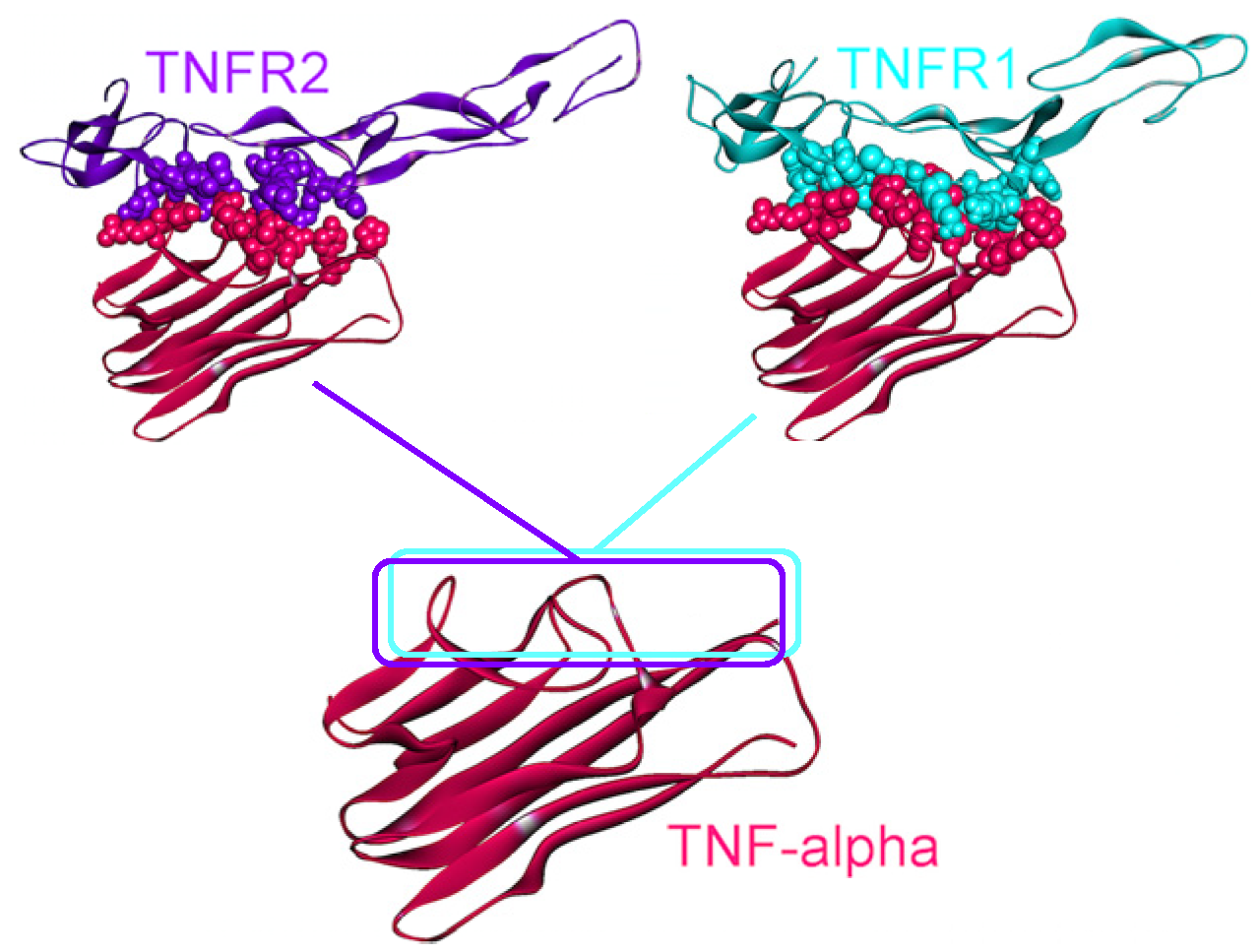
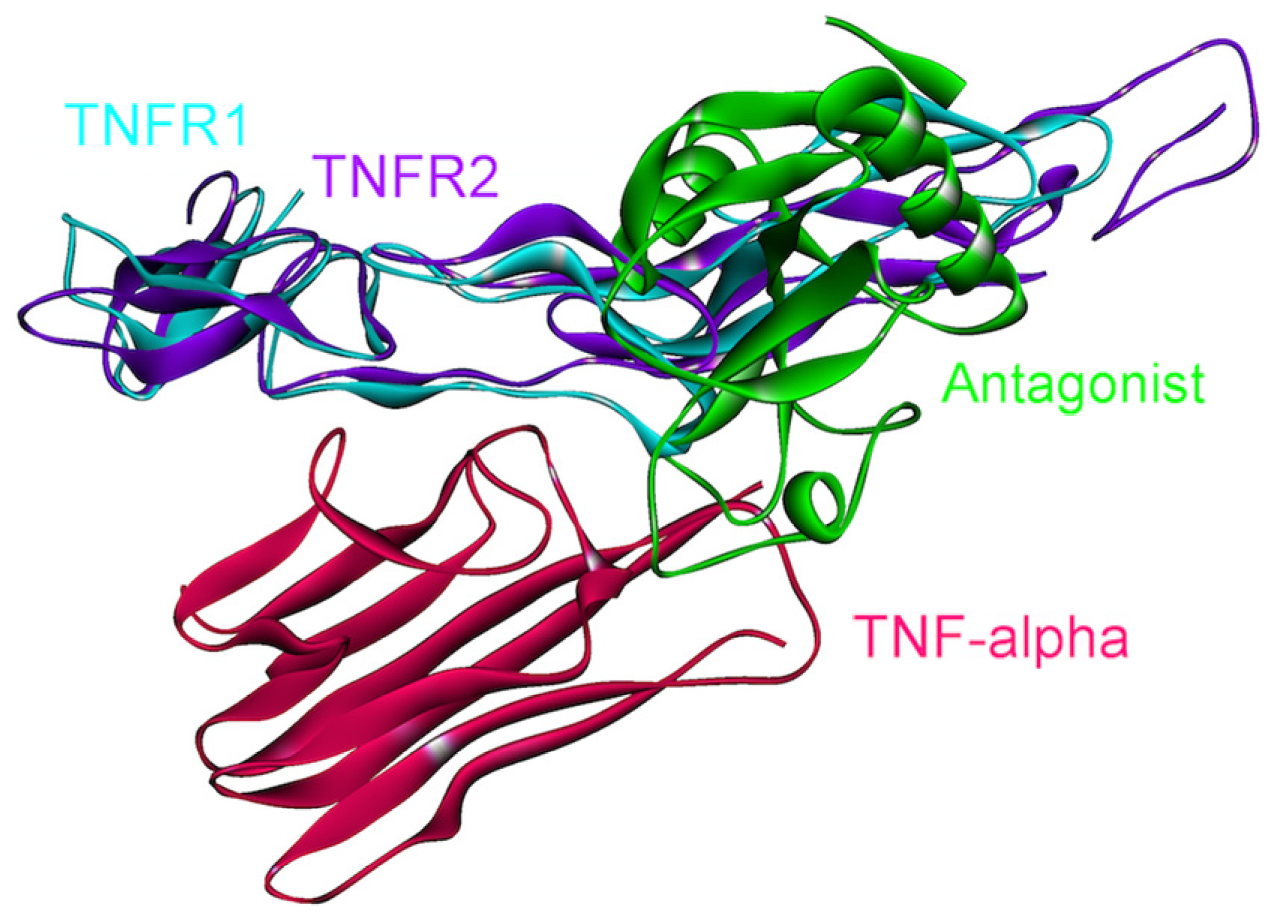
4.2. TNF-α


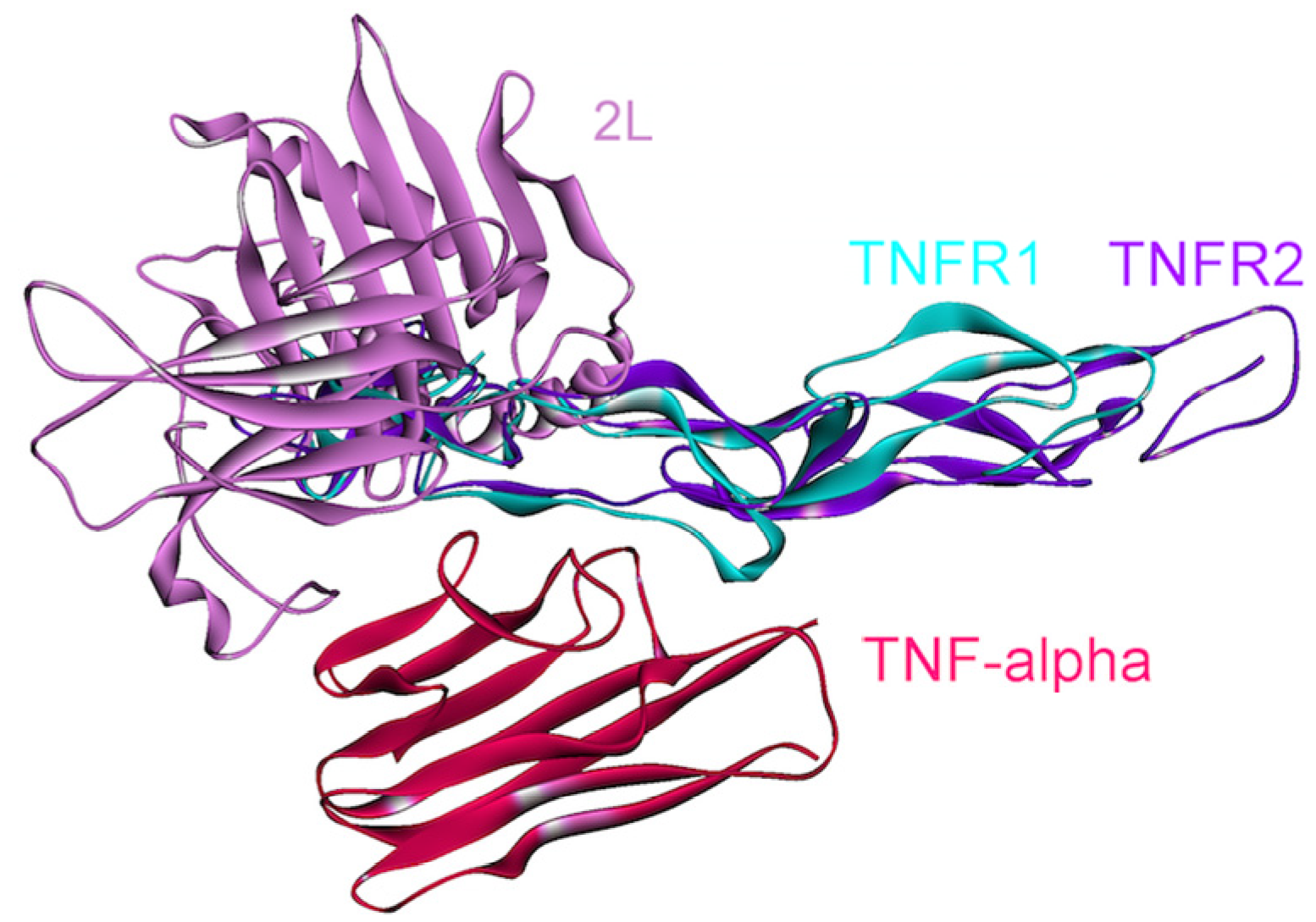
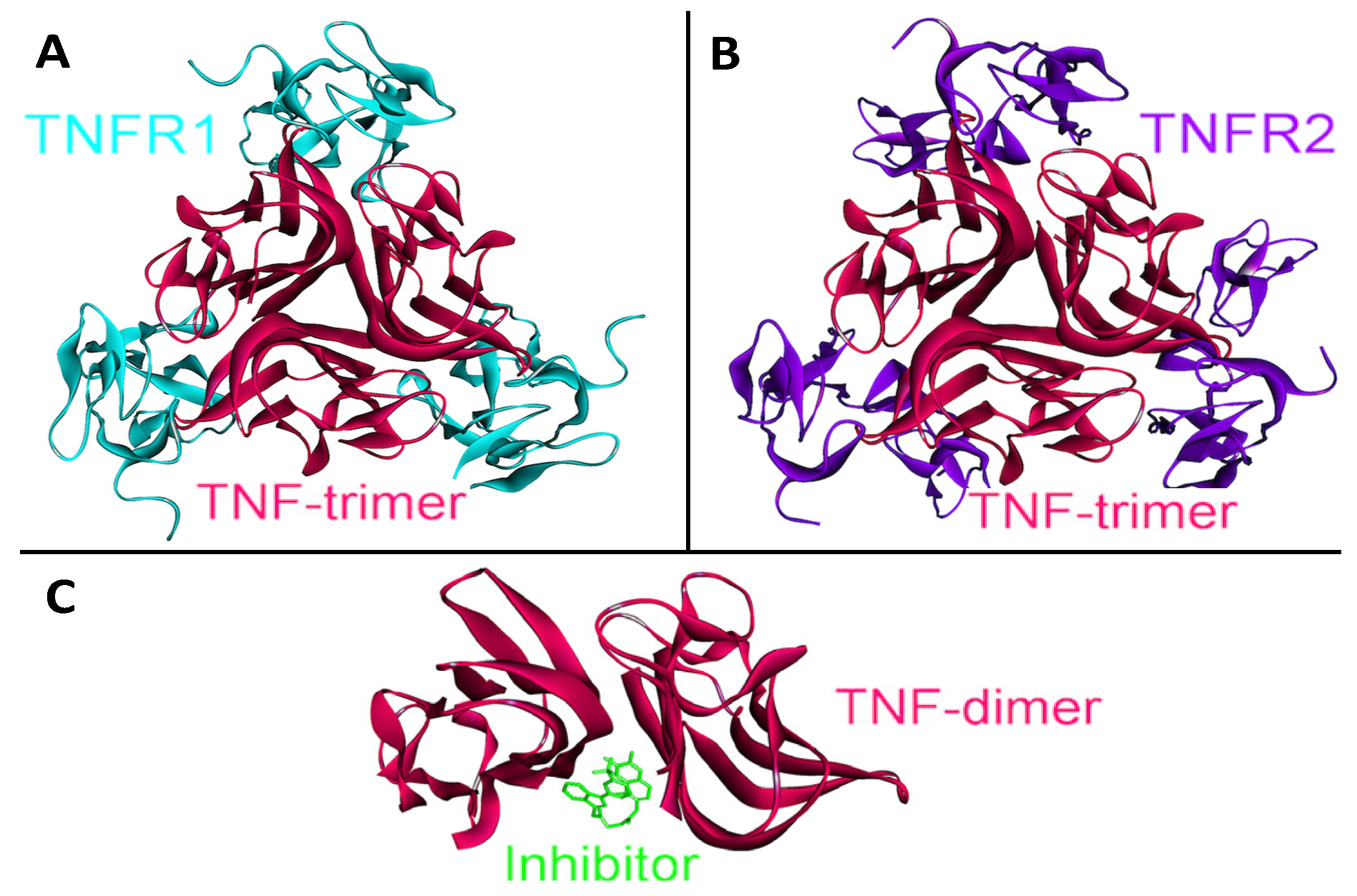
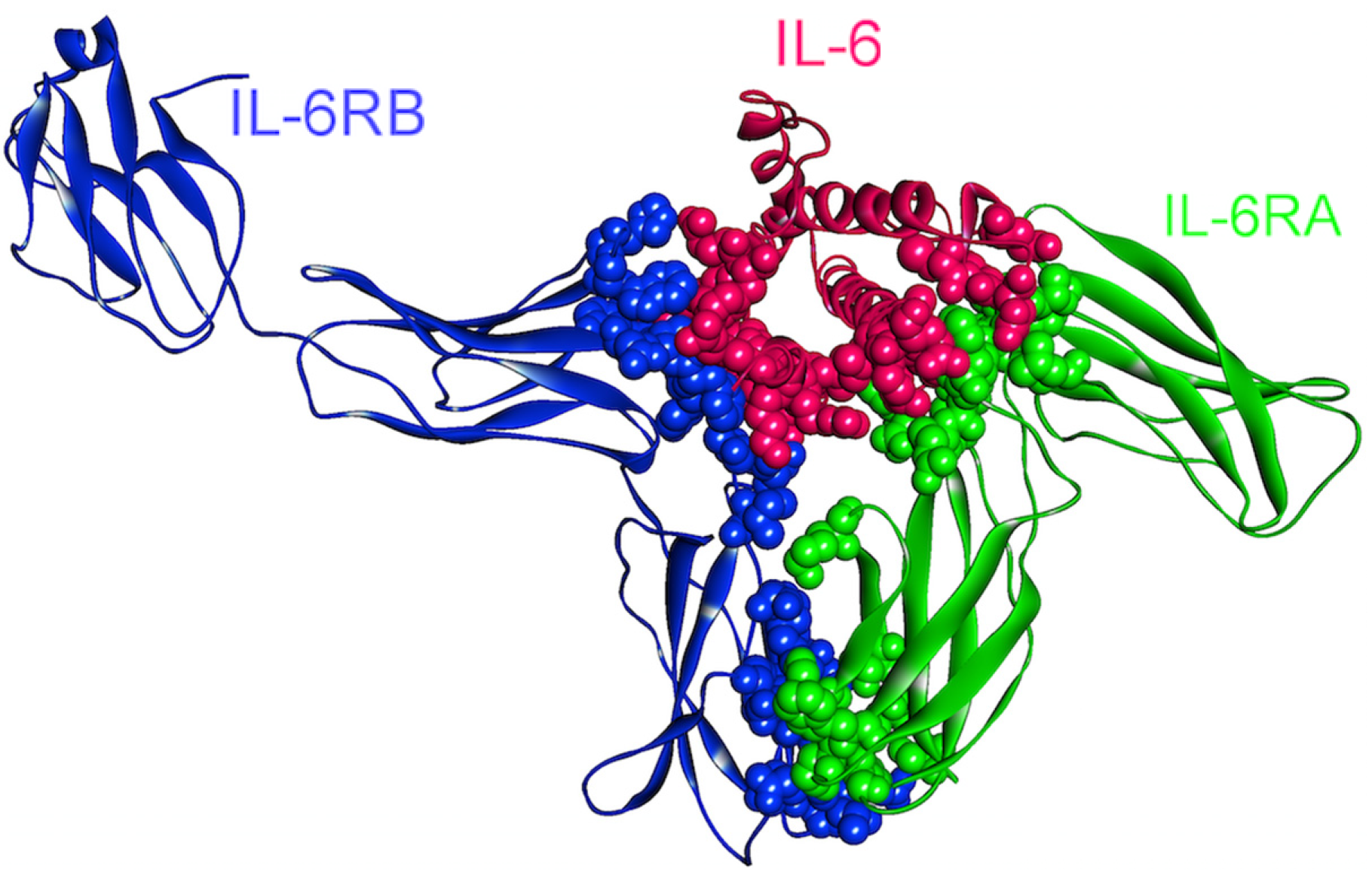
4.3. IL-6
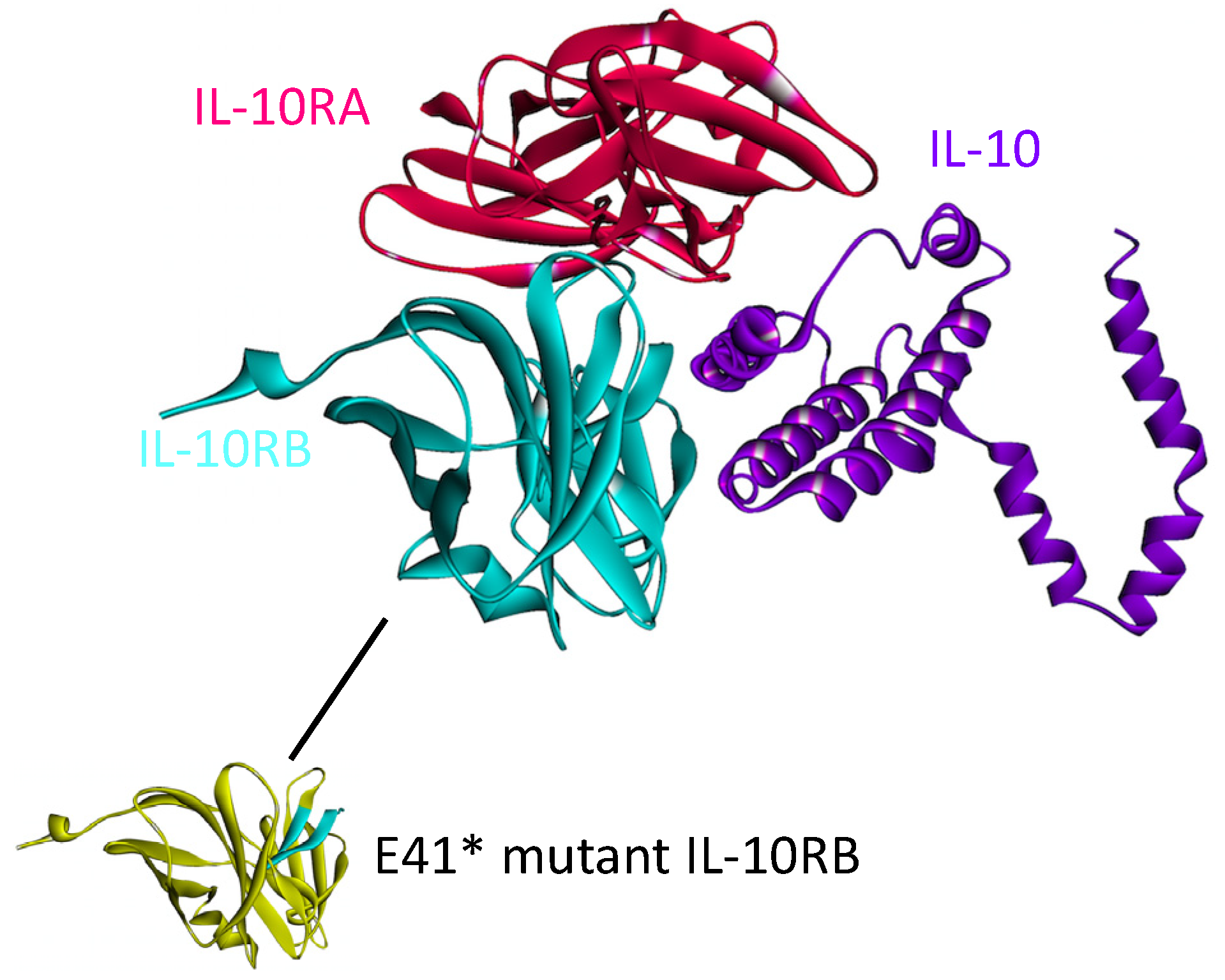
4.4. IL-10

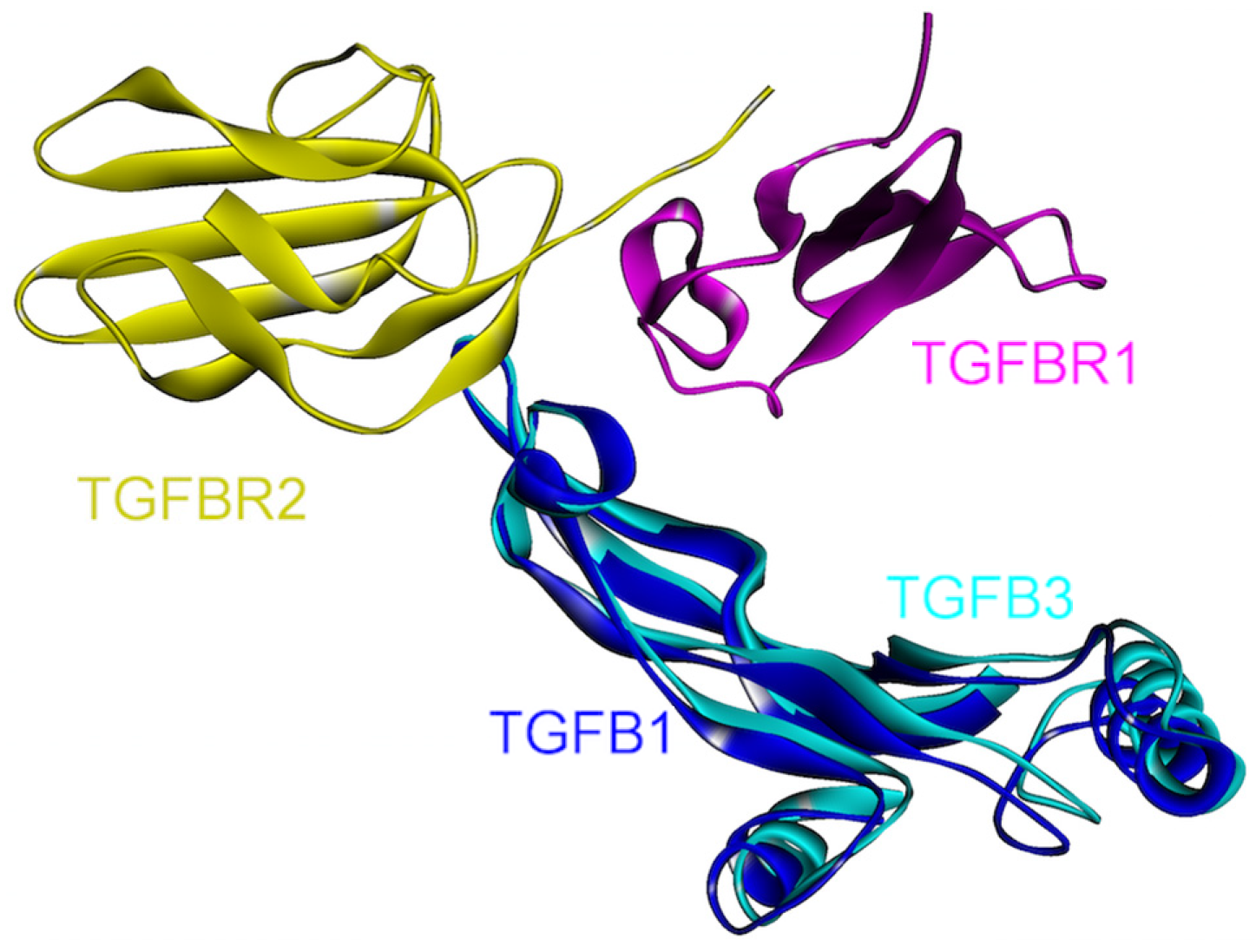
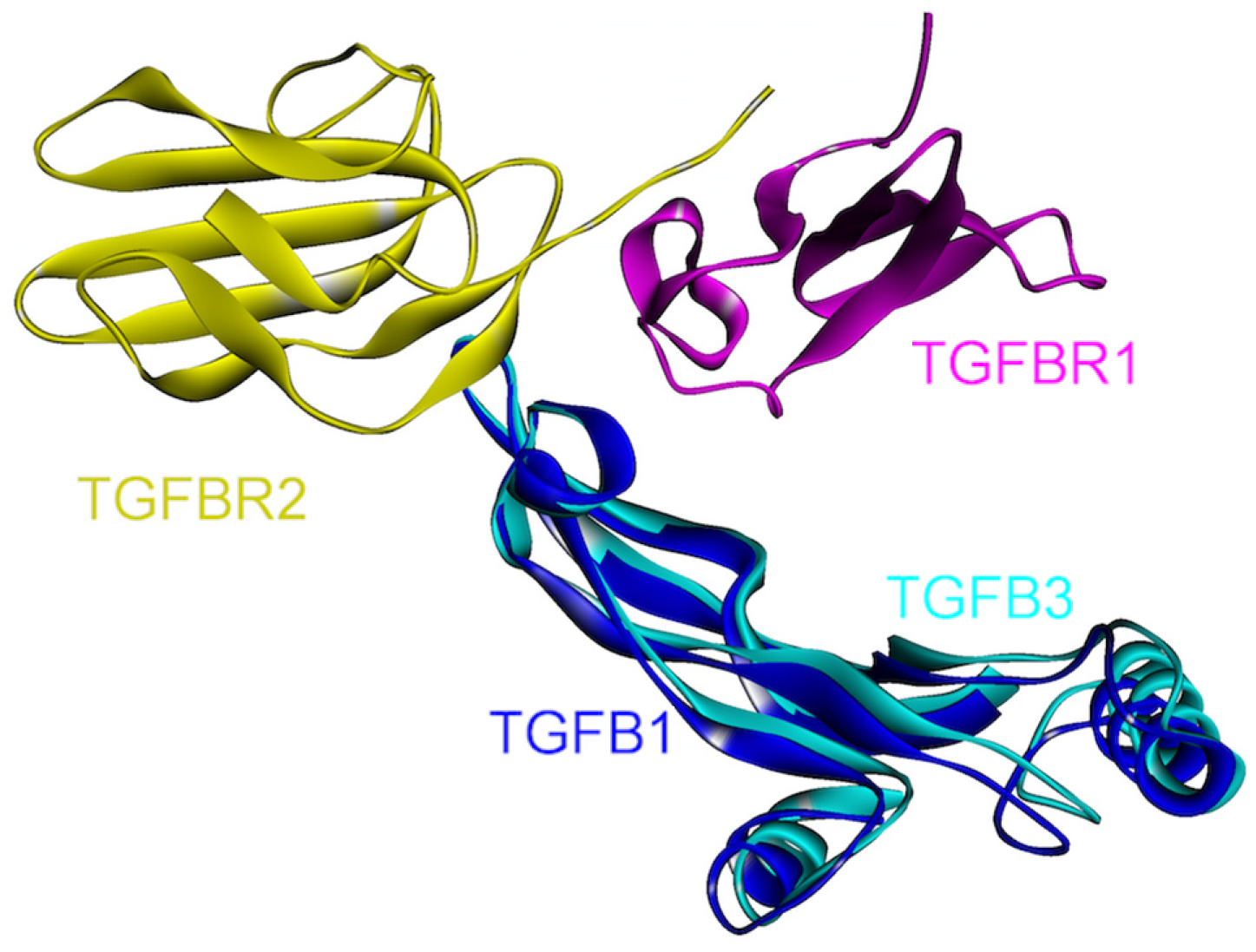
4.5. TGF-β
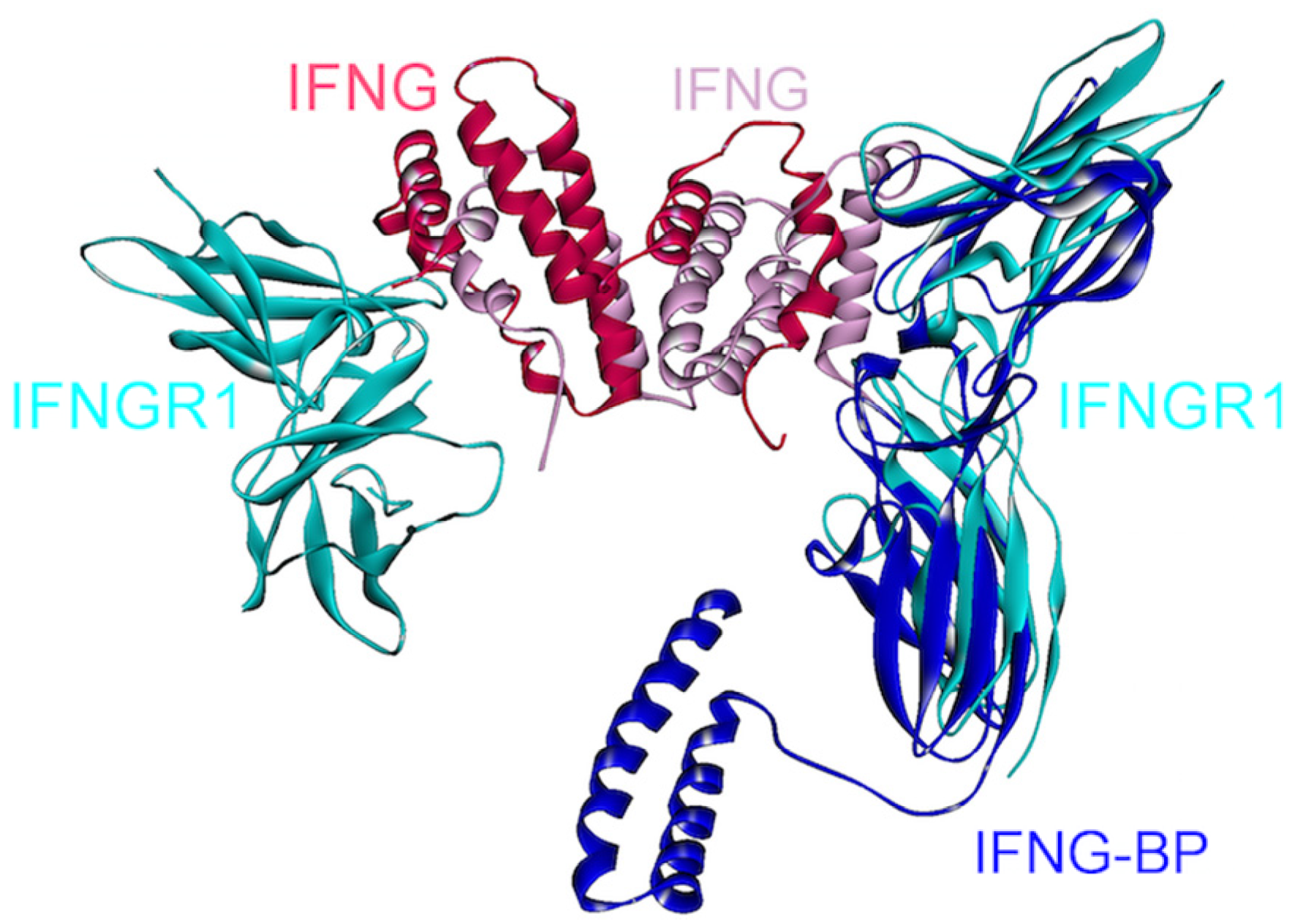
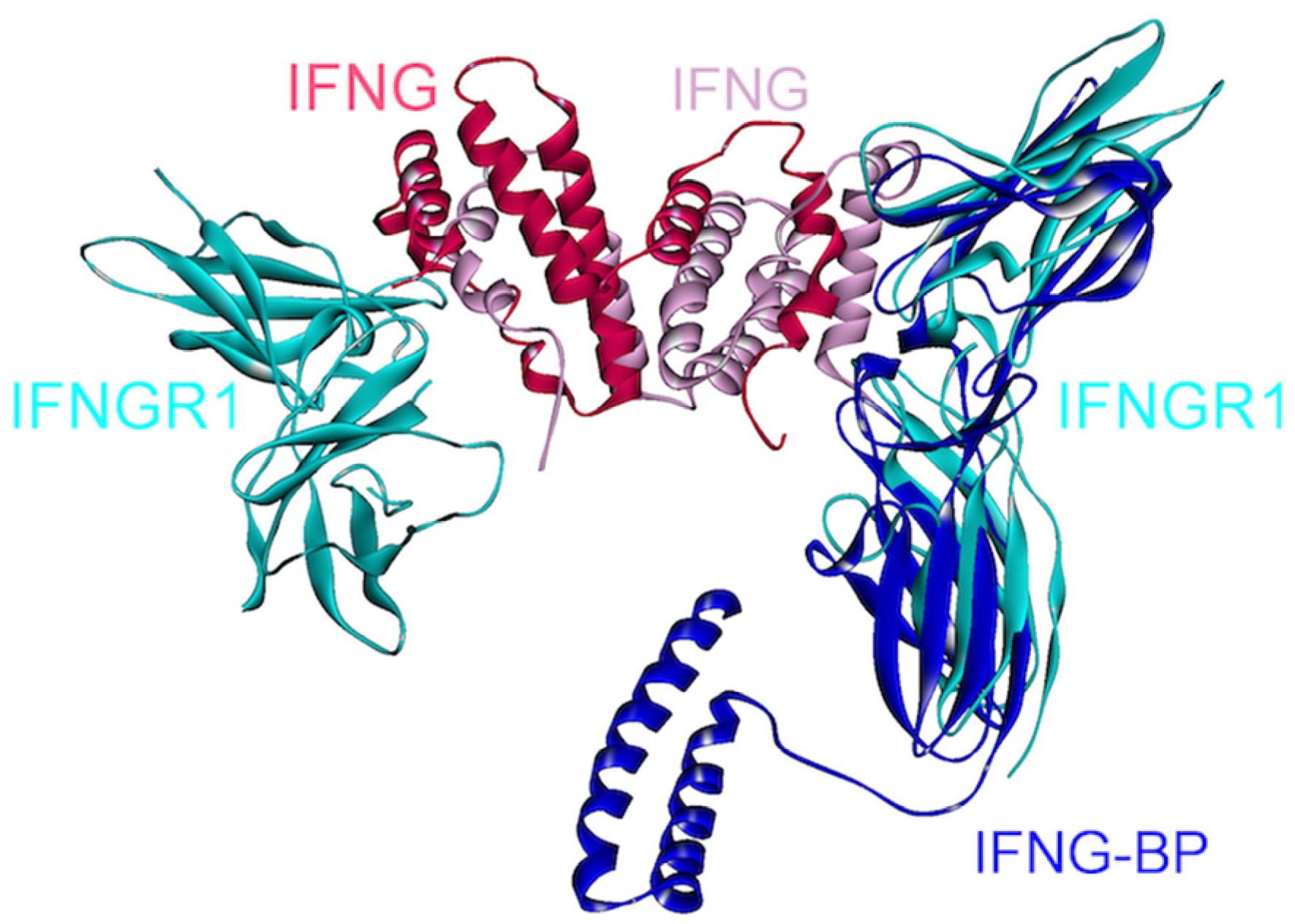
4.6. IFN-γ
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Maiorov, E.G.; Keskin, O.; Gursoy, A.; Nussinov, R. The structural network of inflammation and cancer: Merits and challenges. Semin. Cancer Biol. 2013, 23, 243–251. [Google Scholar] [CrossRef]
- Csermely, P.; Korcsmaros, T. Cancer-related networks: A help to understand, predict and change malignant transformation. Semin. Cancer Biol. 2013, 23, 209–212. [Google Scholar] [CrossRef]
- Tuncbag, N.; Gursoy, A.; Keskin, O. Prediction of protein-protein interactions: Unifying evolution and structure at protein interfaces. Phys. Biol. 2011, 8. [Google Scholar] [CrossRef]
- Ozbabacan, S.E.A.; Gursoy, A.; Nussinov, R.; Keskin, O. The structural pathway of interleukin 1 (IL-1) initiated signaling reveals mechanisms of oncogenic mutations and snps in inflammation and cancer. PLoS Comput. Biol. 2014. [Google Scholar] [CrossRef]
- Kiel, C.; Serrano, L. Structural data in synthetic biology approaches for studying general design principles of cellular signaling networks. Structure 2012, 20, 1806–1813. [Google Scholar] [CrossRef]
- Kuzu, G.; Keskin, O.; Gursoy, A.; Nussinov, R. Constructing structural networks of signaling pathways on the proteome scale. Curr. Opin. Struct. Biol. 2012, 22, 367–377. [Google Scholar] [CrossRef]
- Ozbabacan, S.E.A.; Keskin, O.; Nussinov, R.; Gursoy, A. Enriching the human apoptosis pathway by predicting the structures of protein-protein complexes. J. Struct. Biol. 2012, 179, 338–346. [Google Scholar] [CrossRef]
- Kar, G.; Keskin, O.; Nussinov, R.; Gursoy, A. Human proteome-scale structural modeling of e2–e3 interactions exploiting interface motifs. J. Proteome Res. 2012, 11, 1196–1207. [Google Scholar] [CrossRef]
- Kuzu, G.; Keskin, O.; Nussinov, R.; Gursoy, A. Modeling protein assemblies in the proteome. Mol Cell Proteomics 2014. [Google Scholar] [CrossRef]
- Kar, G.; Keskin, O.; Gursoy, A.; Nussinov, R. Allostery and population shift in drug discovery. Curr. Opin. Pharmacol. 2010, 10, 715–722. [Google Scholar] [CrossRef]
- Leis, S.; Schneider, S.; Zacharias, M. In silico prediction of binding sites on proteins. Curr. Med. Chem. 2010, 17, 1550–1562. [Google Scholar] [CrossRef]
- Kar, G.; Kuzu, G.; Keskin, O.; Gursoy, A. Protein-protein interfaces integrated into interaction networks: Implications on drug design. Curr. Pharm. Des. 2012, 18, 4697–4705. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef]
- Allavena, P.; Germano, G.; Marchesi, F.; Mantovani, A. Chemokines in cancer related inflammation. Exp. Cell Res. 2011, 317, 664–673. [Google Scholar] [CrossRef]
- Vacchelli, E.; Galluzzi, L.; Eggermont, A.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial watch: Immunostimulatory cytokines. Oncoimmunology 2012, 1, 493–506. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Germano, G.; Allavena, P.; Mantovani, A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008, 43, 374–379. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Bayne, L.J. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr. Opin. Immunol. 2013, 25, 200–205. [Google Scholar] [CrossRef]
- Luo, J.L.; Maeda, S.; Hsu, L.C.; Yagita, H.; Karin, M. Inhibition of NF-κb in cancer cells converts inflammation-induced tumor growth mediated by tnfalpha to trail-mediated tumor regression. Cancer Cell 2004, 6, 297–305. [Google Scholar] [CrossRef]
- Sultani, M.; Stringer, A.M.; Bowen, J.M.; Gibson, R.J. Anti-inflammatory cytokines: Important immunoregulatory factors contributing to chemotherapy-induced gastrointestinal mucositis. Chemother. Res. Pract. 2012, 2012, 490804. [Google Scholar]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Trinchieri, G. Inflammation in cancer: A therapeutic target? Oncology 2011, 25, 418–420. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Geng, Y.; Chandrasekaran, S.; Hsu, J.W.; Gidwani, M.; Hughes, A.D.; King, M.R. Phenotypic switch in blood: Effects of pro-inflammatory cytokines on breast cancer cell aggregation and adhesion. PLoS One 2013, 8, e54959. [Google Scholar]
- DuPage, M.; Jacks, T. Genetically engineered mouse models of cancer reveal new insights about the antitumor immune response. Curr. Opin. Immunol. 2013, 25, 192–199. [Google Scholar] [CrossRef]
- Pardoll, D. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 2003, 21, 807–839. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. Ifngamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Davidson, W.F.; Giese, T.; Fredrickson, T.N. Spontaneous development of plasmacytoid tumors in mice with defective fas-fas ligand interactions. J. Exp. Med. 1998, 187, 1825–1838. [Google Scholar] [CrossRef]
- Engle, S.J.; Ormsby, I.; Pawlowski, S.; Boivin, G.P.; Croft, J.; Balish, E.; Doetschman, T. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res. 2002, 62, 6362–6366. [Google Scholar]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999, 5, 828–831. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Bar, D.; Apte, R.N.; Voronov, E.; Dinarello, C.A.; Cohen, S. A continuous delivery system of IL-1 receptor antagonist reduces angiogenesis and inhibits tumor development. FASEB J. 2004, 18, 161–163. [Google Scholar]
- Merlo, P.; Cecconi, F. XIAP: Inhibitor of two worlds. EMBO J. 2013, 32, 2187–2188. [Google Scholar] [CrossRef]
- Massague, J. TGFβ in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. Haddock: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- Keskin, O.; Nussinov, R. Similar binding sites and different partners: Implications to shared proteins in cellular pathways. Structure 2007, 15, 341–354. [Google Scholar] [CrossRef]
- Tsai, C.J.; Lin, S.L.; Wolfson, H.J.; Nussinov, R. Protein-protein interfaces: Architectures and interactions in protein-protein interfaces and in protein cores. Their similarities and differences. Crit. Rev. Biochem. Mol. Biol. 1996, 31, 127–152. [Google Scholar] [CrossRef]
- Tuncbag, N.; Gursoy, A.; Nussinov, R.; Keskin, O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using prism. Nat. Protoc. 2011, 6, 1341–1354. [Google Scholar] [CrossRef]
- Ogmen, U.; Keskin, O.; Aytuna, A.S.; Nussinov, R.; Gursoy, A. Prism: Protein interactions by structural matching. Nucleic Acids Res. 2005, 33, W331–W336. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, S.; Li, L.; Liu, X.; Mei, K.; Wang, X. Structural insights into the assembly and activation of IL-1β with its receptors. Nat. Immunol. 2010, 11, 905–911. [Google Scholar] [CrossRef]
- McMahan, C.J.; Slack, J.L.; Mosley, B.; Cosman, D.; Lupton, S.D.; Brunton, L.L.; Grubin, C.E.; Wignall, J.M.; Jenkins, N.A.; Brannan, C.I.; et al. A novel IL-1 receptor, cloned from B cells by mammalian expression, is expressed in many cell types. EMBO J. 1991, 10, 2821–2832. [Google Scholar]
- Colotta, F.; Re, F.; Muzio, M.; Bertini, R.; Polentarutti, N.; Sironi, M.; Giri, J.G.; Dower, S.K.; Sims, J.E.; Mantovani, A. Interleukin-1 type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science 1993, 261, 472–475. [Google Scholar]
- Colotta, F.; Dower, S.K.; Sims, J.E.; Mantovani, A. The type II “decoy” receptor: A novel regulatory pathway for interleukin 1. Immunol. Today 1994, 15, 562–566. [Google Scholar] [CrossRef]
- Lang, D.; Knop, J.; Wesche, H.; Raffetseder, U.; Kurrle, R.; Boraschi, D.; Martin, M.U. The type II IL-1 receptor interacts with the IL-1 receptor accessory protein: A novel mechanism of regulation of IL-1 responsiveness. J. Immunol. 1998, 161, 6871–6877. [Google Scholar]
- Malinowsky, D.; Lundkvist, J.; Laye, S.; Bartfai, T. Interleukin-1 receptor accessory protein interacts with the type II interleukin-1 receptor. FEBS Lett. 1998, 429, 299–302. [Google Scholar] [CrossRef]
- Ruckert, F.; Dawelbait, G.; Winter, C.; Hartmann, A.; Denz, A.; Ammerpohl, O.; Schroeder, M.; Schackert, H.K.; Sipos, B.; Kloppel, G.; et al. Examination of apoptosis signaling in pancreatic cancer by computational signal transduction analysis. PLoS One 2010, 5, e12243. [Google Scholar] [CrossRef]
- Laios, A.; O’Toole, S.A.; Flavin, R.; Martin, C.; Ring, M.; Gleeson, N.; D’Arcy, T.; McGuinness, E.P.; Sheils, O.; Sheppard, B.L.; et al. An integrative model for recurrence in ovarian cancer. Mol. Cancer 2008, 7. [Google Scholar] [CrossRef]
- Matsuo, Y.; Sawai, H.; Funahashi, H.; Takahashi, H.; Sakamoto, M.; Yamamoto, M.; Okada, Y.; Hayakawa, T.; Manabe, T. Enhanced angiogenesis due to inflammatory cytokines from pancreatic cancer cell lines and relation to metastatic potential. Pancreas 2004, 28, 344–352. [Google Scholar] [CrossRef]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Minowa, K.; Mukai, Y.; Abe, Y.; Taniai, M.; Nomura, T.; Kayamuro, H.; Nabeshi, H.; et al. Creation and x-ray structure analysis of the tumor necrosis factor receptor-1-selective mutant of a tumor necrosis factor-alpha antagonist. J. Biol. Chem. 2008, 283, 998–1007. [Google Scholar] [CrossRef]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef]
- Mukai, Y.; Nakamura, T.; Yoshioka, Y.; Shibata, H.; Abe, Y.; Nomura, T.; Taniai, M.; Ohta, T.; Nakagawa, S.; Tsunoda, S.; et al. Fast binding kinetics and conserved 3D structure underlie the antagonistic activity of mutant TNF: Useful information for designing artificial proteo-antagonists. J. Biochem. 2009, 146, 167–172. [Google Scholar] [CrossRef]
- Byla, P.; Andersen, M.H.; Holtet, T.L.; Jacobsen, H.; Munch, M.; Gad, H.H.; Thogersen, H.C.; Hartmann, R. Selection of a novel and highly specific tumor necrosis factor α (TNFα) antagonist: Insight from the crystal structure of the antagonist-TNFα complex. J. Biol. Chem. 2010, 285, 12096–12100. [Google Scholar] [CrossRef]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-molecule inhibition of tnf-alpha. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef]
- Mukai, Y.; Nakamura, T.; Yoshikawa, M.; Yoshioka, Y.; Tsunoda, S.; Nakagawa, S.; Yamagata, Y.; Tsutsumi, Y. Solution of the structure of the TNF-TNFR2 complex. Sci. Signal 2010, 3. [Google Scholar] [CrossRef]
- Banner, D.W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal structure of the soluble human 55 kD TNF receptor-human TNF β complex: Implications for tnf receptor activation. Cell 1993, 73, 431–445. [Google Scholar] [CrossRef]
- Kuzu, G.; Keskin, O.; Gursoy, A.; Nussinov, R. Expanding the conformational selection paradigm in protein-ligand docking. Methods Mol. Biol. 2012, 819, 59–74. [Google Scholar] [CrossRef]
- Yang, Z.; West, A.P., Jr.; Bjorkman, P.J. Crystal structure of TNFα complexed with a poxvirus MHC-related TNF binding protein. Nat. Struct. Mol. Biol. 2009, 16, 1189–1191. [Google Scholar] [CrossRef]
- Hibi, M.; Murakami, M.; Saito, M.; Hirano, T.; Taga, T.; Kishimoto, T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 1990, 63, 1149–1157. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef]
- Novick, D.; Engelmann, H.; Wallach, D.; Rubinstein, M. Soluble cytokine receptors are present in normal human urine. J. Exp. Med. 1989, 170, 1409–1414. [Google Scholar] [CrossRef]
- Honda, M.; Yamamoto, S.; Cheng, M.; Yasukawa, K.; Suzuki, H.; Saito, T.; Osugi, Y.; Tokunaga, T.; Kishimoto, T. Human soluble IL-6 receptor: Its detection and enhanced release by HIV infection. J. Immunol. 1992, 148, 2175–2180. [Google Scholar]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: Generation and biological function. Biochem. J. 1994, 300, 281–290. [Google Scholar]
- Jones, S.A.; Richards, P.J.; Scheller, J.; Rose-John, S. IL-6 transsignaling: The in vivo consequences. J. Interf. Cytokine Res. 2005, 25, 241–253. [Google Scholar] [CrossRef]
- Scheller, J.; Ohnesorge, N.; Rose-John, S. Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand. J. Immunol. 2006, 63, 321–329. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Dann, S.M.; Spehlmann, M.E.; Hammond, D.C.; Iimura, M.; Hase, K.; Choi, L.J.; Hanson, E.; Eckmann, L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 2008, 180, 6816–6826. [Google Scholar]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-β suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef]
- Barkhausen, T.; Tschernig, T.; Rosenstiel, P.; van Griensven, M.; Vonberg, R.P.; Dorsch, M.; Mueller-Heine, A.; Chalaris, A.; Scheller, J.; Rose-John, S.; et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 2011, 39, 1407–1413. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Ara, T.; Declerck, Y.A. Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer 2010, 46, 1223–1231. [Google Scholar] [CrossRef]
- Jee, S.H.; Chu, C.Y.; Chiu, H.C.; Huang, Y.L.; Tsai, W.L.; Liao, Y.H.; Kuo, M.L. Interleukin-6 induced basic fibroblast growth factor-dependent angiogenesis in basal cell carcinoma cell line via JAK/STAT3 and PI3-kinase/AKT pathways. J. Investig. Dermatol. 2004, 123, 1169–1175. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]
- Kastritis, E.; Charidimou, A.; Varkaris, A.; Dimopoulos, M.A. Targeted therapies in multiple myeloma. Target. Oncol. 2009, 4, 23–36. [Google Scholar] [CrossRef]
- Adachi, Y.; Yoshio-Hoshino, N.; Nishimoto, N. The blockade of IL-6 signaling in rational drug design. Curr. Pharm. Des. 2008, 14, 1217–1224. [Google Scholar] [CrossRef]
- Wallner, L.; Dai, J.; Escara-Wilke, J.; Zhang, J.; Yao, Z.; Lu, Y.; Trikha, M.; Nemeth, J.A.; Zaki, M.H.; Keller, E.T. Inhibition of interleukin-6 with cnto328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006, 66, 3087–3095. [Google Scholar] [CrossRef]
- Savino, R.; Ciapponi, L.; Lahm, A.; Demartis, A.; Cabibbo, A.; Toniatti, C.; Delmastro, P.; Altamura, S.; Ciliberto, G. Rational design of a receptor super-antagonist of human interleukin-6. EMBO J. 1994, 13, 5863–5870. [Google Scholar]
- Sato, K.; Tsuchiya, M.; Saldanha, J.; Koishihara, Y.; Ohsugi, Y.; Kishimoto, T.; Bendig, M.M. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res. 1993, 53, 851–856. [Google Scholar]
- Mihara, M.; Kasutani, K.; Okazaki, M.; Nakamura, A.; Kawai, S.; Sugimoto, M.; Matsumoto, Y.; Ohsugi, Y. Tocilizumab inhibits signal transduction mediated by both mIL-6r and sIL-6r, but not by the receptors of other members of IL-6 cytokine family. Int. Immunopharmacol. 2005, 5, 1731–1740. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. Cosmic: Mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Tanikawa, T.; Wilke, C.M.; Kryczek, I.; Chen, G.Y.; Kao, J.; Nunez, G.; Zou, W. Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Res. 2012, 72, 420–429. [Google Scholar] [CrossRef]
- Sabat, R.; Grutz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef]
- Asadullah, K.; Sterry, W.; Volk, H.D. Interleukin-10 therapy—Review of a new approach. Pharmacol. Rev. 2003, 55, 241–269. [Google Scholar] [CrossRef]
- Acuner Ozbabacan, S.E.; Engin, H.B.; Guven Maiorov, E.; Kuzu, G.; Muratcioglu, S.; Baspinar, A.; Chen, Z.; van Vaes, C.; Gursoy, A.; Keskin, O.; et al. The structural network of interleukin-10 and its implications in inflammation and cancer. BMC Genomics 2014, in press. [Google Scholar]
- Yoon, S.I.; Logsdon, N.J.; Sheikh, F.; Donnelly, R.P.; Walter, M.R. Conformational changes mediate interleukin-10 receptor 2 (IL-10r2) binding to IL-10 and assembly of the signaling complex. J. Biol. Chem. 2006, 281, 35088–35096. [Google Scholar]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022. [Google Scholar] [CrossRef]
- Massague, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β targeted cancer therapy. Int. J. Biolog. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef]
- Randal, M.; Kossiakoff, A.A. The structure and activity of a monomeric interferon-gamma: Alpha-chain receptor signaling complex. Structure 2001, 9, 155–163. [Google Scholar] [CrossRef]
- Thiel, D.J.; le Du, M.H.; Walter, R.L.; D’Arcy, A.; Chene, C.; Fountoulakis, M.; Garotta, G.; Winkler, F.K.; Ealick, S.E. Observation of an unexpected third receptor molecule in the crystal structure of human interferon-gamma receptor complex. Structure 2000, 8, 927–936. [Google Scholar] [CrossRef]
- Nuara, A.A.; Walter, L.J.; Logsdon, N.J.; Yoon, S.I.; Jones, B.C.; Schriewer, J.M.; Buller, R.M.; Walter, M.R. Structure and mechanism of IFN-gamma antagonism by an orthopoxvirus IFN-gamma-binding protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1861–1866. [Google Scholar] [CrossRef]
- Tarasova, N.I.; Trinchieri, G.; Young, H.A.; Stewart, C.A.; Cardone, M.A.; Perantoni, A.O. Peptide-based inhibitor of interleukin-10 or interferon-gamma signaling. Google Patents US 20130109619 A1, 2 May 2013. [Google Scholar]
- Ghosh, S.; Chaudhary, R.; Carpani, M.; Playford, R. Interfering with interferons in inflammatory bowel disease. Gut 2006, 55, 1071–1073. [Google Scholar]
- Bernstein, C.N.; Blanchard, J.F.; Kliewer, E.; Wajda, A. Cancer risk in patients with inflammatory bowel disease: A population-based study. Cancer 2001, 91, 854–862. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guven-Maiorov, E.; Acuner-Ozbabacan, S.E.; Keskin, O.; Gursoy, A.; Nussinov, R. Structural Pathways of Cytokines May Illuminate Their Roles in Regulation of Cancer Development and Immunotherapy. Cancers 2014, 6, 663-683. https://doi.org/10.3390/cancers6020663
Guven-Maiorov E, Acuner-Ozbabacan SE, Keskin O, Gursoy A, Nussinov R. Structural Pathways of Cytokines May Illuminate Their Roles in Regulation of Cancer Development and Immunotherapy. Cancers. 2014; 6(2):663-683. https://doi.org/10.3390/cancers6020663
Chicago/Turabian StyleGuven-Maiorov, Emine, Saliha Ece Acuner-Ozbabacan, Ozlem Keskin, Attila Gursoy, and Ruth Nussinov. 2014. "Structural Pathways of Cytokines May Illuminate Their Roles in Regulation of Cancer Development and Immunotherapy" Cancers 6, no. 2: 663-683. https://doi.org/10.3390/cancers6020663