Replicative Stress and the FHIT Gene: Roles in Tumor Suppression, Genome Stability and Prevention of Carcinogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
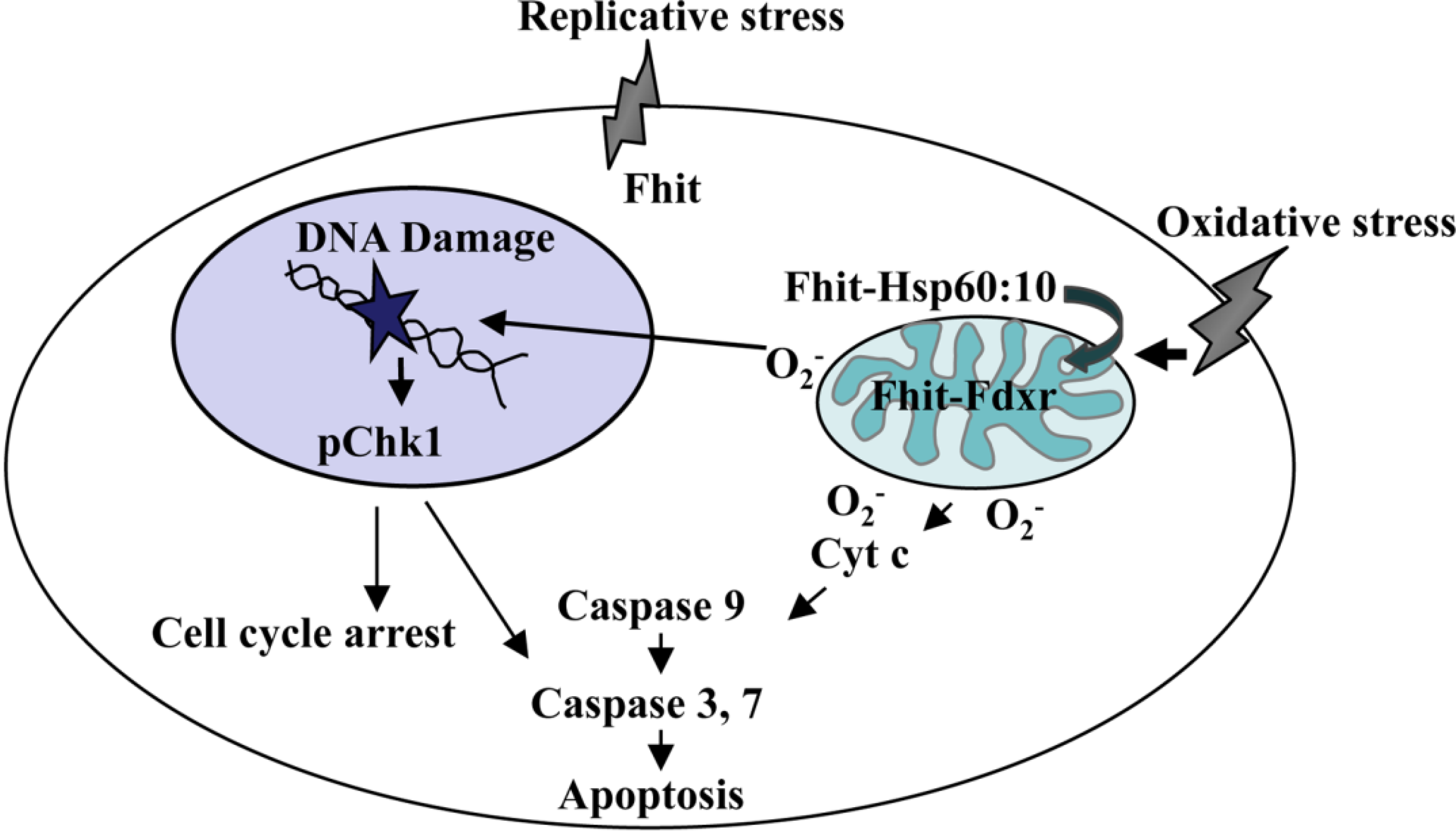
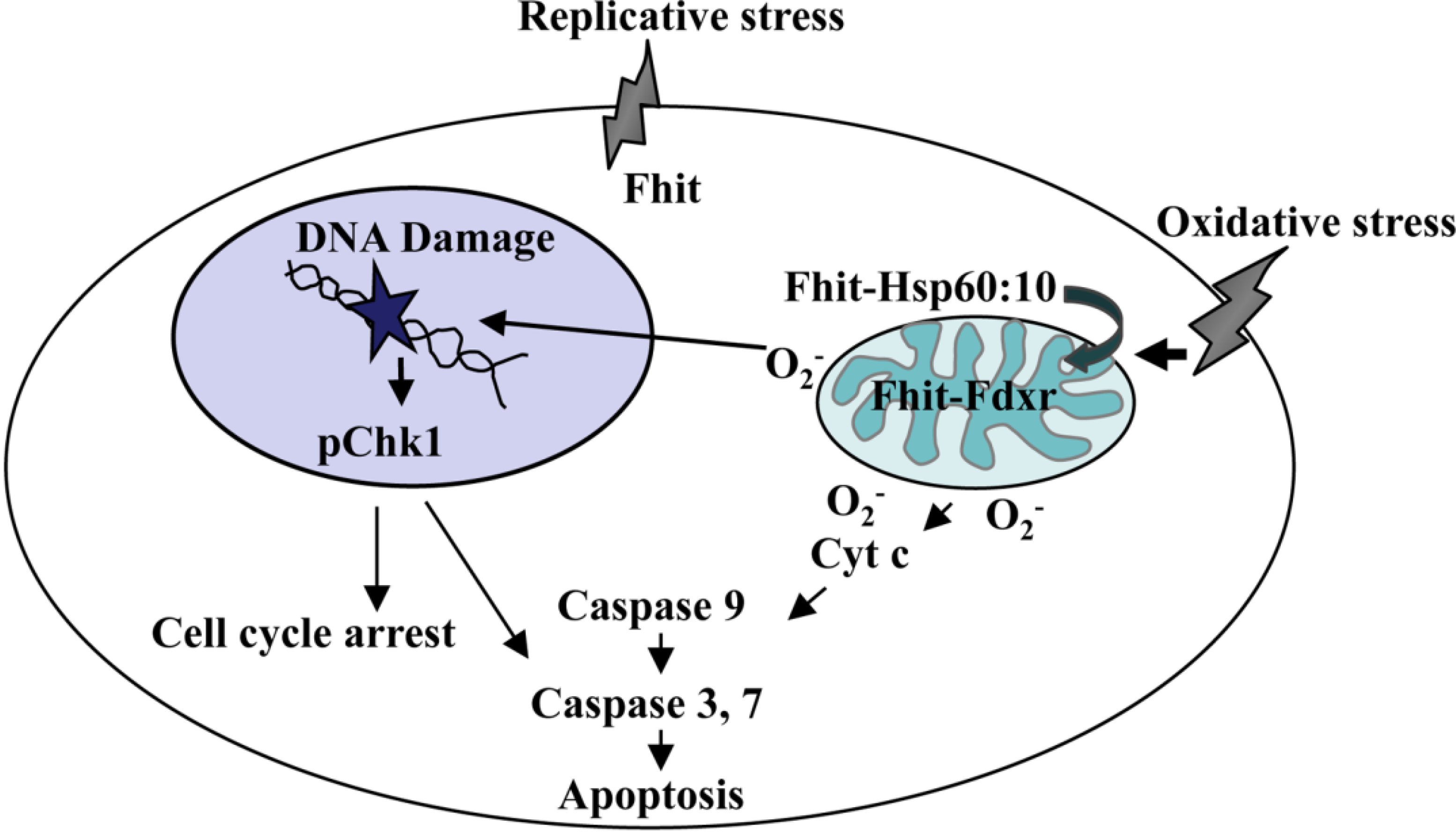
2. Oxidative Stress and the Mitochondrial Fraction of FHIT Protein
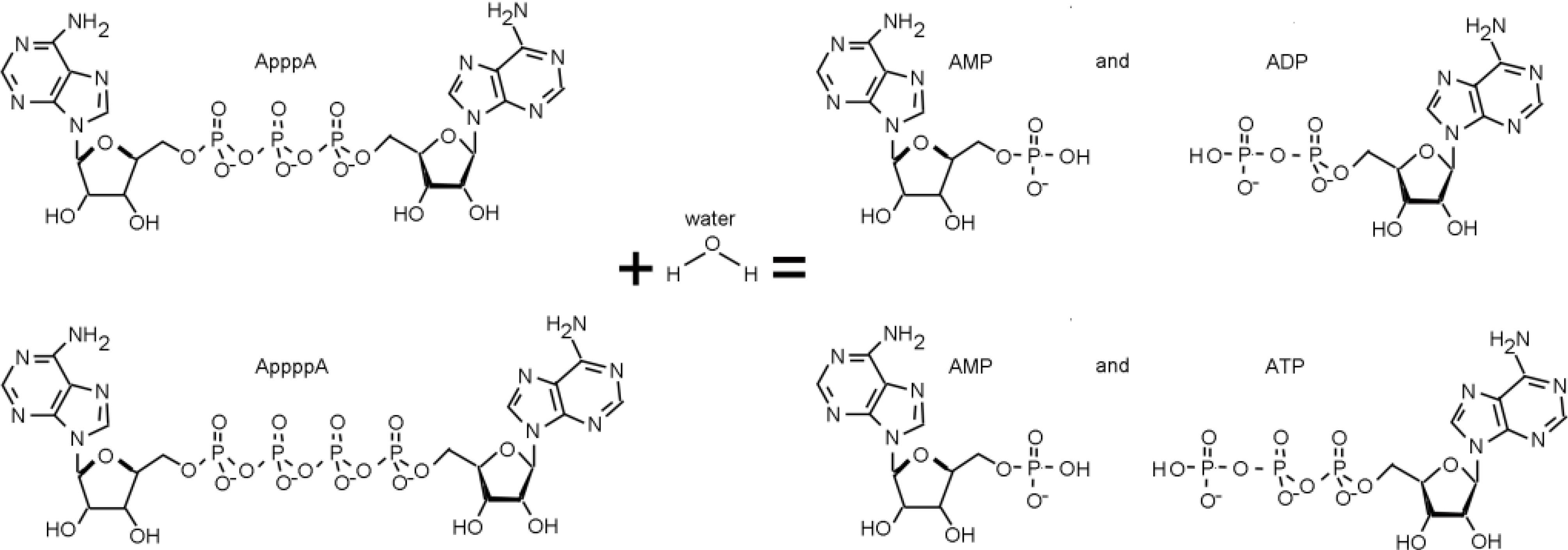
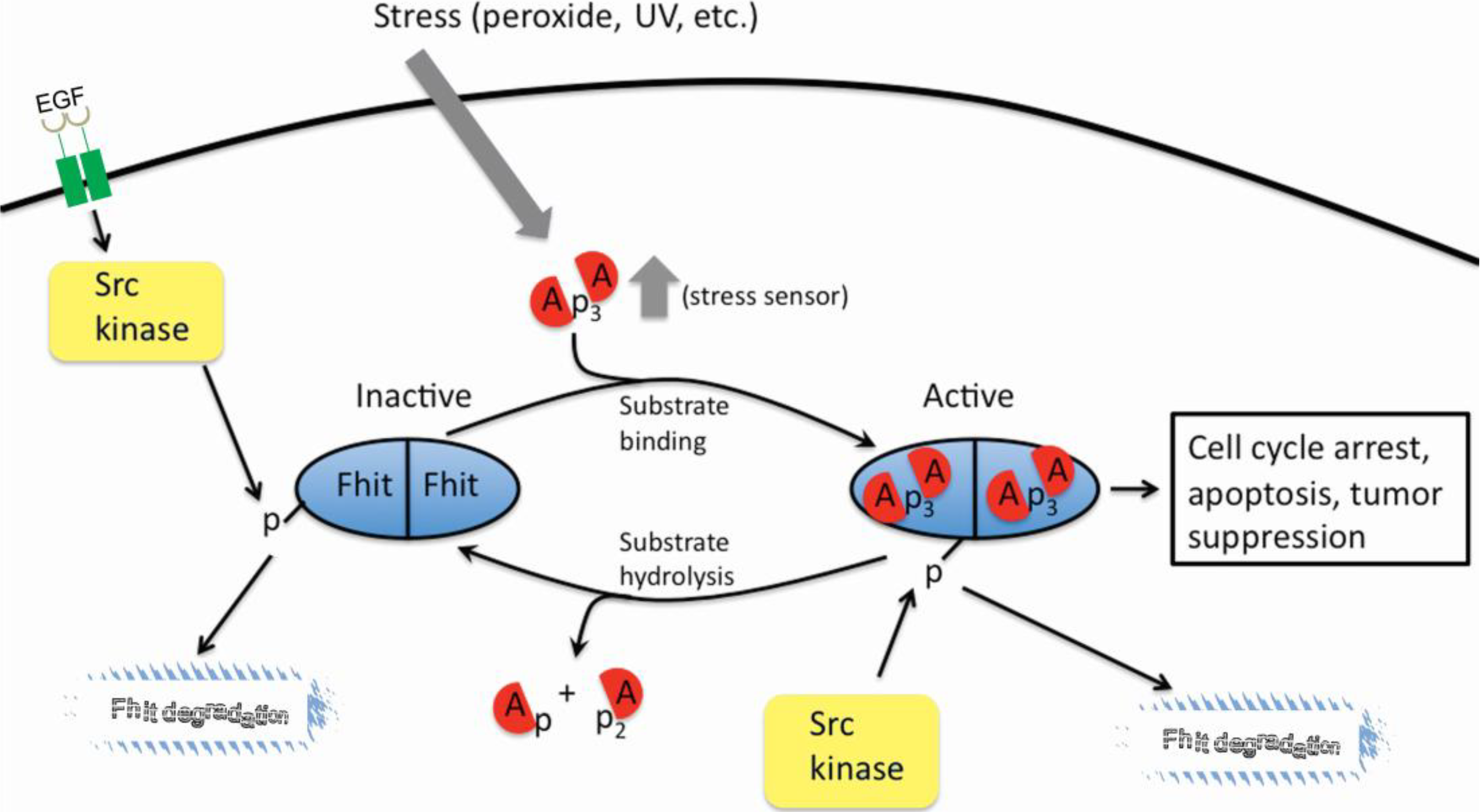
3. The FHIT-Substrate Complex
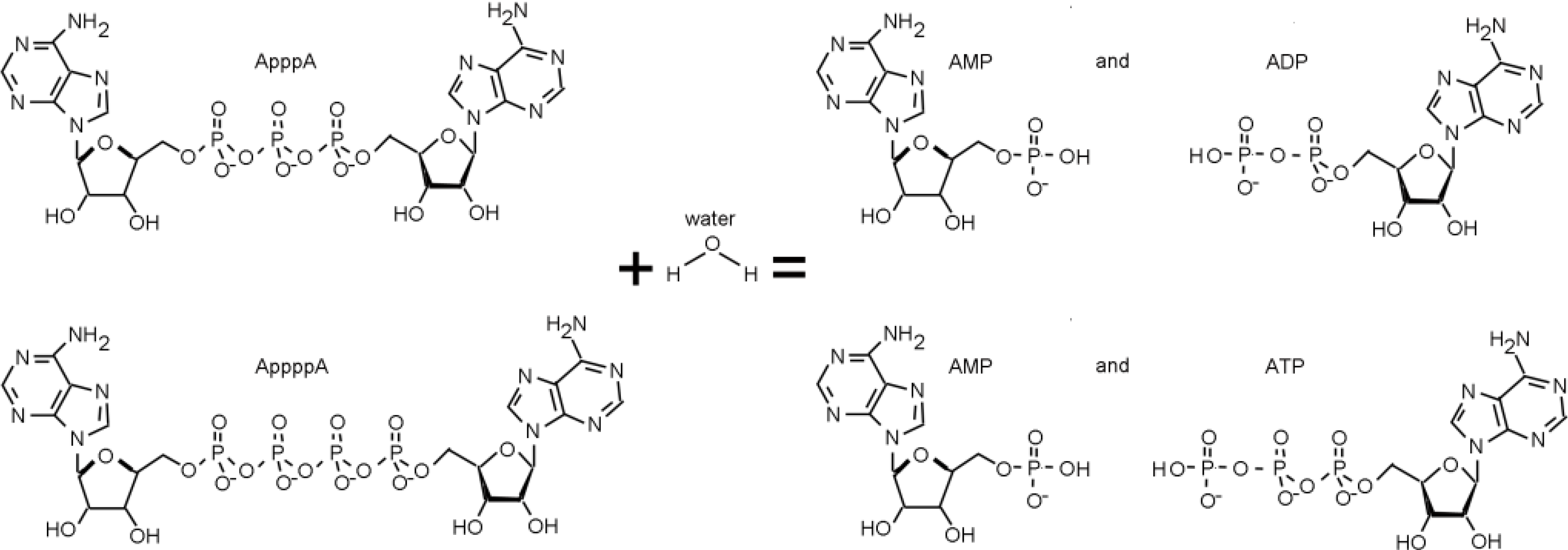

4. FHIT and Genome Stability

5. Oxidative Stress, ROS, and FHIT
6. ROS, Mutations and FHIT
7. Discussion
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bayani, J.; Paderova, J.; Murphy, J.; Rosen, B.; Zielenska, M.; Squire, J.A. Distinct patterns of structural and numerical chromosomal instability characterize sporadic ovarian cancer. Neoplasia 2008, 10, 1057–1065. [Google Scholar]
- Gagos, S.; Irminger-Finger, I. Chromosome instability in neoplasia: Chaotic roots to continuous growth. Int. J. Biochem. Cell Biol. 2005, 37, 1014–1033. [Google Scholar] [CrossRef]
- Huebner, K.; Garrison, P.N.; Barnes, L.D.; Croce, C.M. The role of the FHIT/FRA3B locus in cancer. Annu. Rev. Genet. 1998, 32, 7–31. [Google Scholar] [CrossRef]
- Huebner, K.; Croce, C.M. FRA3B and other common fragile sites: The weakest links. Nat. Rev. Cancer 2001, 1, 214–221. [Google Scholar] [CrossRef]
- Palumbo, E.; Tosoni, E.; Matricardi, L.; Russo, A. Genetic instability of the tumor suppressor gene FHIT in normal human cells. Genes Chromosomes Cancer 2013, 52, 832–844. [Google Scholar] [CrossRef]
- Siprashvili, Z.; Sozzi, G.; Barnes, L.D.; McCue, P.; Robinson, A.K.; Eryomin, V.; Sard, L.; Tagliabue, E.; Greco, A.; Fusetti, L.; et al. Replacement of FHIT in cancer cells suppresses tumorigenicity. Proc. Natl. Acad. Sci. USA 1997, 94, 13771–13776. [Google Scholar] [CrossRef]
- Sozzi, G.; Pastorino, U.; Moiraghi, L.; Tagliabue, E.; Pezzella, F.; Ghirelli, C.; Tornielli, S.; Sard, L.; Huebner, K.; Pierotti, M.A.; et al. Loss of FHIT function in lung cancer and preinvasive bronchial lesions. Cancer Res. 1998, 58, 5032–5037. [Google Scholar]
- Saldivar, J.C.; Miuma, S.; Bene, J.; Hosseini, S.A.; Shibata, H.; Sun, J.; Wheeler, L.J.; Mathews, C.K.; Huebner, K. Initiation of genome instability and preneoplastic processes through loss of FHIT expression. PLoS Genet. 2012, 8, e1003077. [Google Scholar]
- Trapasso, F.; Pichiorri, F.; Gaspari, M.; Palumbo, T.; Aqeilan, R.I.; Gaudio, E.; Okumura, H.; Iuliano, R.; di Leva, G.; Fabbri, M.; et al. FHIT interaction with ferredoxin reductase triggers generation of reactive oxygen species and apoptosis of cancer cells. J. Biol. Chem. 2008, 283, 13736–13744. [Google Scholar] [CrossRef]
- Miuma, S.; Saldivar, J.C.; Karras, J.R.; Waters, C.E.; Paisie, C.A.; Wang, Y.; Jin, V.; Sun, J.; Druck, T.; Zhang, J.; et al. FHIT deficiency-induced global genome instability promotes mutation and clonal expansion. PLoS One 2013, 8, e80730. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Pichiorri, F.; Palumbo, T.; Suh, S.S.; Okamura, H.; Trapasso, F.; Ishii, H.; Huebner, K.; Croce, C.M. FHIT tumor suppressor: Guardian of the preneoplastic genome. Future Oncol. 2008, 4, 815–824. [Google Scholar] [CrossRef]
- Ohta, M.; Inoue, H.; Cotticelli, M.G.; Kastury, K.; Baffa, R.; Palazzo, J.; Siprashvili, Z.; Mori, M.; McCue, P.; Druck, T.; et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996, 84, 587–597. [Google Scholar]
- Croce, C.M.; Sozzi, G.; Huebner, K. Role of FHIT in human cancer. J. Clin. Oncol. 1999, 17, 1618–1624. [Google Scholar]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef]
- Zanesi, N.; Fidanza, V.; Fong, L.Y.; Mancini, R.; Druck, T.; Valtieri, M.; Rüdiger, T.; McCue, P.A.; Croce, C.M.; Huebner, K. The tumor spectrum in FHIT-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10250–10255. [Google Scholar] [CrossRef]
- Fong, L.Y.; Fidanza, V.; Zanesi, N.; Lock, L.F.; Siracusa, L.D.; Mancini, R.; Siprashvili, Z.; Ottey, M.; Martin, S.E.; Druck, T.; et al. Muir-Torre-like syndrome in FHIT-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4742–4747. [Google Scholar] [CrossRef]
- Dumon, K.R.; Ishii, H.; Fong, L.Y.; Zanesi, N.; Fidanza, V.; Mancini, R.; Vecchione, A.; Baffa, R.; Trapasso, F.; During, M.J.; et al. FHIT gene therapy prevents tumor development in FHIT-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3346–3351. [Google Scholar] [CrossRef]
- Ishii, H.; Zanesi, N.; Vecchione, A.; Trapasso, F.; Yendamuri, S.; Sarti, M.; Baffa, R.; During, M.J.; Huebner, K.; Fong, L.Y.; et al. Regression of upper gastric cancer in mice by FHIT gene delivery. FASEB J. 2003, 17, 1768–1770. [Google Scholar]
- Rimessi, A.; Marchi, S.; Fotino, C.; Romagnoli, A.; Huebner, K.; Croce, C.M.; Pinton, P.; Rizzuto, R. Intramitochondrial calcium regulation by the FHIT gene product sensitizes to apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 12753–12758. [Google Scholar] [CrossRef]
- Pichiorri, F.; Okumura, H.; Nakamura, T.; Garrison, P.N.; Gasparini, P.; Suh, S.S.; Druck, T.; McCorkell, K.A.; Barnes, L.D.; Croce, C.M.; et al. Correlation of fragile histidine triad (FHIT) protein structural features with effector interactions and biological functions. J. Biol. Chem. 2009, 284, 1040–1049. [Google Scholar] [CrossRef]
- Barnes, L.D.; Garrison, P.N.; Siprashvili, Z.; Guranowski, A.; Robinson, A.K.; Ingram, S.W.; Croce, C.M.; Ohta, M.; Huebner, K. FHIT, a putative tumor suppressor in humans, is a dinucleoside 5',5"'-P1,P3-triphosphate hydrolase. Biochemistry 1996, 35, 11529–11535. [Google Scholar] [CrossRef]
- Brenner, C.; Pace, H.C.; Garrison, P.N.; Robinson, A.K.; Rosler, A.; Liu, X.H.; Blackburn, G.M.; Croce, C.M.; Huebner, K.; Barnes, L.D. Purification and crystallization of complexes modeling the active state of the fragile histidine triad protein. Protein Eng. 1997, 10, 1461–1463. [Google Scholar] [CrossRef]
- Trapasso, F.; Krakowiak, A.; Cesari, R.; Arkles, J.; Yendamuri, S.; Ishii, H.; Vecchione, A.; Kuroki, T.; Bieganowski, P.; Pace, H.C.; et al. Designed FHIT alleles establish that FHIT-induced apoptosis in cancer cells is limited by substrate binding. Proc. Natl. Acad. Sci. USA 2003, 100, 1592–1597. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Garrison, P.N.; Palamarchuk, A.; Zanesi, N.; Aqeilan, R.I.; Huebner, K.; Barnes, L.D.; Croce, C.M. FHIT is a physiological target of the protein kinase Src. Proc. Natl. Acad. Sci. USA 2004, 101, 3775–3779. [Google Scholar] [CrossRef]
- Bianchi, F.; Magnifico, A.; Olgiati, C.; Zanesi, N.; Pekarsky, Y.; Tagliabue, E.; Croce, C.M.; Menard, S.; Campiglio, M. FHIT-proteasome degradation caused by mitogenic stimulation of the EGF receptor family in cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 18981–18986. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Bene, J.; Hosseini, S.A.; Miuma, S.; Horton, S.; Heerema, N.A.; Huebner, K. Characterization of the role of FHIT in suppression of DNA damage. Adv. Biol. Regul. 2013, 53, 77–85. [Google Scholar] [CrossRef]
- Nisman, B.; Kadouri, L.; Allweis, T.; Maly, B.; Hamburger, T.; Gronowitz, S.; Peretz, T. Increased proliferative background in healthy women with BRCA1/2 haploinsufficiency is associated with high risk for breast cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2110–2115. [Google Scholar] [CrossRef]
- Okumura, H.; Ishii, H.; Pichiorri, F.; Croce, C.M.; Mori, M.; Huebner, K. Fragile gene product, FHIT, in oxidative and replicative stress responses. Cancer Sci. 2009, 100, 1145–1150. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar]
- Bacolla, A.; Cooper, D.N.; Vasquez, K.M. Mechanisms of base substitution mutagenesis in cancer genomes. Genes 2014, 5, 108–146. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Li, J.; Braganza, A.; Sobol, R.W. Base excision repair facilitates a functional relationship between Guanine oxidation and histone demethylation. Antioxid. Redox Signal. 2013, 18, 2429–2443. [Google Scholar] [CrossRef]
- Jascur, T.; Boland, C.R. Structure and function of the components of the human DNA mismatch repair system. Int. J. Cancer 2006, 119, 2030–2035. [Google Scholar] [CrossRef]
- Weill, J.C.; Reynaud, C.A. DNA polymerases in adaptive immunity. Nat. Rev. Immunol. 2008, 8, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Couronne, L.; Ruminy, P.; Waultier-Rascalou, A.; Rainville, V.; Cornic, M.; Picquenot, J.M.; Figeac, M.; Bastard, C.; Tilly, H.; Jardin, F. Mutation mismatch repair gene deletions in diffuse large B-cell lymphoma. Leuk. Lymphoma 2013, 54, 1079–1086. [Google Scholar] [CrossRef]
- Kreutzer, D.A.; Essigmann, J.M. Oxidized, deaminated cytosines are a source of C → T transitions in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 3578–3582. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Karras, J.R.; Paisie, C.A.; Huebner, K. Replicative Stress and the FHIT Gene: Roles in Tumor Suppression, Genome Stability and Prevention of Carcinogenesis. Cancers 2014, 6, 1208-1219. https://doi.org/10.3390/cancers6021208
Karras JR, Paisie CA, Huebner K. Replicative Stress and the FHIT Gene: Roles in Tumor Suppression, Genome Stability and Prevention of Carcinogenesis. Cancers. 2014; 6(2):1208-1219. https://doi.org/10.3390/cancers6021208
Chicago/Turabian StyleKarras, Jenna R., Carolyn A. Paisie, and Kay Huebner. 2014. "Replicative Stress and the FHIT Gene: Roles in Tumor Suppression, Genome Stability and Prevention of Carcinogenesis" Cancers 6, no. 2: 1208-1219. https://doi.org/10.3390/cancers6021208