Inflammatory Cell Distribution in Primary Merkel Cell Carcinoma

Abstract
:1. Introduction
2. Results and Discussion
2.1. Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Age at diagnosis | Site of
primary tumour | MCC stage | Tumour MCPyV LT status | Serum IgG VP1/LTA | Survival time (m) |
|---|---|---|---|---|---|---|---|
| P16 | F | 73 | Upper limb | I | + | nd | 101 a |
| P17 | F | 79 | Lower limb | I | − | − | 78 |
| P21 | F | 79 | Lower limb | I | − | nd | 11 |
| P22 | M | 77 | Lower limb | II | + | nd | 99 |
| P25 | F | 84 | Head&neck | III | − | nd | 22 |
| P26 | F | 78 | Head&neck | II | + | nd | 136 |
| P27 | F | 84 | Lower limb | II | − | nd | 41 |
| P30 | M | 87 | Head&neck | II | − | nd | 11 |
| P43 | M | 82 | Lower limb | II | − | + | 14 |
| P45 | M | 63 | Lower limb | III | − | nd | 10 |
| P46 | M | 56 | Lower limb | II | + | + | 12 |
| P50 | F | 90 | Head&neck | I | − | − | 3 |
| P53 | F | 70 | Upper limb | II | + | + | 46 a |
| P58 | M | 83 | Upper limb | I/II | − | nd | 19 |
| P60 | F | 75 | Upper limb | I/II | − | nd | 40 |
| P72 | M | 59 | Upper limb | II | + | + | 30 |
| P74 | F | 72 | Head&neck | I | − | nd | 40 |
| P77 | M | 66 | Trunk | II | + | + | 36 a |
| P79 | F | 68 | Upper limb | II | + | + | 30 a |
| P82 | M | 61 | Upper limb | I | + | + | 14 |
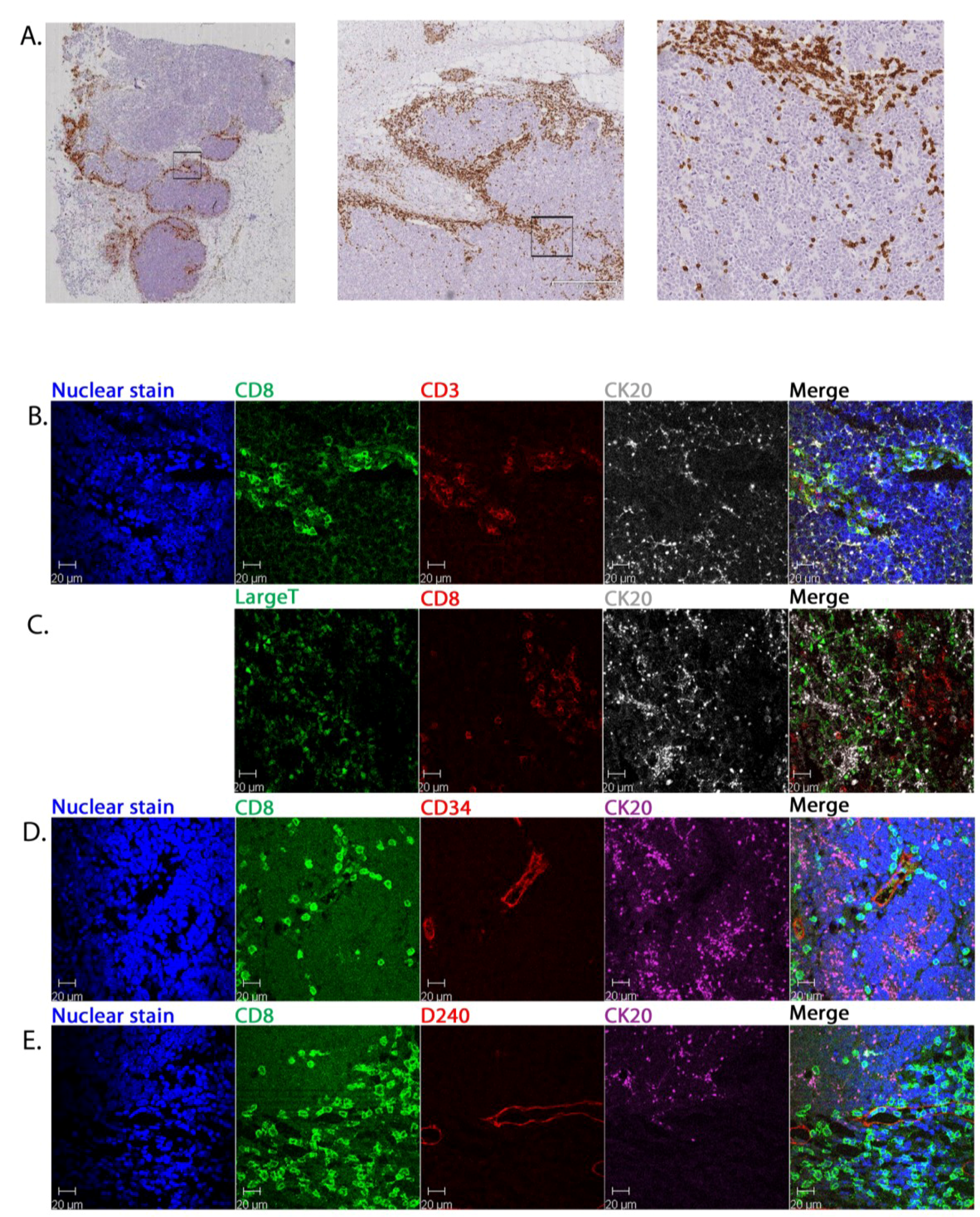
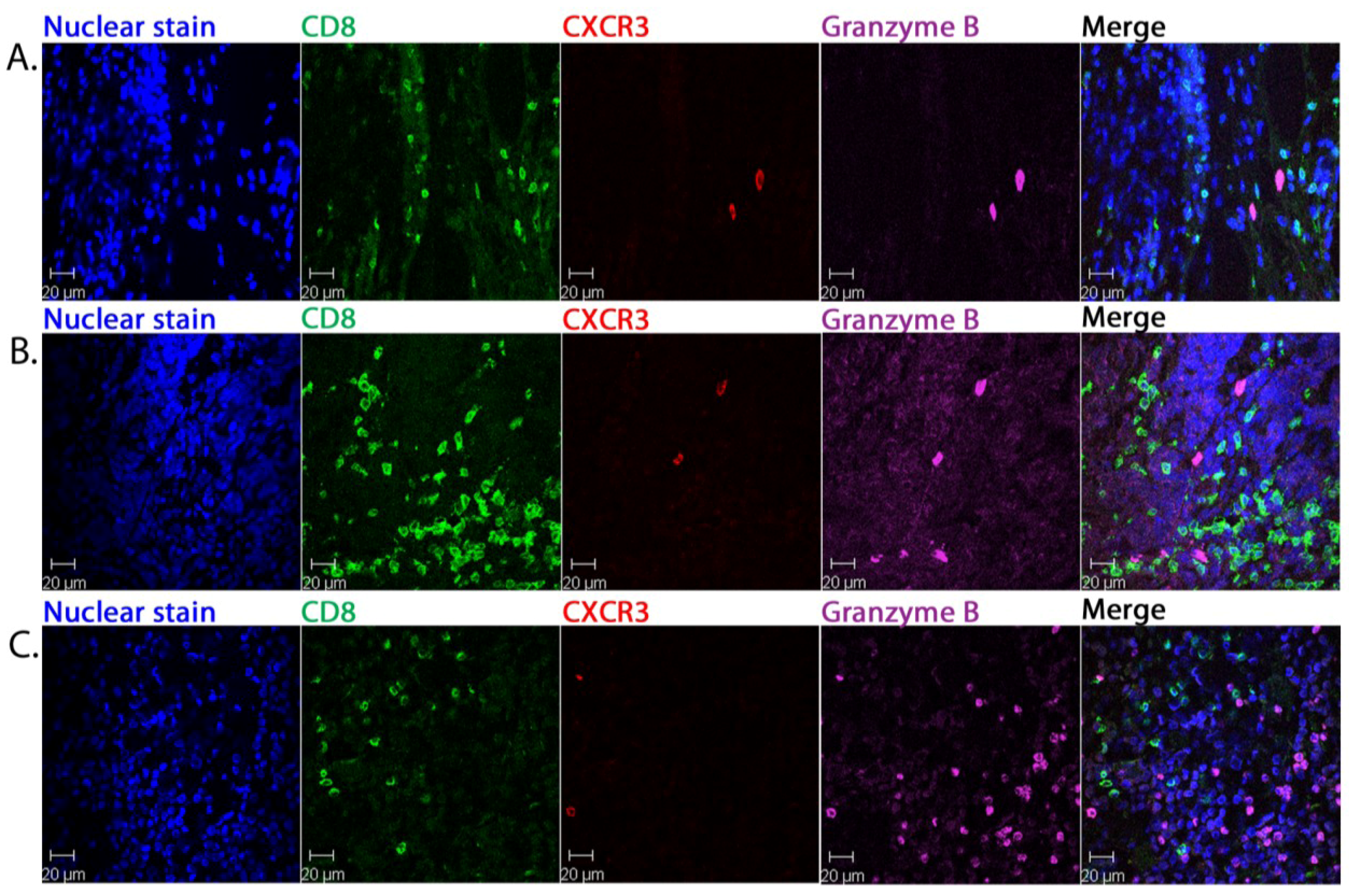
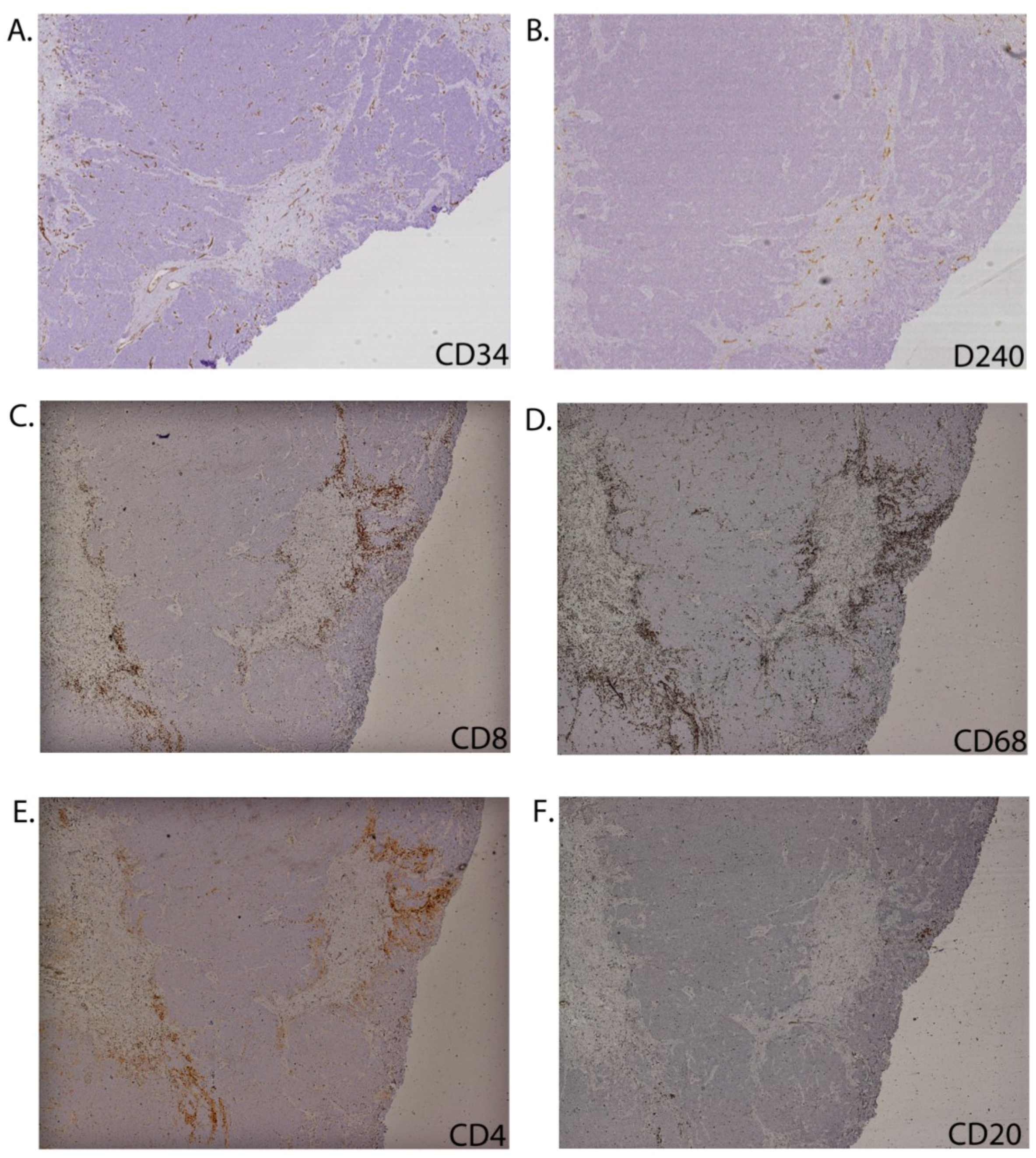
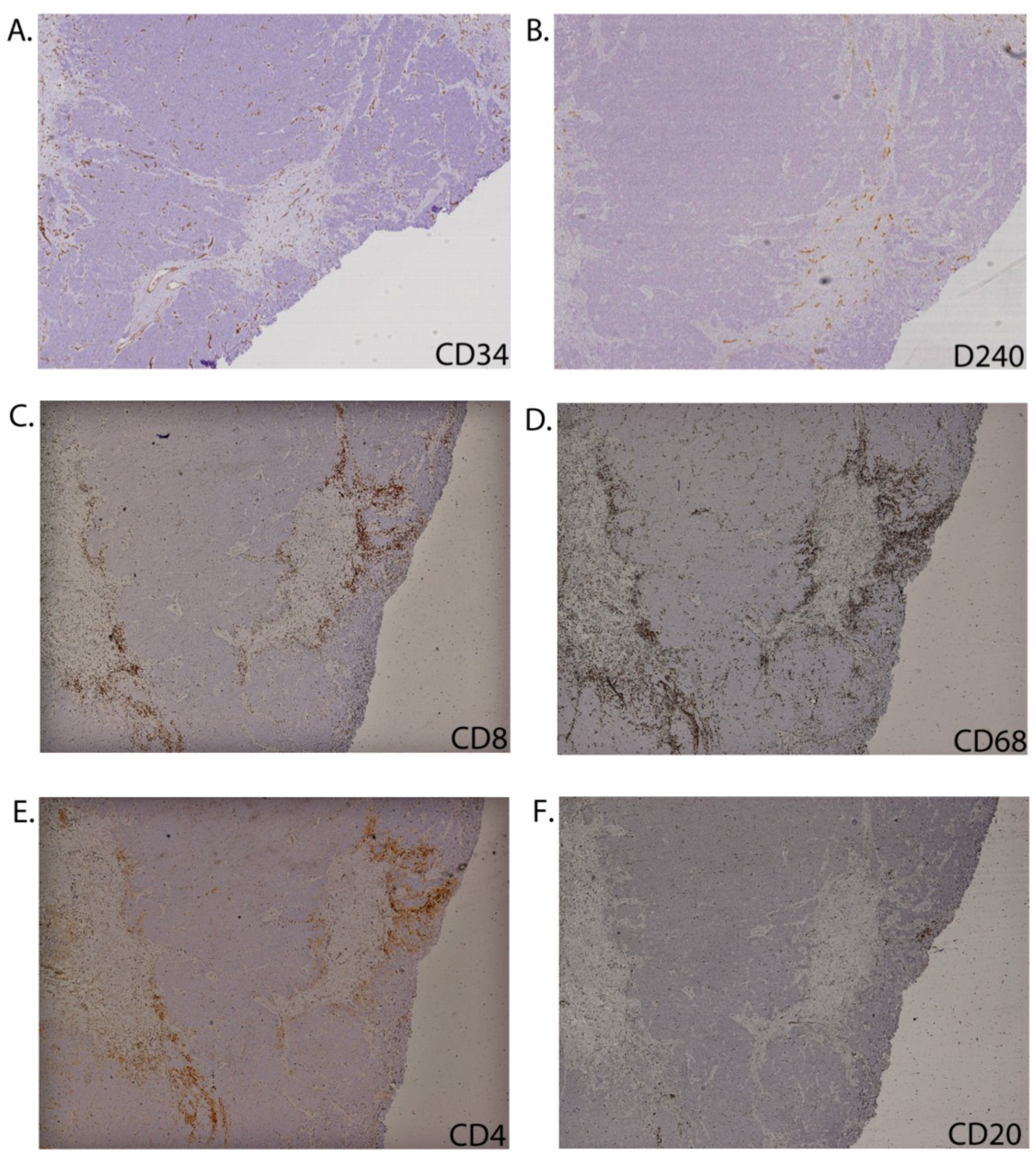
2.2. CD8+ Cell Phenotype
| ID | Vessels within malignant aggregates | Inflammatory cell abundance and distribution throughout tumour specimen | Functional properties of inflammatory cells | CXCL12/CXCR4 expression | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD34 blood | D240 lymphatic | CD8 | CD68 | CD4 | CD20 | Fox P3 | Dominant pattern of distribution | CXCR3 | GrB | CD8+GrB+ | CXCL12 | CXCR4 | |
| P16 | + | − | ++ | + | + | + | −/+ | M/Tr | |||||
| P17 | −/+ | − | −/+ | −/+ | − | − | − | M/Tr | −/+ | −/+ | − | ||
| P21 | + | − | − | − | −/+ | − | −/+ | M/Tr | |||||
| P22 | + | Focally + | + | + | + | + | −/+ | M/Tr | |||||
| P25 | + | − | + | + | + | − | −/+ | M | |||||
| P26 | + | − | + | + | + | + | −/+ | M | |||||
| P27 | + | − | −/+ | + | −/+ | − | −/+ | M | |||||
| P30 | + | − | + | + | + | + | −/+ | M/Tr | |||||
| P43 | + | − | −/+ | −/+ | + | −/+ | − | M | −/+ | −/+ | − | ||
| P45 | −/+ | − | + | + | M/Tr | − | + | − | |||||
| P46 | + | − | −/+ | + | −/+ | − | − | M | + | + | − | ||
| P50 | Focally + | − | + | + | Tr | −/+ | −/+ | − | |||||
| P53 | + | − | ++ | + | M | −/+ | −/+ | − | M | IT | |||
| P58 | + | − | ++ | ++ | M/Tr/IT | − | −/+ | − | M | IT | |||
| P60 | Focally + | − | + | + | M/Tr | −/+ | −/+ | − | |||||
| P72 | + | −/+ | + | + | M | −/+ | + | − | M | IT | |||
| P74 | −/+ | Focally + | + | + | M/Tr | − | −/+ | − | |||||
| P77 | + | − | + | + | M | −/+ | −/+ | − | M | IT | |||
| P79 | −/+ | − | + | + | M | −/+ | −/+ | − | |||||
| P82 | + | −/+ | ++ | + | M/Tr/IT | −/+ | −/+ | − | |||||
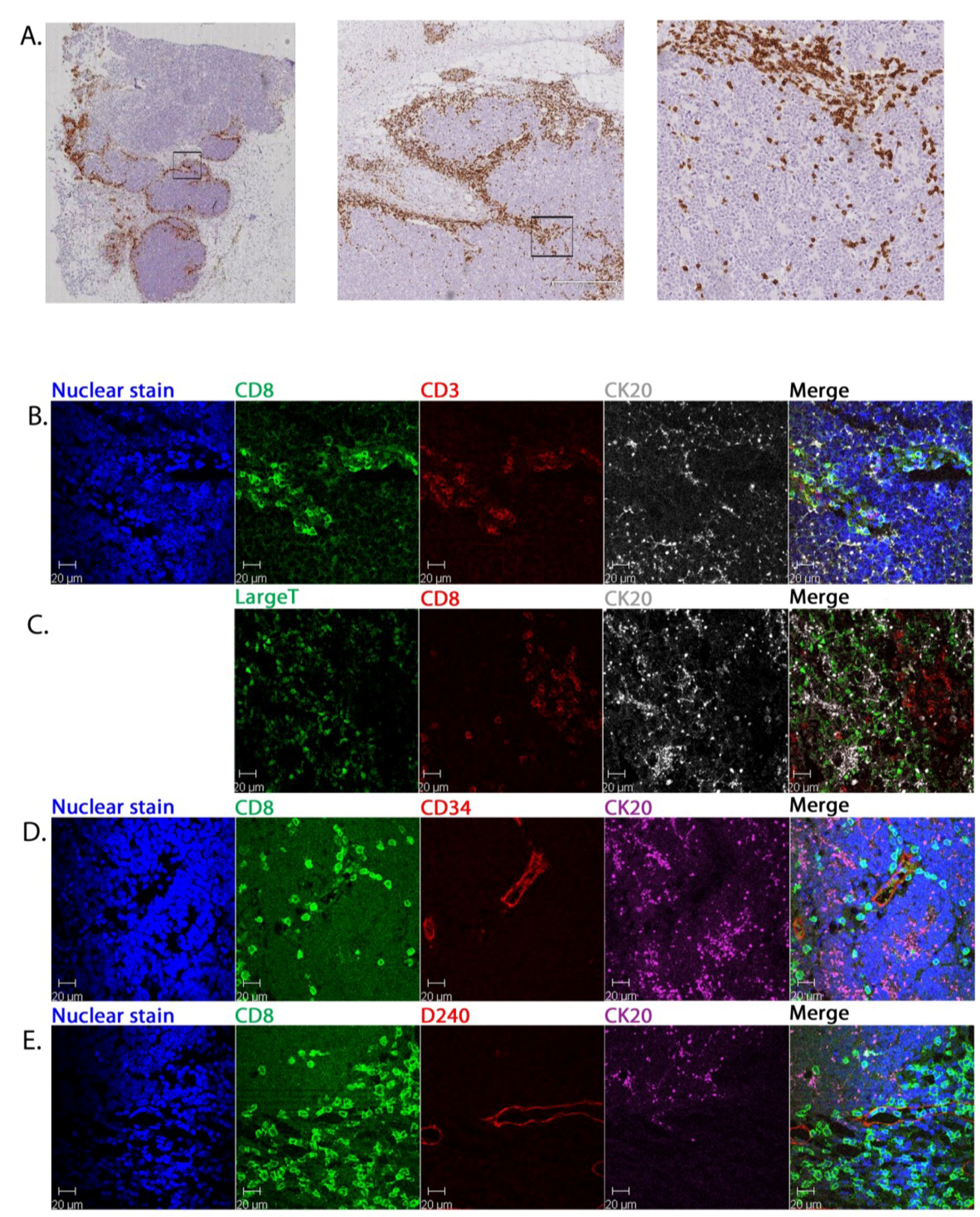

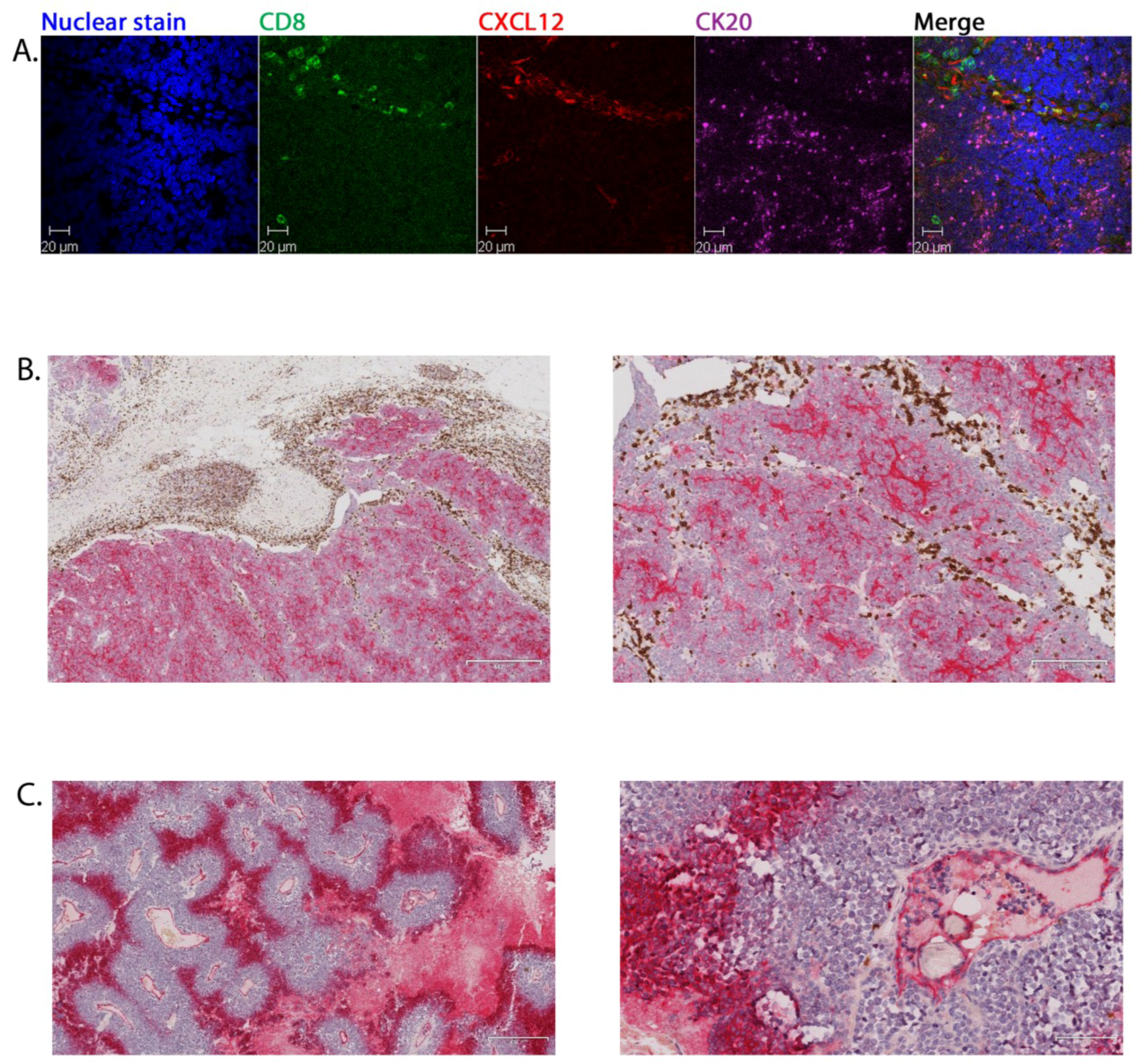
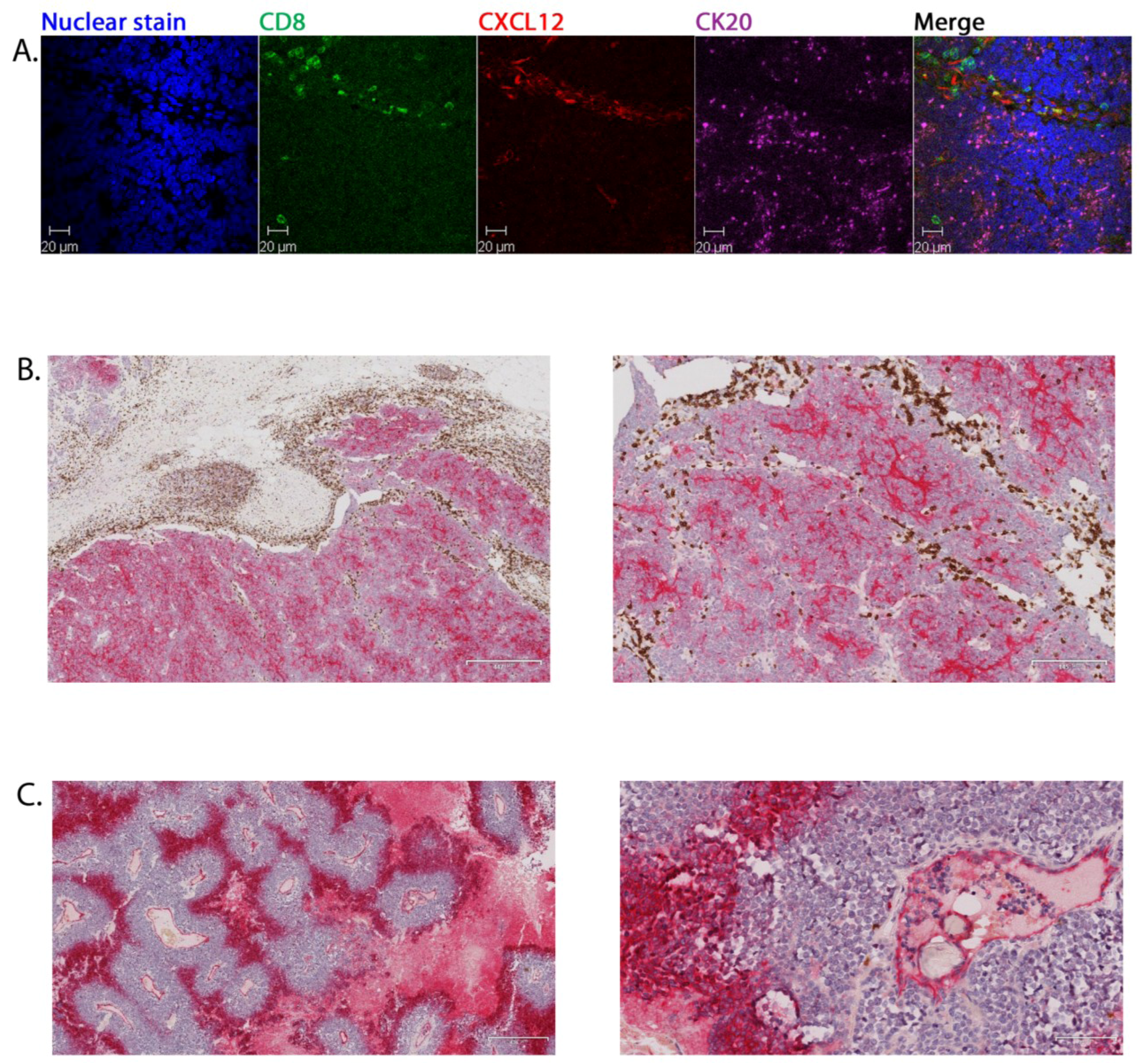
2.3. Tumour-Associated Inflammatory Cells
3. Experimental
3.1. Patients and Samples
3.2. Haematoxylin and Eosin (H&E) Staining
3.3. Conventional Immunohistochemistry (IHC)
3.4. Multicolour Immunohistochemistry
3.5. Dual-Colour Immunohistochemistry
3.6. MCPyV Serology
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bichakjian, C.K.; Lowe, L.; Lao, C.D.; Sandler, H.M.; Bradford, C.R.; Johnson, T.M.; Wong, S.L. Merkel cell carcinoma: Critical review with guidelines for multidisciplinary management. Cancer 2007, 110, 1–12. [Google Scholar] [CrossRef]
- Albores-Saavedra, J.; Batich, K.; Chable-Montero, F.; Sagy, N.; Schwartz, A.M.; Henson, D.E. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: A population based study. J. Cutan. Pathol. 2010, 37, 20–27. [Google Scholar] [CrossRef]
- Lemos, B.D.; Storer, B.E.; Iyer, J.G.; Phillips, J.L.; Bichakjian, C.K.; Fang, L.C.; Johnson, T.M.; Liegeois-Kwon, N.J.; Otley, C.C.; Paulson, K.G.; et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: Analysis of 5823 cases as the basis of the first consensus staging system. J. Am. Acad. Dermatol. 2010, 63, 751–761. [Google Scholar] [CrossRef]
- Allen, P.J.; Bowne, W.B.; Jaques, D.P.; Brennan, M.F.; Busam, K.; Coit, D.G. Merkel cell carcinoma: Prognosis and treatment of patients from a single institution. J. Clin. Oncol. 2005, 23, 2300–2309. [Google Scholar] [CrossRef]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef]
- Miller, R.W.; Rabkin, C.S. Merkel cell carcinoma and melanoma: Etiological similarities and differences. Cancer Epidemiol. Biol. Prev. 1999, 8, 153–158. [Google Scholar]
- Rodig, S.J.; Cheng, J.; Wardzala, J.; DoRosario, A.; Scanlon, J.J.; Laga, A.C.; Martinez-Fernandez, A.; Barletta, J.A.; Bellizzi, A.M.; Sadasivam, S.; et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J. Clin. Investig. 2012, 122, 4645–4653. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, F.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1049–1050. [Google Scholar] [CrossRef]
- Foulongne, V.; Kluger, N.; Dereure, O.; Brieu, N.; Guillot, B.; Segondy, M. Merkel cell polyomavirus and Merkel cell carcinoma, France. Emerg. Infect. Dis. 2008, 14, 1491–1493. [Google Scholar] [CrossRef]
- Garneski, K.M.; Warcola, A.H.; Feng, Q.; Kiviat, N.B.; Leonard, J.H.; Nghiem, P. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J. Investig. Dermatol. 2009, 129, 246–248. [Google Scholar] [CrossRef]
- Kantola, K.; Sadeghi, M.; Lahtinen, A.; Koskenvuo, M.; Aaltonen, L.M.; Mottonen, M.; Rahiala, J.; Saarinen-Pihkala, U.; Riikonen, P.; Jartti, T.; et al. Merkel cell polyomavirus DNA in tumor-free tonsillar tissues and upper respiratory tract samples: Implications for respiratory transmission and latency. J. Clin. Virol. 2009, 45, 292–295. [Google Scholar] [CrossRef]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.G.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.J.; Lai, I.; et al. Merkel cell polyomavirus-specific CD8+ and CD4+ T-cell responses identified in Merkel cell carcinomas and blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar] [CrossRef]
- Robertson, D.; Savage, K.; Reis-Filho, J.S.; Isacke, C.M. Multiple immunofluorescence labelling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell. Biol. 2008. [Google Scholar] [CrossRef]
- Paulson, K.G.; Iyer, J.G.; Tegeder, A.R.; Thibodeau, R.; Schelter, J.; Koba, S.; Schrama, D.; Simonson, W.T.; Lemos, B.D.; Byrd, D.R.; et al. Transcriptome-wide studies of Merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J. Clin. Oncol. 2011, 29, 1539–1546. [Google Scholar] [CrossRef]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in cancer and immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef]
- Kjaerheim, K.; Roe, O.D.; Waterboer, T.; Sehr, P.; Rizk, R.; Dai, H.Y.; Sandeck, H.; Larsson, E.; Andersen, A.; Boffetta, P.; et al. Absence of SV40 antibodies or DNA fragments in prediagnostic mesothelioma serum samples. Int. J. Cancer 2007, 120, 2459–2465. [Google Scholar] [CrossRef]
- Rollison, D.E.; Giuliano, A.R.; Messina, J.L.; Fenske, N.A.; Cherpelis, B.S.; Sondak, V.K.; Roetzheim, R.G.; Iannacone, M.R.; Michael, K.M.; Gheit, T.; et al. Case-control study of Merkel cell polyomavirus infection and cutaneous squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2012, 21, 74–81. [Google Scholar] [CrossRef]
- Inoue, T.; Yoneda, K.; Manabe, M.; Demitsu, T. Spontaneous regression of merkel cell carcinoma: A comparative study of TUNEL index and tumor-infiltrating lymphocytes between spontaneous regression and non-regression group. J. Dermatol. Sci. 2000, 24, 203–211. [Google Scholar] [CrossRef]
- Junquera, L.; Torre, A.; Vicente, J.C.; Garcia-Consuegra, L.; Fresno, M.F. Complete spontaneous regression of Merkel cell carcinoma. Ann. Otol. Rhinol. Laryngol. 2005, 114, 376–380. [Google Scholar]
- Richetta, A.G.; Mancini, M.; Torroni, A.; Lore, B.; Iannetti, G.; Sardella, B.; Calvieri, S. Total spontaneous regression of advanced merkel cell carcinoma after biopsy: Review and a new case. Dermatol. Surg. 2008, 34, 815–822. [Google Scholar] [CrossRef]
- Connelly, T. Regarding complete spontaneous regression of Merkel cell carcinoma. Dermatol. Surg. 2009, 35, 721. [Google Scholar] [CrossRef]
- Sihto, H.; Bohling, T.; Kavola, H.; Koljonen, V.; Salmi, M.; Jalkanen, S.; Joensuu, H. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: A population-based study. Clin. Cancer Res. 2012, 18, 2872–2881. [Google Scholar] [CrossRef]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar]
- Gomez, B.P.; Wang, C.; Viscidi, R.P.; Peng, S.; He, L.; Wu, T.C.; Hung, C.F. Strategy for eliciting antigen-specific CD8+ T cell-mediated immune response against a cryptic CTL epitope of merkel cell polyomavirus large T antigen. Cell Biosci. 2012, 2, 36. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar]
- Ewen, C.L.; Kane, K.P.; Bleackley, R.C. A quarter century of granzymes. Cell Death Differ. 2012, 19, 28–35. [Google Scholar] [CrossRef]
- Mullins, I.M.; Slingluff, C.L.; Lee, J.K.; Garbee, C.F.; Shu, J.; Anderson, S.G.; Mayer, M.E.; Knaus, W.A.; Mullins, D.W. CXC chemokine receptor 3 expression by activated CD8+ T cells is associated with survival in melanoma patients with stage III disease. Cancer Res. 2004, 64, 7697–7701. [Google Scholar] [CrossRef]
- Gorbachev, A.V.; Kobayashi, H.; Kudo, D.; Tannenbaum, C.S.; Finke, J.H.; Shu, S.; Farber, J.M.; Fairchild, R.L. CXC chemokine ligand 9/monokine induced by IFN-gamma production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors. J. Immunol. 2007, 178, 2278–2286. [Google Scholar] [CrossRef]
- Oldham, K.A.; Parsonage, G.; Bhatt, R.I.; Wallace, D.M.; Deshmukh, N.; Chaudhri, S.; Adams, D.H.; Lee, S.P. T lymphocyte recruitment into renal cell carcinoma tissue: A role for chemokine receptors CXCR3, CXCR6, CCR5, and CCR6. Eur. Urol. 2012, 61, 385–394. [Google Scholar] [CrossRef]
- Popple, A.; Durrant, L.G.; Spendlove, I.; Rolland, P.; Scott, I.V.; Deen, S.; Ramage, J.M. The chemokine, CXCL12, is an independent predictor of poor survival in ovarian cancer. Br. J. Can. 2012, 106, 1306–1313. [Google Scholar] [CrossRef]
- Faint, J.M.; Annels, N.E.; Curnow, S.J.; Shields, P.; Pilling, D.; Hislop, A.D.; Wu, L.; Akbar, A.N.; Buckley, C.D.; Moss, P.A.; et al. Memory T cells constitute a subset of the human CD8+CD45RA+ pool with distinct phenotypic and migratory characteristics. J. Immunol. 2001, 167, 212–220. [Google Scholar] [CrossRef]
- Zhang, T.; Somasundaram, R.; Berencsi, K.; Caputo, L.; Rani, P.; Guerry, D.; Furth, E.; Rollins, B.J.; Putt, M.; Gimotty, P.; et al. CXC chemokine ligand 12 (stromal cell-derived factor 1 alpha) and CXCR4-dependent migration of CTLs toward melanoma cells in organotypic culture. J. Immunol. 2005, 174, 5856–5863. [Google Scholar] [CrossRef]
- Goddard, S.; Williams, A.; Morland, C.; Qin, S.; Gladue, R.; Hubscher, S.G.; Adams, D.H. Differential expression of chemokines and chemokine receptors shapes the inflammatory response in rejecting human liver transplants. Transplantation 2001, 72, 1957–1967. [Google Scholar] [CrossRef]
- Poznansky, M.C.; Olszak, I.T.; Foxall, R.; Evans, R.H.; Luster, A.D.; Scadden, D.T. Active movement of T cells away from a chemokine. Nat. Med. 2000, 6, 543–548. [Google Scholar] [CrossRef]
- Burman, A.; Haworth, O.; Hardie, D.L.; Amft, E.N.; Siewert, C.; Jackson, D.G.; Salmon, M.; Buckley, C.D. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J. Immunol. 2005, 174, 1693–1700. [Google Scholar] [CrossRef]
- Zagzag, D.; Krishnamachary, B.; Yee, H.; Okuyama, H.; Chiriboga, L.; Ali, M.A.; Melamed, J.; Semenza, G.L. Stromal cell-derived factor-1alpha and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: Von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res. 2005, 65, 6178–6188. [Google Scholar] [CrossRef]
- Tang, X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013, 332, 3–10. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wheat, R.; Roberts, C.; Waterboer, T.; Steele, J.; Marsden, J.; Steven, N.M.; Blackbourn, D.J. Inflammatory Cell Distribution in Primary Merkel Cell Carcinoma. Cancers 2014, 6, 1047-1064. https://doi.org/10.3390/cancers6021047
Wheat R, Roberts C, Waterboer T, Steele J, Marsden J, Steven NM, Blackbourn DJ. Inflammatory Cell Distribution in Primary Merkel Cell Carcinoma. Cancers. 2014; 6(2):1047-1064. https://doi.org/10.3390/cancers6021047
Chicago/Turabian StyleWheat, Rachel, Claudia Roberts, Tim Waterboer, Jane Steele, Jerry Marsden, Neil M. Steven, and David J. Blackbourn. 2014. "Inflammatory Cell Distribution in Primary Merkel Cell Carcinoma" Cancers 6, no. 2: 1047-1064. https://doi.org/10.3390/cancers6021047