The Complex Relationship between Liver Cancer and the Cell Cycle: A Story of Multiple Regulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Liver and Cancer
1.1. Liver Anatomy and Regeneration
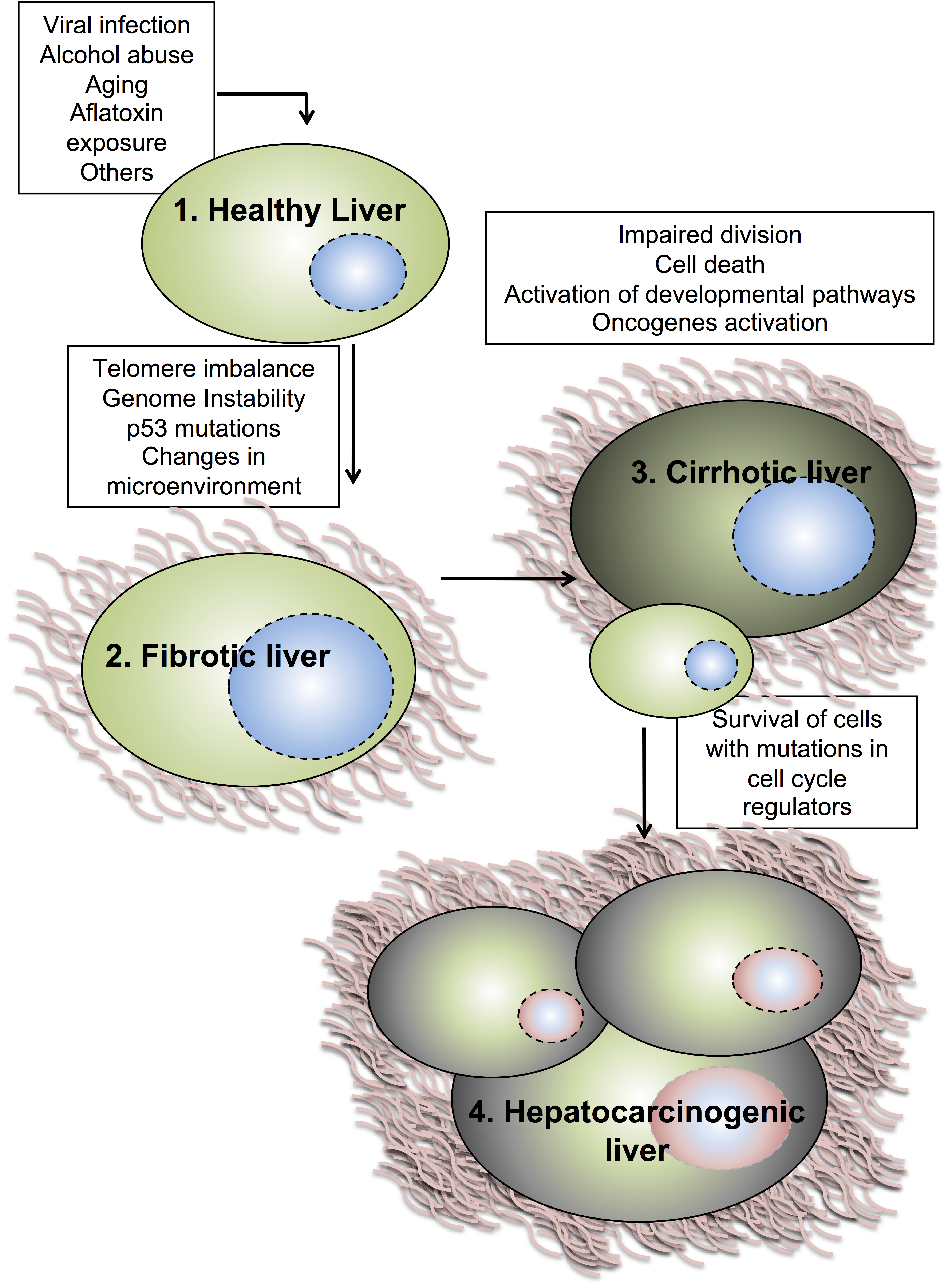
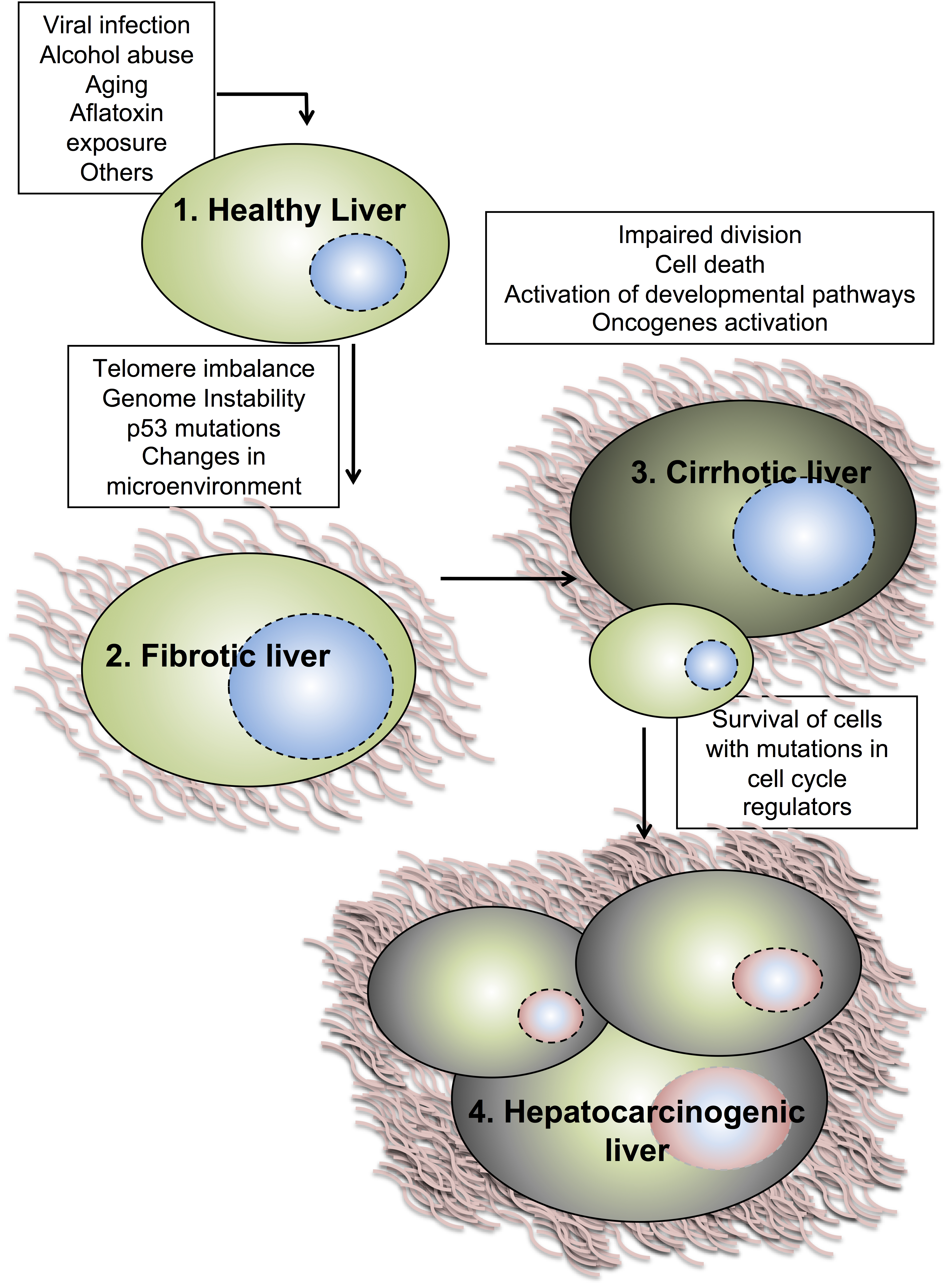
1.2. Molecular Mechanisms of Hepatocellular Carcinoma
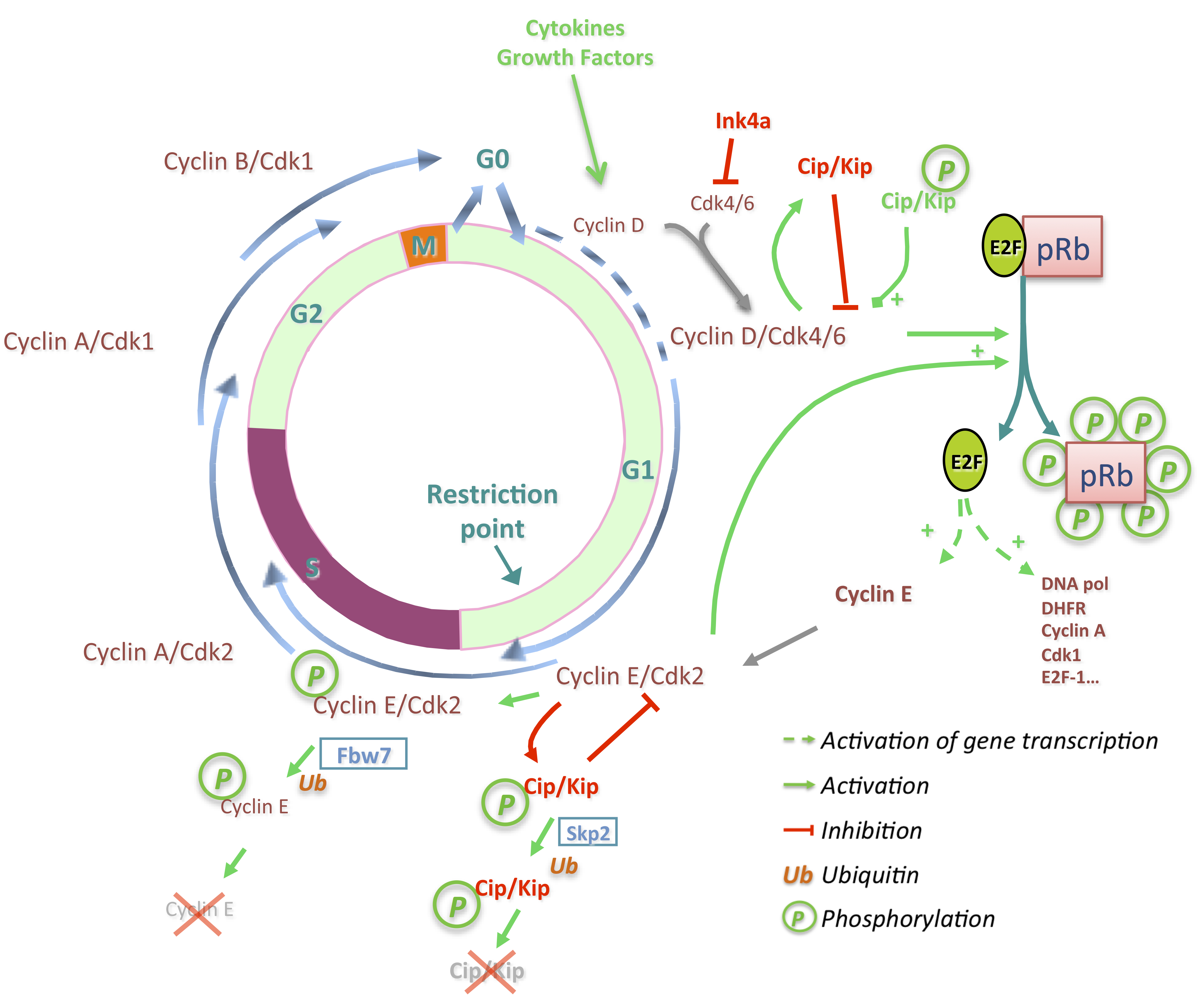
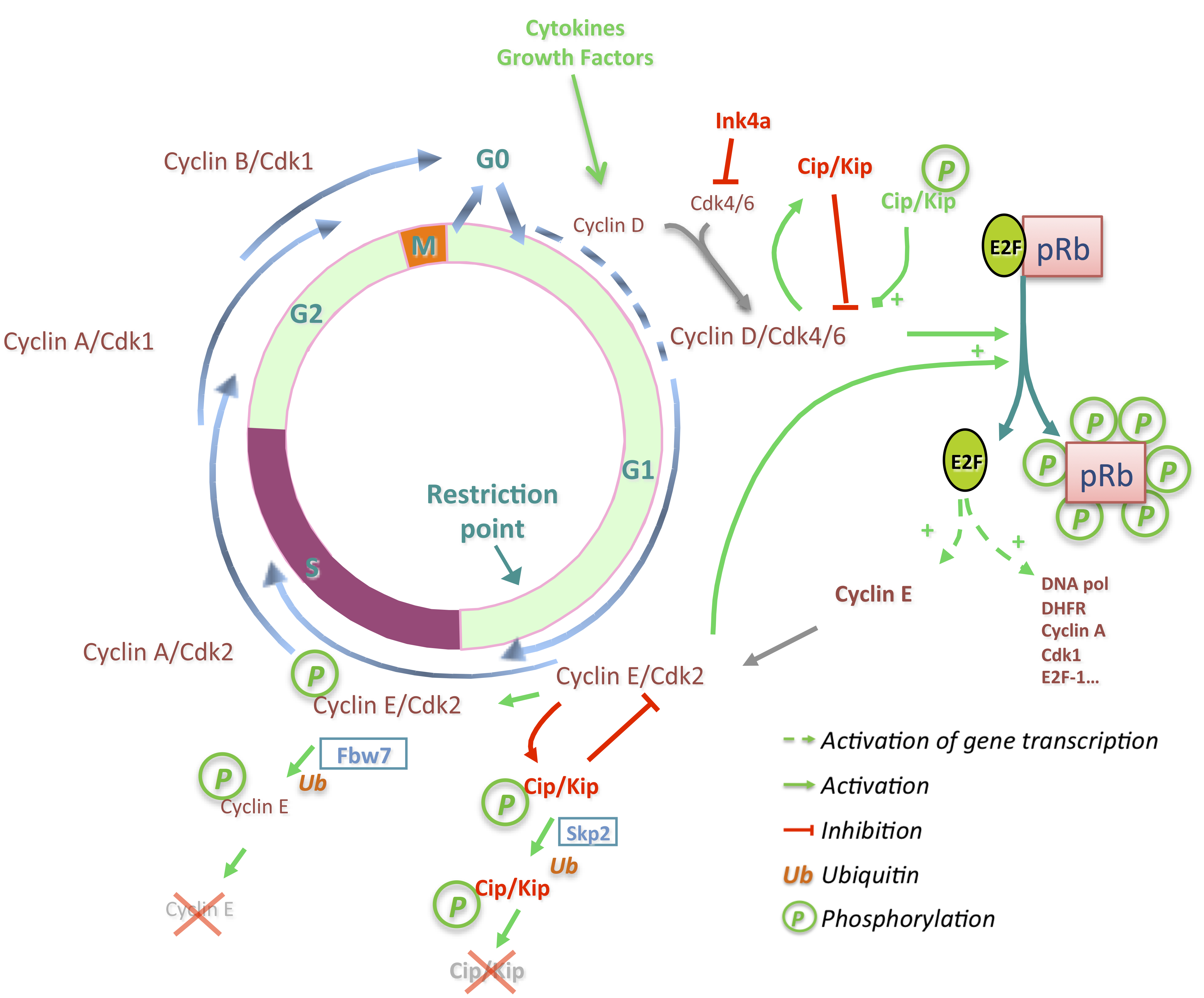
2. Hepatocyte Cell Cycle Progression
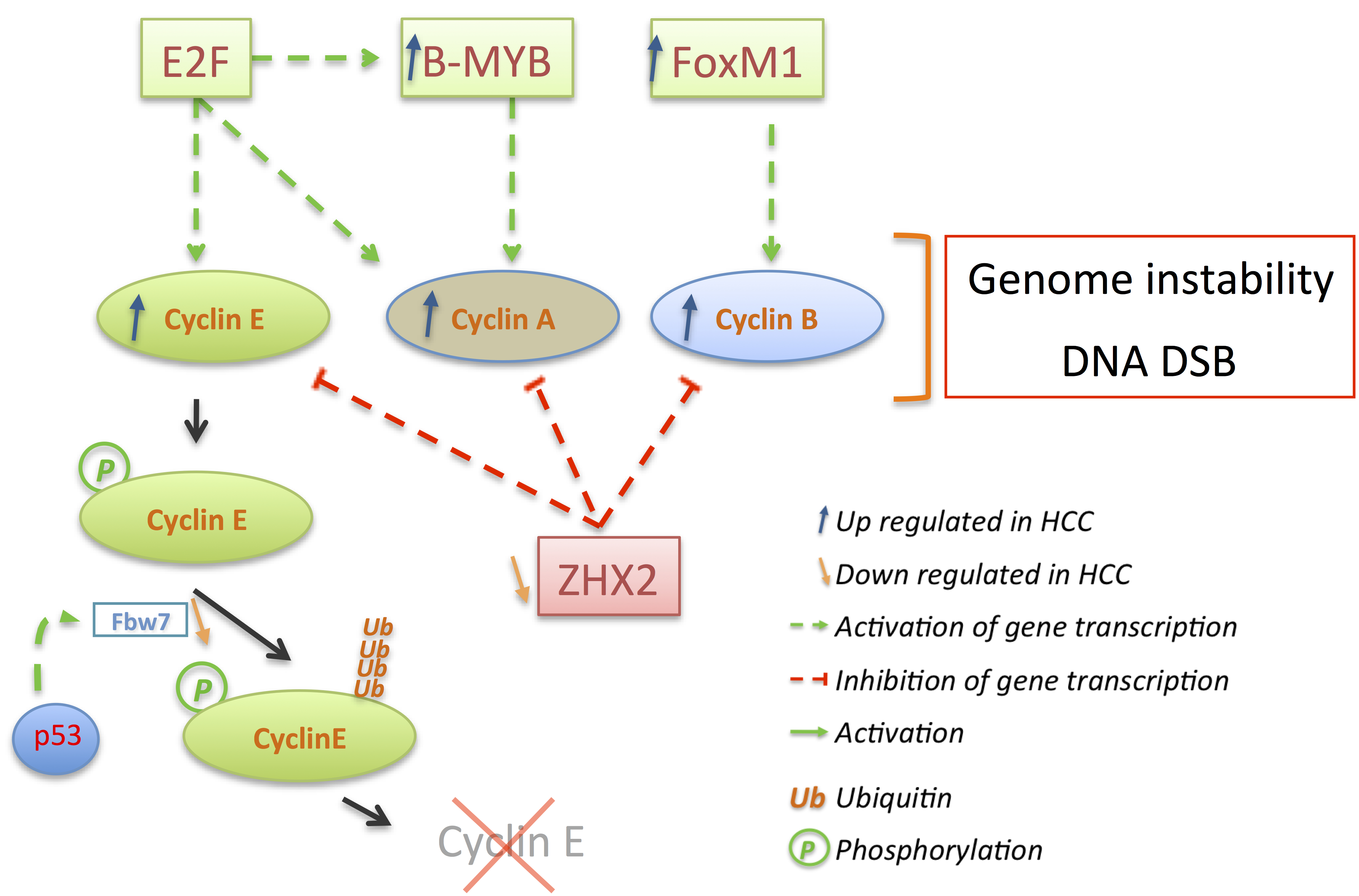
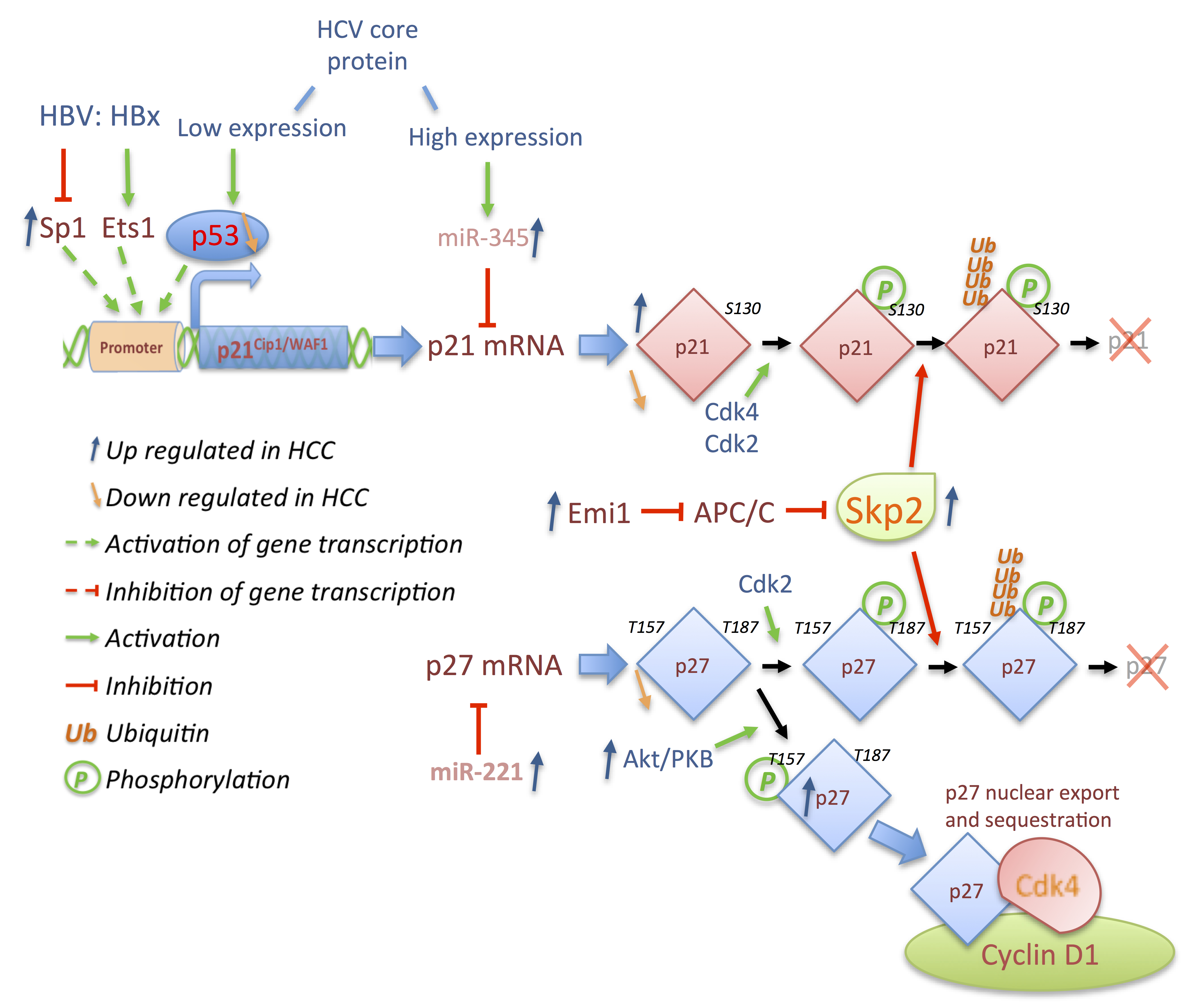
3. Deregulations of Cell Cycle Genes in HCC
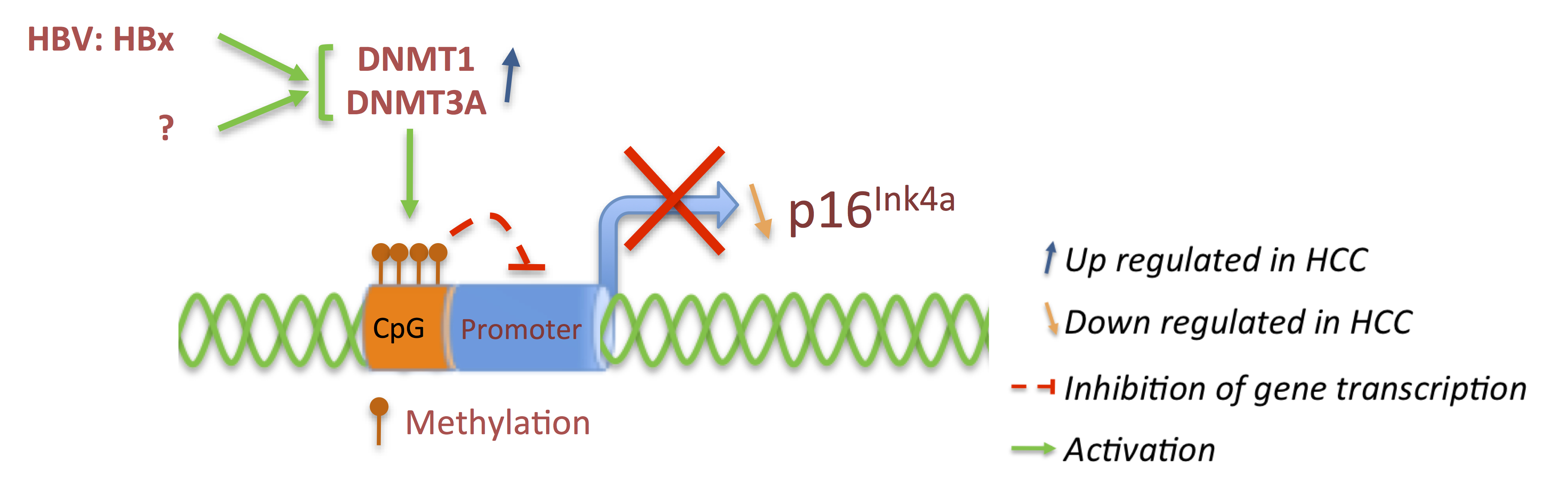
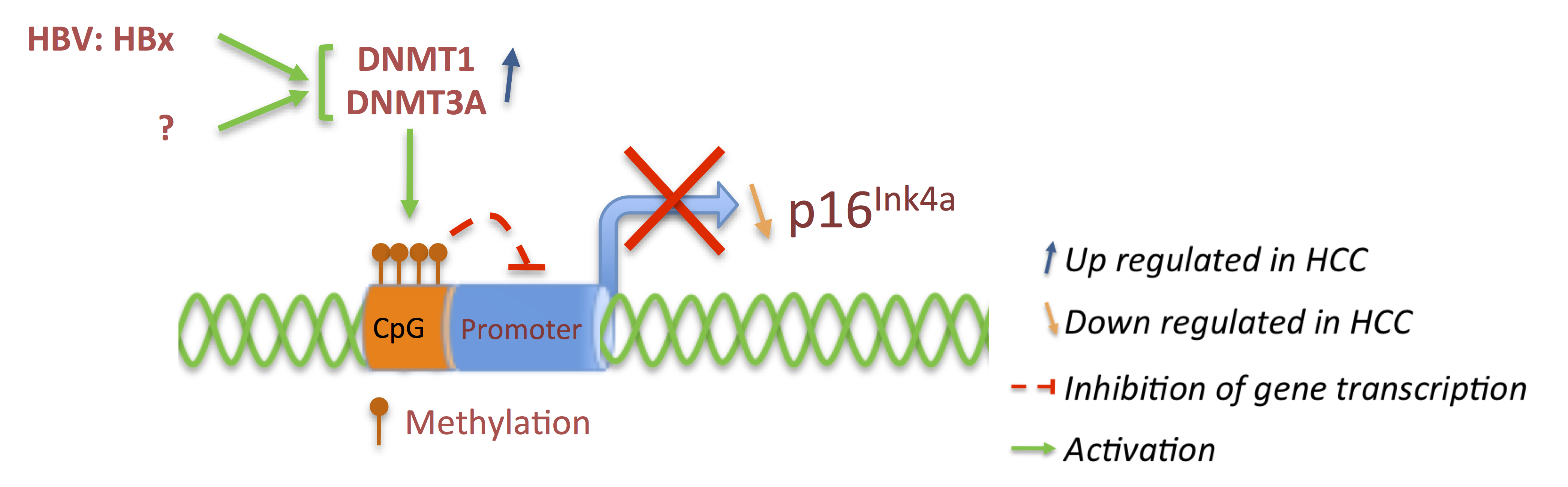
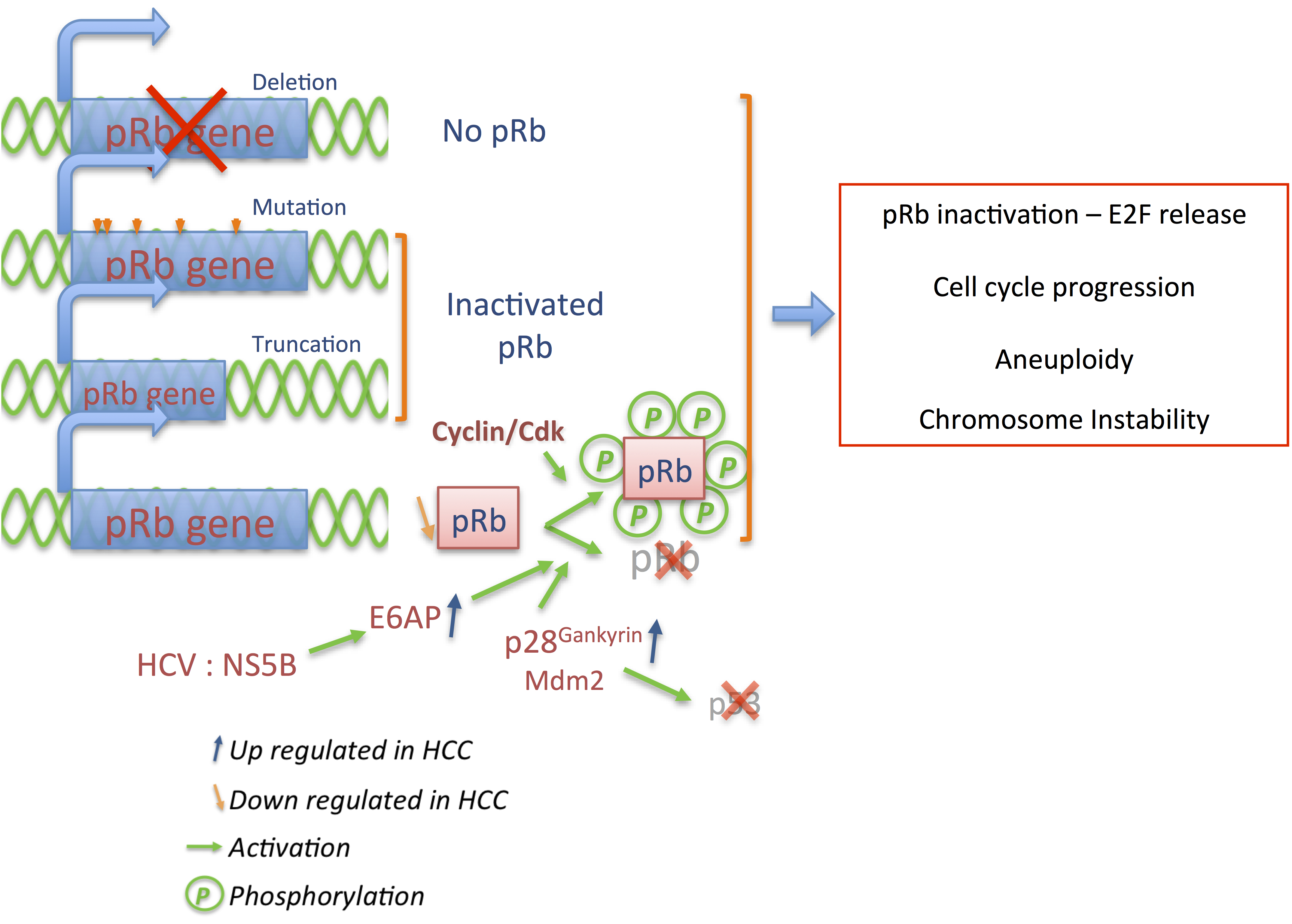
3.1. p16 and pRb: Tumor Suppressors
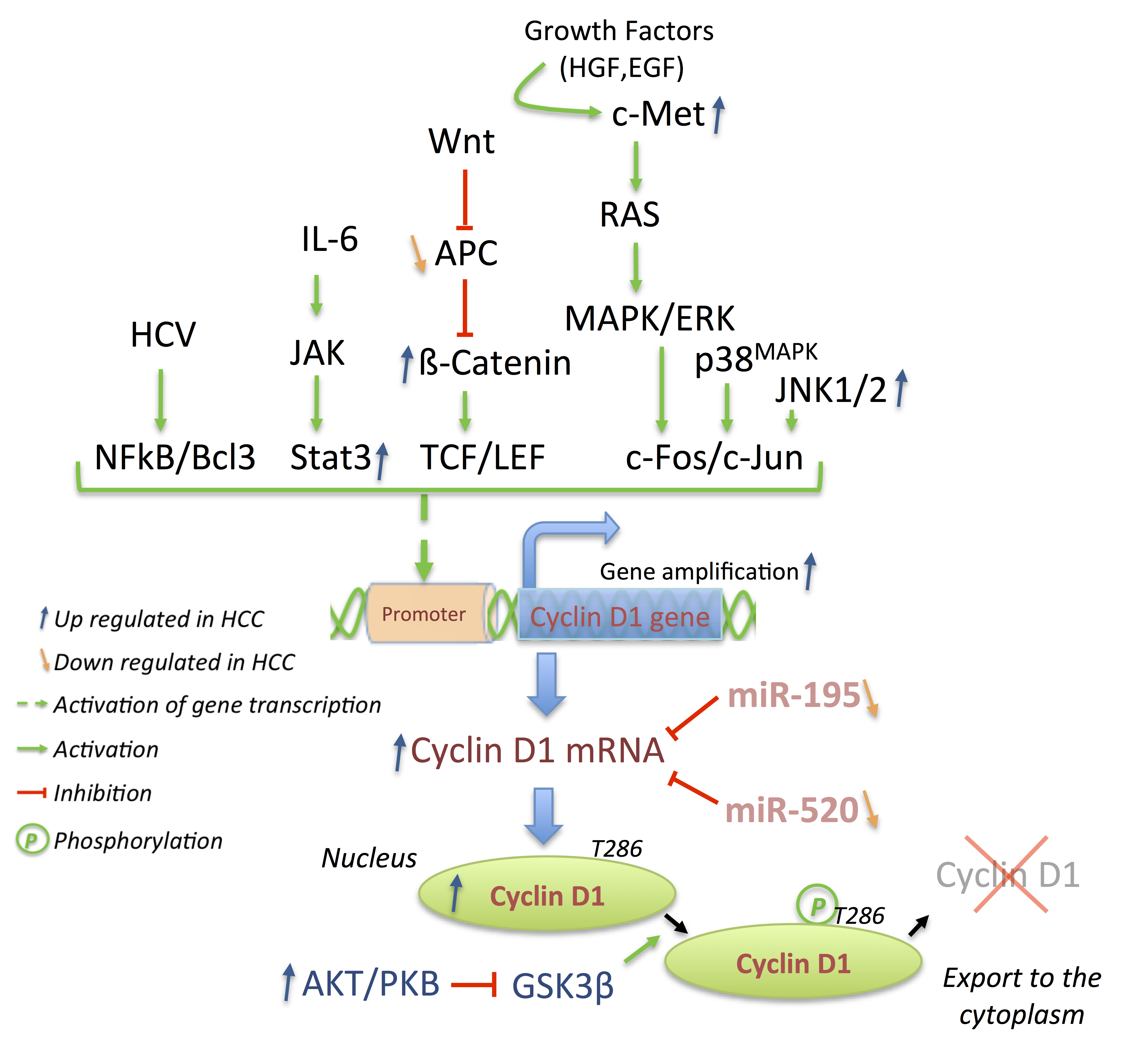
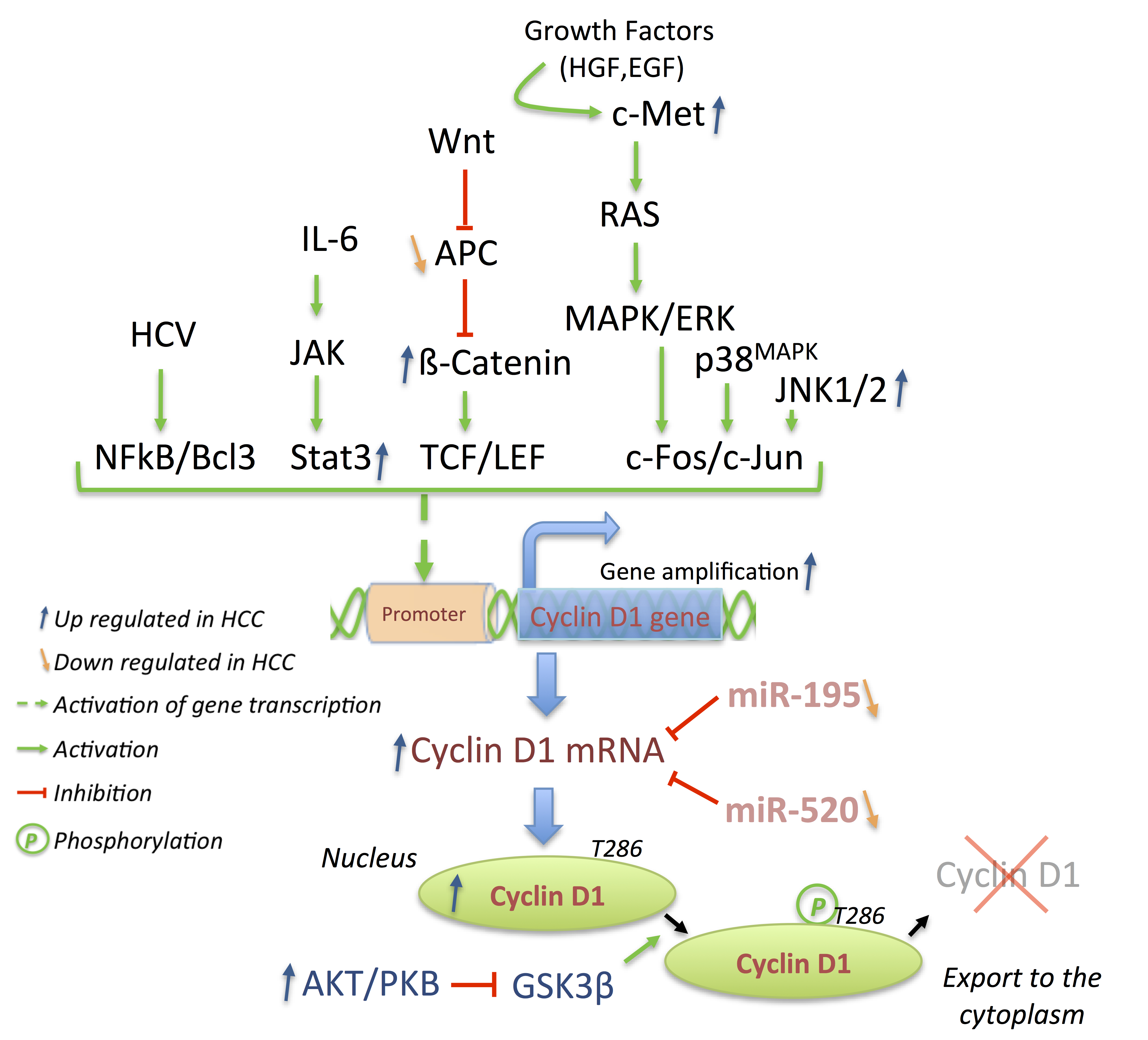
3.2. Cyclins: The Oncogenes
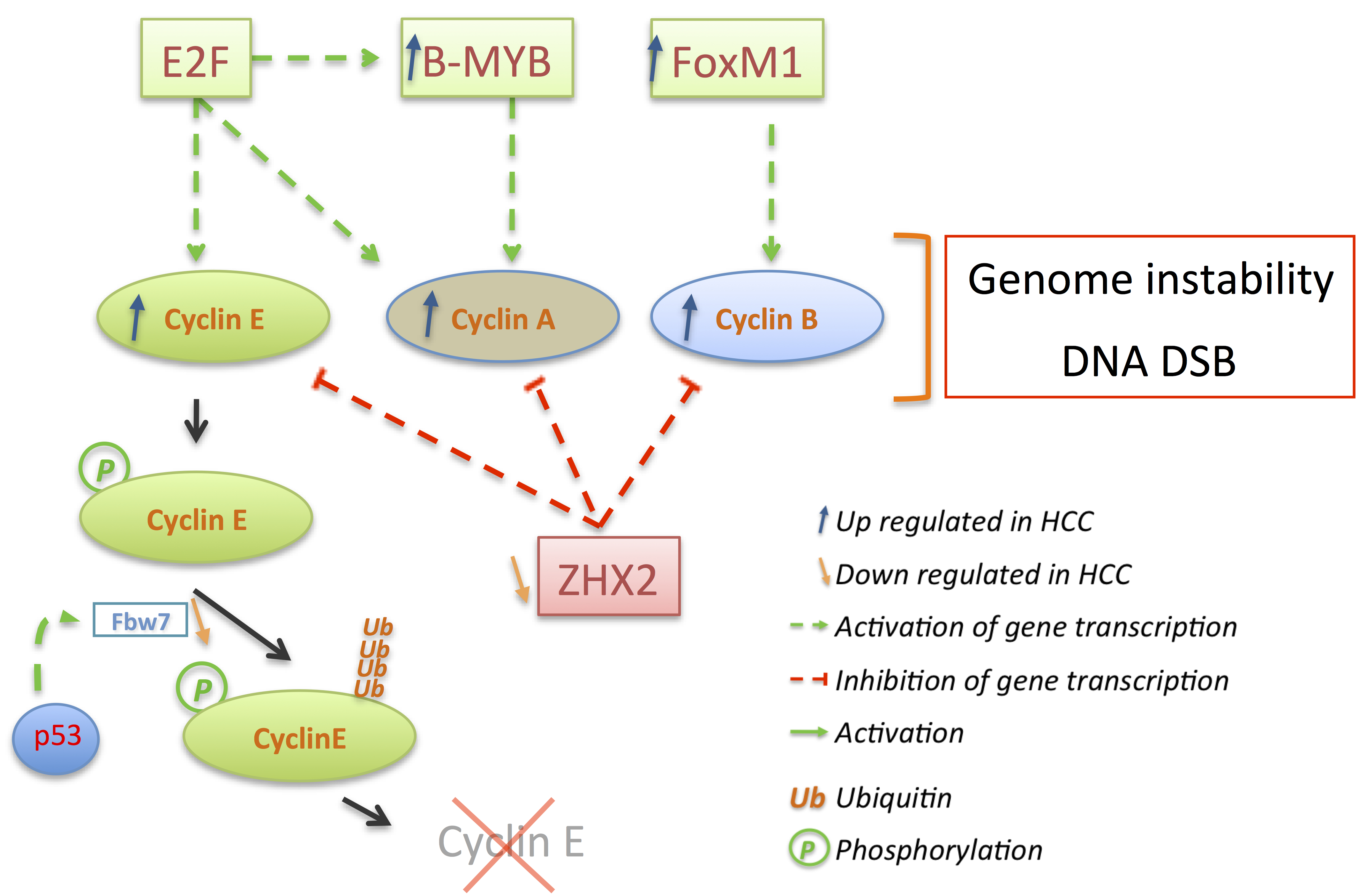
3.3. p21 and p27: A Complex Connection
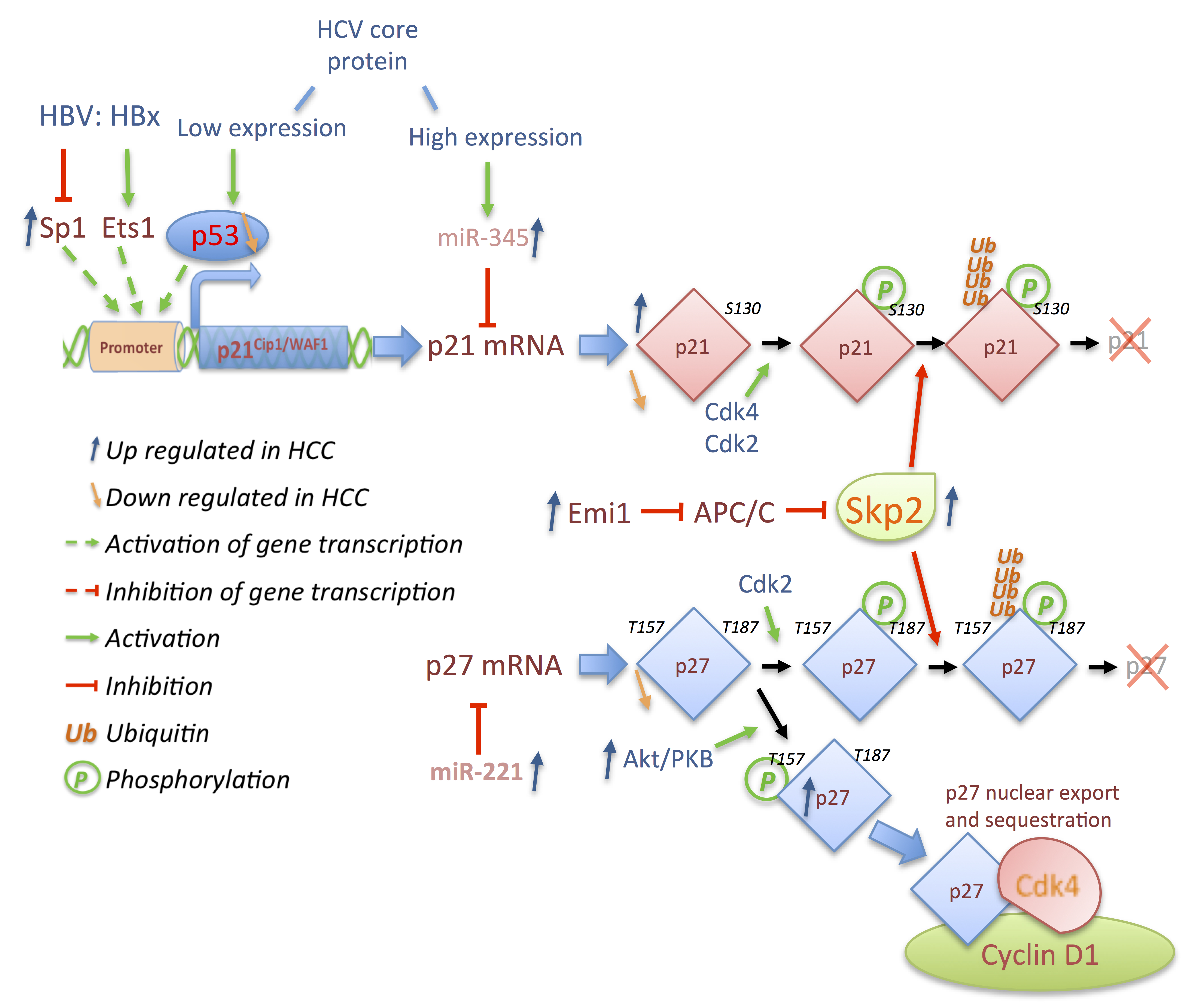
4. Outlook and Conclusions
Acknowledgments
Conflicts of Interest
References
- Washabau, R.J. Chapter 61—Liver. In Canine and Feline Gastroenterology; Chandlers, M.L., Steiner, J.M., Syring, R.J., Twedt, D.C., Wahsabau, R.J., Willard, M.D., Eds.; Saunders-Elsevier Inc.: St. Louis, MO, USA, 2013; pp. 849–957. [Google Scholar]
- Hayat, M.A. Liver Carcinoma. In Handbook of Immunohistochemistry and in Situ Hybridization of Human Carcinomas; Elsevier Academia Press: Burlington, MA, USA, 2005; Volume 3, pp. 131–151. [Google Scholar]
- Rissler, P.; Torndal, U.B.; Eriksson, L.C. Induced drug resistance inhibits selection of initiated cells and cancer development. Carcinogenesis 1997, 18, 649–655. [Google Scholar] [CrossRef]
- Yusuf, A.; Rao, P.M.; Rajalakshmi, S.; Sarma, D.S. Development of resistance during the early stages of experimental liver carcinogenesis. Carcinogenesis 1999, 20, 1641–1644. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Tabor, E. Hepatocellular carcinoma: Global epidemiology. Dig. Liver Dis. 2001, 33, 115–117. [Google Scholar] [CrossRef]
- Costanza, C.; Selim, K. Hepatocellular Carcinoma (HCC). In Encyclopedia of Gastroenterology; Johnson, L., Ed.; Elsevier Inc.: New York, NY, USA, 2004; pp. 340–346. [Google Scholar]
- Kew, M.C. Liver cancer. Int. Encycl. Public Health 2008, 4, 105–114. [Google Scholar]
- McGlynn, K.A.; London, W.T. Epidemiology and natural history of hepatocellular carcinoma. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 3–23. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef]
- Hassan, M.M.; Hwang, L.-Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Bluteau, O.; Beaudoin, J.-C.; Pasturaud, P.; Belghiti, J.; Franco, D.; Bioulac-Sage, P.; Laurent-Puig, P.; Zucman Rossi, J. Specific association between alcohol intake, high grade of differentiation and 4q34-q35 deletions in hepatocellular carcinomas identified by high resolution allelotyping. Oncogene 2002, 21, 1225–1232. [Google Scholar] [CrossRef]
- Yakicier, M.C.; Legoix, P.; Vaury, C.; Gressin, L.; Tubacher, E.; Capron, F.; Bayer, J.; Degott, C.; Balabaud, C.; Zucman-Rossi, J. Identification of homozygous deletions at chromosome 16q23 in aflatoxin B1 exposed hepatocellular carcinoma. Oncogene 2001, 20, 5232–5238. [Google Scholar] [CrossRef]
- Fattovich, G.; Bortolotti, F.; Donato, F. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J. Hepatol. 2008, 48, 335–352. [Google Scholar] [CrossRef]
- Chuang, S.-C.; la Vecchia, C.; Boffetta, P. Liver cancer: Descriptive epidemiology and risk factors other than HBV and HCV infection. Cancer Lett. 2009, 286, 9–14. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Lechel, A.; Rudolph, K.L. Epidemiology and Molecular Mechanisms of Hepatocarcinogenesis. In Zakim and Boyer’s Hepatology: A Textbook of Liver Disease, 6th ed.; Boyer, T.D., Manns, M.P., Sanyal, A.J., Eds.; Saunders-Elsevier Inc.: Philadelphia, PA, USA, 2012; pp. 142–156.e4. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ishibashi, H.; Nakamura, M.; Komori, A.; Migita, K.; Shimoda, S. Liver architecture, cell function, and disease. Semin. Immunopathol. 2009, 31, 399–409. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am. J. Pathol. 2010, 176, 2–13. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef]
- Mitchell, C.; Willenbring, H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 2008, 3, 1167–1170. [Google Scholar] [CrossRef]
- Roncalli, M.; Park, Y.N.; di Tommaso, L. Histopathological classification of hepatocellular carcinoma. Dig. Liver Dis. 2010, 42, S228–S234. [Google Scholar] [CrossRef]
- Okuda, K. Hepatocellular carcinoma. J. Hepatol. 2000, 32, 225–237. [Google Scholar] [CrossRef]
- Asia-Pacific Working Party on Prevention of Hepatocellular Carcinoma Prevention of hepatocellular carcinoma in the Asia-Pacific region: Consensus statements. J. Gastroenterol. Hepatol. 2010, 25, 657–663. [CrossRef]
- Bruix, J.; Sherman, M.; Llovet, J.M.; Beaugrand, M.; Lencioni, R.; Burroughs, A.K.; Christensen, E.; Pagliaro, L.; Colombo, M.; Rodés, J. EASL Panel of Experts on HCC Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J. Hepatol. 2001, 35, 421–430. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. European Organisation for Research and Treatment of Cancer EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar]
- Marrero, J.A.; Fontana, R.J.; Barrat, A.; Askari, F.; Conjeevaram, H.S.; Su, G.L.; Lok, A.S. Prognosis of hepatocellular carcinoma: Comparison of 7 staging systems in an American cohort. Hepatology 2005, 41, 707–716. [Google Scholar] [CrossRef]
- Varela, M.; Bruix, J. Hepatocellular carcinoma in the United States. Lessons from a population-based study in Medicare recipients. J. Hepatol. 2006, 44, 8–10. [Google Scholar] [CrossRef]
- Beyoğlu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman Rossi, J.; Dufour, J.-F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar] [CrossRef]
- Lee, J.S.; Thorgeirsson, S.S. Comparative and integrative functional genomics of HCC. Oncogene 2006, 25, 3801–3809. [Google Scholar] [CrossRef]
- Pei, Y.; Zhang, T.; Renault, V.; Zhang, X. An overview of hepatocellular carcinoma study by omics-based methods. Acta Biochim. Biophys. Sin. 2009, 41, 1–15. [Google Scholar] [CrossRef]
- Uehara, T.; Ainslie, G.R.; Kutanzi, K.; Pogribny, I.P.; Muskhelishvili, L.; Izawa, T.; Yamate, J.; Kosyk, O.; Shymonyak, S.; Bradford, B.U.; et al. Molecular mechanisms of fibrosis-associated promotion of liver carcinogenesis. Toxicol. Sci. 2013, 132, 53–63. [Google Scholar] [CrossRef]
- Borzio, M.; Bruno, S.; Roncalli, M.; Mels, G.C.; Ramella, G.; Borzio, F.; Leandro, G.; Servida, E.; Podda, M. Liver cell dysplasia is a major risk factor for hepatocellular carcinoma in cirrhosis: A prospective study. Gastroenterology 1995, 108, 812–817. [Google Scholar] [CrossRef]
- Nishida, N.; Goel, A. Genetic and epigenetic signatures in human hepatocellular carcinoma: A systematic review. Curr. Genomics 2011, 12, 130. [Google Scholar] [CrossRef]
- Arzumanyan, A.; Reis, H.M.G.P.V.; Feitelson, M.A. Pathogenic mechanisms in HBV-and HCV-associated hepatocellularcarcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef]
- Breuhahn, K.; Longerich, T.; Schirmacher, P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006, 25, 3787–3800. [Google Scholar] [CrossRef]
- Van Malenstein, H.; van Pelt, J.; Verslype, C. Molecular classification of hepatocellular carcinoma anno 2011. Eur. J. Cancer 2011, 47, 1789–1797. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Benhamouche, S.; Godard, C.; Boyault, S.; Grimber, G.; Balabaud, C.; Cunha, A.S.; Bioulac-Sage, P.; Perret, C. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene 2007, 26, 774–780. [Google Scholar] [CrossRef]
- Micsenyi, A.; Tan, X.; Sneddon, T.; Luo, J.-H.; Michalopoulos, G.K.; Monga, S.P. β-Catenin is temporally regulated during normal liver development. Gastroenterology 2004, 126, 1134–1146. [Google Scholar] [CrossRef]
- Monga, S.P.; Pediaditakis, P.; Mule, K.; Stolz, D.B.; Michalopoulos, G.K. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology 2001, 33, 1098–1109. [Google Scholar] [CrossRef]
- Pez, F.; Lopez, A.; Kim, M.; Wands, J.R.; Caron de Fromentel, C.; Merle, P. Wnt signaling and hepatocarcinogenesis: Molecular targets for the development of innovative anticancer drugs. J. Hepatol. 2013, 59, 1107–1117. [Google Scholar] [CrossRef]
- Edamoto, Y.; Hara, A.; Biernat, W.; Terracciano, L.; Cathomas, G.; Riehle, H.-M.; Matsuda, M.; Fujii, H.; Scoazec, J.-Y.; Ohgaki, H. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int. J. Cancer 2003, 106, 334–341. [Google Scholar] [CrossRef]
- Austinat, M.; Dunsch, R.; Wittekind, C.; Tannapfel, A.; Gebhardt, R.; Gaunitz, F. Correlation between beta-catenin mutations and expression of Wnt-signaling target genes in hepatocellular carcinoma. Mol. Cancer 2008, 7, 21. [Google Scholar] [CrossRef]
- Schlaeger, C.; Longerich, T.; Schiller, C.; Bewerunge, P.; Mehrabi, A.; Toedt, G.; Kleeff, J.; Ehemann, V.; Eils, R.; Lichter, P.; et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 2008, 47, 511–520. [Google Scholar]
- Yuen, M.F.; Wu, P.C.; Lai, V.C.; Lau, J.Y.; Lai, C.L. Expression of c-Myc, c-Fos, and c-jun in hepatocellular carcinom. Cancer 2001, 91, 106–112. [Google Scholar] [CrossRef]
- Chan, K.-L.; Guan, X.-Y.; Ng, I.O.-L. High-throughput tissue microarray analysis of c-myc activation in chronic liver diseases and hepatocellular carcinoma. Hum. Pathol. 2004, 35, 1324–1331. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Lin, C.-P.; Liu, C.-R.; Lee, C.-N.; Chan, T.-S.; Liu, H.E. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J. Hepatol. 2010, 2, 16–20. [Google Scholar]
- Osada, S.; Kanematsu, M.; Imai, H.; Goshima, S.; Sugiyama, Y. Evaluation of extracellular signal regulated kinase expression and its relation to treatment of hepatocellular carcinoma. J. Am. Coll. Surg. 2005, 201, 405–411. [Google Scholar] [CrossRef]
- Christensen, J.G.; Burrows, J.; Salgia, R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005, 225, 1–26. [Google Scholar] [CrossRef]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.-H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef]
- Gopinathan, L.; Ratnacaram, C.K.; Kaldis, P. Established and novel Cdk/cyclin complexes regulating the cell cycle and development. Results Probl. Cell Differ. 2011, 53, 365–389. [Google Scholar] [CrossRef]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 2002, 1602, 73–87. [Google Scholar]
- Albrecht, J.H.; Poon, R.Y.; Ahonen, C.L.; Rieland, B.M.; Deng, C.; Crary, G.S. Involvement of p21 and p27 in the regulation of CDK activity and cell cycle progression in the regenerating liver. Oncogene 1998, 16, 2141–2150. [Google Scholar]
- Pujol, M.J.; Jaime, M.; Serratosa, J.; Jaumot, M.; Agell, N.; Bachs, O. Differential association of p21Cip1 and p27Kip1 with cyclin E-CDK2 during rat liver regeneration. J. Hepatol. 2000, 33, 266–274. [Google Scholar] [CrossRef]
- Ilyin, G.P.; Glaise, D.; Gilot, D.; Baffet, G.; Guguen-Guillouzo, C. Regulation and role of p21 and p27 cyclin-dependent kinase inhibitors during hepatocyte differentiation and growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G115–G127. [Google Scholar]
- Kurinna, S.; Barton, M.C. Cascades of transcription regulation during liver regeneration. Int. J. Biochem. Cell Biol. 2011, 43, 189–197. [Google Scholar]
- Rickheim, D.G.; Nelsen, C.J.; Fassett, J.T.; Timchenko, N.A.; Hansen, L.K.; Albrecht, J.H. Differential regulation of cyclins D1 and D3 in hepatocyte proliferation. Hepatology 2002, 36, 30–38. [Google Scholar] [CrossRef]
- Albrecht, J.H.; Hu, M.Y.; Cerra, F.B. Distinct patterns of cyclin D1 regulation in models of liver regeneration and human liver. Biochem. Biophys. Res. Commun. 1995, 209, 648–655. [Google Scholar] [CrossRef]
- Albrecht, J.H.; Hansen, L.K. Cyclin D1 promotes mitogen-independent cell cycle progression in hepatocytes. Cell Growth Differ. 1999, 10, 397. [Google Scholar]
- Loyer, P.; Cariou, S.; Glaise, D.; Bilodeau, M.; Baffet, G.; Guguen-Guillouzo, C. Growth factor dependence of progression through G and S phases of adult rat hepatocytes in vitro. J. Biol. Chem. 1996, 271, 11484–11492. [Google Scholar] [CrossRef]
- Boylan, J.M.; Gruppuso, P.A. D-type cyclins and G1 progression during liver development in the rat. Biochem. Biophys. Res. Commun. 2005, 330, 722–730. [Google Scholar] [CrossRef]
- Kato, J.; Matsuoka, M.; Strom, D. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol. Cell. Biol. 1994, 14, 2713–2721. [Google Scholar] [CrossRef]
- Jaumot, M.; Estanyol, J.M.; Serratosa, J.; Agell, N.; Bachs, O. Activation of cdk4 and cdk2 during rat liver regeneration is associated with intranuclear rearrangements of cyclin-cdk complexes. Hepatology 1999, 29, 385–395. [Google Scholar] [CrossRef]
- Bisteau, X.; Paternot, S.; Colleoni, B.; Ecker, K.; Coulonval, K.; de Groote, P.; Declercq, W.; Hengst, L.; Roger, P.P. CDK4 T172 phosphorylation is central in a CDK7-dependent bidirectional CDK4/CDK2 interplay mediated by p21 phosphorylation at the restriction point. PLoS Genet. 2013, 9, e1003546. [Google Scholar] [CrossRef] [Green Version]
- Henley, S.A.; Dick, F.A. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012, 7, 10. [Google Scholar] [CrossRef]
- Dick, F.A. Structure-function analysis of the retinoblastoma tumor suppressor protein—Is the whole a sum of its parts? Cell Div. 2007, 2, 26. [Google Scholar] [CrossRef]
- Rubin, S.M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38, 12–19. [Google Scholar] [CrossRef]
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 1998, 18, 753–761. [Google Scholar]
- Ezhevsky, S.A.; Nagahara, H.; Vocero-Akbani, A.M.; Gius, D.R.; Wei, M.C.; Dowdy, S.F. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D: Cdk4/6 complexes results in active pRb. Proc. Natl. Acad. Sci. USA 1997, 94, 10699–10704. [Google Scholar] [CrossRef]
- Yao, G.; Lee, T.J.; Mori, S.; Nevins, J.R.; You, L. A bistable Rb–E2F switch underlies the restriction point. Nat. Cell Biol. 2008, 10, 476–482. [Google Scholar] [CrossRef]
- Albrecht, J.H.; Meyer, A.H.; Hu, M.Y. Regulation of cyclin-dependent kinase inhibitor p21WAF1/Cip1/Sdi1 gene expression in hepatic regeneration. Hepatology 1997, 25, 557–563. [Google Scholar] [CrossRef]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef]
- Zhang, H.; Hannon, G.J.; Beach, D. p21-containing cyclin kinases exist in both active and inactive states. Genes Dev. 1994, 8, 1750–1758. [Google Scholar] [CrossRef]
- Cheng, M.; Olivier, P.; Diehl, J.; Fero, M. The p21Cip1 and p27Kip1 CDK “inhibitors” are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Zhu, H.; Nie, L.; Maki, C.G. Cdk2-dependent Inhibition of p21 stability via a C-terminal cyclin-binding motif. J. Biol. Chem. 2005, 280, 29282–29288. [Google Scholar] [CrossRef]
- Hayashi, E.; Yasui, A.; Oda, K.; Nagino, M.; Nimura, Y.; Nakanishi, M.; Motoyama, N.; Ikeda, K.; Matsuura, A. Loss of p27Kip1 accelerates DNA replication after partial hepatectomy in mice. J. Surg. Res. 2003, 111, 196–202. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T. Ubiquitylation and proteasomal degradation of the p21Cip1, p27Kip1 and p57Kip2 CDK inhibitors. Cell Cycle 2010, 9, 2342–2352. [Google Scholar] [CrossRef]
- Starostina, N.G.; Kipreos, E.T. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012, 22, 33–41. [Google Scholar] [CrossRef]
- Welcker, M.; Singer, J.; Loeb, K.R.; Grim, J.; Bloecher, A.; Gurien-West, M.; Clurman, B.E.; Roberts, J.M. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell 2003, 12, 381–392. [Google Scholar] [CrossRef]
- Hao, B.; Oehlmann, S.; Sowa, M.E.; Harper, J.W.; Pavletich, N.P. Structure of a Fbw7-Skp1-cyclin E complex: Multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol. Cell 2007, 26, 131–143. [Google Scholar] [CrossRef]
- Merrick, K.A.; Larochelle, S.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Fisher, R.P. Distinct activation pathways confer cyclin-binding specificity on Cdk1 and Cdk2 in human cells. Mol. Cell 2008, 32, 662–672. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, H.J.; Murakami, Y.; Sachse, R.; Yashima, K.; Hirohashi, S.; Hu, S.X.; Benedict, W.F.; Sekiya, T. Deletions of chromosome 13q, mutations in Retinoblastoma 1, and retinoblastoma protein state in human hepatocellular carcinoma. Cancer Res. 1994, 54, 4177–4182. [Google Scholar]
- Nakamura, T.; Iwamura, Y.; Kaneko, M.; Nakagawa, K.; Kawai, K.; Mitamura, K.; Futagawa, T.; Hayashi, H. Deletions and rearrangements of the retinoblastoma gene in hepatocellular carcinoma, insulinoma and some neurogenic tumors as found in a study of 121 tumors. Jpn. J. Clin. Oncol. 1991, 21, 325–329. [Google Scholar]
- Murakami, Y.; Hayashi, K.; Hirohashi, S.; Sekiya, T. Aberrations of the tumor suppressor p53 and retinoblastoma genes in human hepatocellular carcinomas. Cancer Res. 1991, 51, 5520–5525. [Google Scholar]
- Seki, S.; Kawakita, N.; Yanai, A.; Kitada, T.; Sakai, Y.; Nakatani, K.; Yamada, T.; Sakaguchi, H.; Kuroki, T. Expression of the retinoblastoma gene product in human hepatocellular carcinoma. Hum. Pathol. 1995, 26, 366–374. [Google Scholar] [CrossRef]
- Liew, C.T.; Li, H.-M.; Lo, K.-W.; Leow, C.K.; Chan, J.Y.; Hin, L.Y.; Lau, W.Y.; Lai, P.B.; Lim, B.K.; Huang, J. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene 1999, 18, 789–795. [Google Scholar] [CrossRef]
- Suh, S.I.; Pyun, H.Y.; Cho, J.W.; Baek, W.K.; Park, J.B.; Kwon, T.; Park, J.W.; Suh, M.H.; Carson, D.A. 5-Aza-2'-deoxycytidine leads to down-regulation of aberrant p16INK4A RNA transcripts and restores the functional retinoblastoma protein pathway in hepatocellular carcinoma cell lines. Cancer Lett. 2000, 160, 81–88. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.I.; Lapidus, R.G.; Issa, J. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar]
- Chaubert, P.; Gayer, R.; Zimmermann, A.; Fontolliet, C.; Stamm, B.; Bosman, F.; Shaw, P. Germ-line mutations of the p16INK4/MTS1 gene occur in a subset of patients with hepatocellular carcinoma. Hepatology 1997, 25, 1376–1381. [Google Scholar] [CrossRef]
- Biden, K.; Young, J.; Buttenshaw, R.; Searle, J.; Cooksley, G.; Xu, D.B.; Leggett, B. Frequency of mutation and deletion of the tumor suppressor gene CDKN2A (MTS1/p16) in hepatocellular carcinoma from an Australian population. Hepatology 1997, 25, 593–597. [Google Scholar] [CrossRef]
- Zhu, Y.-Z.; Zhu, R.; Shi, L.-G.; Mao, Y.; Zheng, G.-J.; Chen, Q.; Zhu, H.-G. Hepatitis B virus X protein promotes hypermethylation of p16INK4A promoter through upregulation of DNA methyltransferases in hepatocarcinogenesis. Exp. Mol. Path. 2010, 89, 268–275. [Google Scholar] [CrossRef]
- Zhu, Y.Z.; Zhu, R.; Fan, J.; Pan, Q.; Li, H.; Chen, Q.; Zhu, H.G. Hepatitis B virus X protein induces hypermethylation of p16INK4A promoter via DNA methyltransferases in the early stage of HBV-associated hepatocarcinogenesis. J. Viral Hepat. 2010, 17, 98–107. [Google Scholar] [CrossRef]
- Kaneto, H.; Sasaki, S.; Yamamoto, H.; Itoh, F.; Toyota, M.; Suzuki, H.; Ozeki, I.; Iwata, N.; Ohmura, T.; Satoh, T. Detection of hypermethylation of thep16INK4A gene promoter in chronic hepatitis and cirrhosis associated with hepatitis B or C virus. Gut 2001, 48, 372–377. [Google Scholar] [CrossRef]
- Shim, Y.-H.; Yoon, G.-S.; Choi, H.-J.; Chung, Y.H.; Yu, E. p16 Hypermethylation in the early stage of hepatitis B virus-associated hepatocarcinogenesis. Cancer Lett. 2003, 190, 213–219. [Google Scholar] [CrossRef]
- Li, X.; Hui, A.-M.; Sun, L.; Hasegawa, K.; Torzilli, G.; Minagawa, M.; Takayama, T.; Makuuchi, M. p16INK4A hypermethylation is associated with hepatitis virus infection, age, and gender in hepatocellular carcinoma. Clin. Cancer Res. 2004, 10, 7484–7489. [Google Scholar] [CrossRef]
- Zhu, R.; Li, B.-Z.; Li, H.; Ling, Y.-Q.; Hu, X.-Q.; Zhai, W.-R.; Zhu, H.-G. Association of p16INK4A hypermethylation with hepatitis B virus X protein expression in the early stage of HBV-associated hepatocarcinogenesis. Pathol. Int. 2007, 57, 328–336. [Google Scholar] [CrossRef]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007, 3, e139. [Google Scholar] [CrossRef]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef]
- Higashitsuji, H.; Itoh, K.; Nagao, T.; Dawson, S.; Nonoguchi, K.; Kido, T.; Mayer, R.J.; Arii, S.; Fujita, J. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat. Med. 2000, 6, 96–99. [Google Scholar] [CrossRef]
- Tan, L.; Fu, X.-Y.; Liu, S.-Q.; Li, H.-H.; Hong, Y.; Wu, M.-C.; Wang, H.-Y. Expression of p28GANK and its correlation with RB in human hepatocellular carcinoma. Liver Int. 2005, 25, 667–676. [Google Scholar] [CrossRef]
- Fu, J.; Chen, Y.; Cao, J.; Luo, T.; Qian, Y.-W.; Yang, W.; Ren, Y.-B.; Su, B.; Cao, G.-W.; Yang, Y.; et al. p28GANK overexpression accelerates hepatocellular carcinoma invasiveness and metastasis via phosphoinositol 3-kinase/AKT/hypoxia-inducible factor-1α pathways. Hepatology 2010, 53, 181–192. [Google Scholar]
- Chen, Y.; Li, H.H.; Fu, J.; Wang, X.F.; Ren, Y.-B.; Dong, L.-W.; Tang, S.H.; Liu, S.Q.; Wu, M.C.; Wang, H.-Y. Oncoprotein p28GANK binds to RelA and retains NF-κB in the cytoplasm through nuclear export. Cell Res. 2007, 17, 1020–1029. [Google Scholar] [CrossRef]
- Dong, L.-W.; Yang, G.-Z.; Pan, Y.-F.; Chen, Y.; Tan, Y.-X.; Dai, R.-Y.; Ren, Y.-B.; Fu, J.; Wang, H.-Y. The oncoprotein p28. Cell Res. 2011, 21, 1248–1261. [Google Scholar] [CrossRef]
- Liu, Y.; Higashitsuji, H.; Higashitsuji, H.; Itoh, K.; Sakurai, T.; Koike, K.; Hirota, K.; Fukumoto, M.; Fujita, J. Overexpression of gankyrin in mouse hepatocytes induces hemangioma by suppressing factor inhibiting hypoxia-inducible factor-1 (FIH-1) and activating hypoxia-inducible factor-1. Biochem. Biophys. Res. Commun. 2013, 432, 22–27. [Google Scholar] [CrossRef]
- Higashitsuji, H.; Higashitsuji, H.; Itoh, K.; Sakurai, T.; Nagao, T.; Sumitomo, Y.; Sumitomo, H.; Masuda, T.; Dawson, S.; Shimada, Y.; et al. The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer Cell 2005, 8, 75–87. [Google Scholar] [CrossRef]
- Dawson, S.; Higashitsuji, H.; Wilkinson, A.J.; Fujita, J.; Mayer, R.J. Gankyrin: A new oncoprotein and regulator of pRb and p53. Trends Cell Biol. 2006, 16, 229–233. [Google Scholar] [CrossRef]
- Qiu, W.; Wu, J.; Walsh, E.M.; Zhang, Y.; Chen, C.-Y.; Fujita, J.; Xiao, Z.-X.J. Retinoblastoma protein modulates gankyrin-MDM2 in regulation of p53 stability and chemosensitivity in cancer cells. Oncogene 2008, 27, 4034–4043. [Google Scholar] [CrossRef]
- Morel, A.P.; Unsal, K.; Cagatay, T.; Ponchel, F.; Carr, B.; Ozturk, M. p53 but not p16INK4a induces growth arrest in retinoblastoma-deficient hepatocellular carcinoma cells. J. Hepatol. 2000, 33, 254–265. [Google Scholar] [CrossRef]
- Hui, A.M.; Li, X.; Makuuchi, M.; Takayama, T.; Kubota, K. Over-expression and lack of retinoblastoma protein are associated with tumor progression and metastasis in hepatocellular carcinoma. Int. J. Cancer 1999, 84, 604–608. [Google Scholar] [CrossRef]
- Hui, A.-M.; Shi, Y.-Z.; Li, X.; Takayama, T.; Makuuchi, M. Loss of p16INK4 protein, alone and together with loss of retinoblastoma protein, correlate with hepatocellular carcinoma progression. Cancer Lett. 2000, 154, 93–99. [Google Scholar] [CrossRef]
- Mayhew, C.N.; Bosco, E.E.; Fox, S.R.; Okaya, T.; Tarapore, P.; Schwemberger, S.J.; Babcock, G.F.; Lentsch, A.B.; Fukasawa, K.; Knudsen, E.S. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005, 65, 4568–4577. [Google Scholar] [CrossRef]
- Viatour, P.; Ehmer, U.; Saddic, L.A.; Dorrell, C.; Andersen, J.B.; Lin, C.; Zmoos, A.-F.; Mazur, P.K.; Schaffer, B.E.; Ostermeier, A.; et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J. Exp. Med. 2011, 208, 1963–1976. [Google Scholar] [CrossRef]
- Mayhew, C.N.; Carter, S.L.; Fox, S.R.; Sexton, C.R.; Reed, C.A.; Srinivasan, S.V.; Liu, X.; Wikenheiser Brokamp, K.; Boivin, G.P.; Lee, J.-S.; et al. RB loss abrogates cell cycle control and genome integrity to promote liver tumorigenesis. Gastroenterology 2007, 133, 976–984. [Google Scholar] [CrossRef]
- Reed, C.A.; Mayhew, C.N.; McClendon, A.K.; Yang, X.; Witkiewicz, A.; Knudsen, E.S. RB has a critical role in mediating the in vivo checkpoint response, mitigating secondary DNA damage and suppressing liver tumorigenesis initiated by aflatoxin B1. Oncogene 2009, 28, 4434–4443. [Google Scholar] [CrossRef]
- McClendon, A.K.; Dean, J.L.; Ertel, A.; Fu, Z.; Rivadeneira, D.B.; Reed, C.A.; Bourgo, R.J.; Witkiewicz, A.; Addya, S.; Mayhew, C.N.; et al. RB and p53 cooperate to prevent liver tumorigenesis in response to tissue damage. Gastroenterology 2011, 141, 1439–1450. [Google Scholar] [CrossRef]
- Zhao, H.; Bauzon, F.; Fu, H.; Lu, Z.; Cui, J.; Nakayama, K.; Nakayama, K.I.; Locker, J.; Zhu, L. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 2013, 24, 645–659. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Jiang, W.; Chen, C.J.; Lee, C.S.; Kahn, S.M.; Santella, R.M.; Weinstein, I.B. Amplification and overexpression of cyclin D1 in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 1993, 196, 1010–1016. [Google Scholar] [CrossRef]
- Nishida, N.; Fukuda, Y.; Komeda, T.; Kita, R.; Sando, T.; Furukawa, M.; Amenomori, M.; Shibagaki, I.; Nakao, K.; Ikenaga, M. Amplification and overexpression of the cyclin D1 gene in aggressive human hepatocellular carcinoma. Cancer Res. 1994, 54, 3107–3110. [Google Scholar]
- Wang, K.; Lim, H.Y.; Shi, S.; Lee, J.; Deng, S.; Xie, T.; Zhu, Z.; Wang, Y.; Pocalyko, D.; Yang, W.J.; et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013, 58, 706–717. [Google Scholar] [CrossRef]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef]
- Woo, H.G.; Park, E.S.; Thorgeirsson, S.S.; Kim, Y.J. Exploring genomic profiles of hepatocellular carcinoma. Mol. Carcinog. 2011, 50, 235–243. [Google Scholar] [CrossRef]
- Joo, M.; Kang, Y.K.; Kim, M.R.; Lee, H.K.; Jang, J.J. Cyclin D1 overexpression in hepatocellular carcinoma. Liver 2001, 21, 89–95. [Google Scholar] [CrossRef]
- Choi, Y.J.; Anders, L. Signaling through cyclin D-dependent kinases. Oncogene 2013, in press. [Google Scholar]
- Klein, E.A.; Assoian, R.K. Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell Sci. 2008, 121, 3853–3857. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Tetsu, O.; McCormick, F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar] [CrossRef]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef]
- Cui, J.; Zhou, X.; Liu, Y.; Tang, Z.; Romeih, M. Wnt signaling in hepatocellular carcinoma: Analysis of mutation and expression of beta-catenin, T-cell factor-4 and glycogen synthase kinase 3-beta genes. J. Gastroenterol. Hepatol. 2003, 18, 280–287. [Google Scholar] [CrossRef]
- Joo, M.; Lee, H.K.; Kang, Y.K. Expression of beta-catenin in hepatocellular carcinoma in relation to tumor cell proliferation and cyclin D1 expression. J. Korean Med. Sci. 2003, 18, 211–217. [Google Scholar]
- Patil, M.A.; Lee, S.A.; Macias, E.; Lam, E.T.; Xu, C.; Jones, K.D.; Ho, C.; Rodriguez-Puebla, M.; Chen, X. Role of cyclin D1 as a mediator of c-Met- and beta-catenin-induced hepatocarcinogenesis. Cancer Res. 2009, 69, 253–261. [Google Scholar] [CrossRef]
- Nejak-Bowen, K.N.; Thompson, M.D.; Singh, S.; Bowen, W.C.; Dar, M.J.; Khillan, J.; Dai, C.; Monga, S.P. Accelerated liver regeneration and hepatocarcinogenesis in mice overexpressing serine-45 mutant β-catenin. Hepatology 2010, 51, 1603–1613. [Google Scholar] [CrossRef]
- Luo, D.; Liu, Q.-F.; Gove, C.; Naomov, N.V.; Su, J.-J.; Williams, R. Analysis of N-ras gene mutation and p53 gene expression in human hepatocellular carcinomas. World J. Gastroenterol. 1998, 4, 97–99. [Google Scholar]
- Newell, P.; Toffanin, S.; Villanueva, A.; Chiang, D.Y.; Minguez, B.; Cabellos, L.; Savic, R.; Hoshida, Y.; Lim, K.H.; Melgar-Lesmes, P.; et al. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J. Hepatol. 2009, 51, 725–733. [Google Scholar] [CrossRef]
- Colombino, M.; Sperlongano, P.; Izzo, F.; Tatangelo, F.; Botti, G.; Lombardi, A.; Accardo, M.; Tarantino, L.; Sordelli, I.; Agresti, M.; et al. BRAF and PIK3CA genes are somatically mutated in hepatocellular carcinoma among patients from South Italy. Cell Death Dis. 2012, 3, e259. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.-S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef]
- Huynh, H.; Do, P.T.; Nguyen, T.H.; Chow, P.; Tan, P.H.; Quach, T.H.; Van, T.; Soo, K.C.; Tran, E. Extracellular signal-regulated kinase induces cyclin D1 and Cdk-2 expression and phosphorylation of retinoblastoma in hepatocellular carcinoma. Int. J. Oncol. 2004, 25, 1839–1847. [Google Scholar]
- Parekh, P.; Rao, K. Overexpression of cyclin D1 is associated with elevated levels of MAP kinases, Akt and Pak1 during diethylnitrosamine-induced progressive liver carcinogenesis. Cell Biol. Int. 2007, 31, 35–43. [Google Scholar] [CrossRef]
- Lau, C.K.; Yang, Z.F.; Lam, S.P.; Lam, C.T.; Ngai, P.; Tam, K.H.; Poon, R.T.-P.; Fan, S.-T. Inhibition of Stat3 activity by YC-1 enhances chemo-sensitivity in hepatocellular carcinoma. Cancer Biol. Ther. 2007, 6, 1900–1907. [Google Scholar] [CrossRef]
- Won, C.; Lee, C.S.; Lee, J.-K.; Kim, T.-J.; Lee, K.-H.; Yang, Y.M.; Kim, Y.-N.; Ye, S.-K.; Chung, M.-H. CADPE suppresses cyclin D1 expression in hepatocellular carcinoma by blocking IL-6-induced STAT3 activation. Anticancer Res. 2010, 30, 481–488. [Google Scholar]
- Feng, D.Y.; Zheng, H.; Tan, Y.; Cheng, R.X. Effect of phosphorylation of MAPK and Stat3 and expression of c-fos and c-jun proteins on hepatocarcinogenesis and their clinical significance. World J. Gastroenterol. 2001, 7, 33–36. [Google Scholar]
- Su, H.; Yang, J.-R.; Xu, T.; Huang, J.; Xu, L.; Yuan, Y.; Zhuang, S.-M. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009, 69, 1135–1142. [Google Scholar] [CrossRef]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar] [CrossRef]
- Xu, T.; Zhu, Y.; Xiong, Y.; Ge, Y.-Y.; Yun, J.-P.; Zhuang, S.-M. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology 2009, 50, 113–121. [Google Scholar] [CrossRef]
- Zhang, W.; Kong, G.; Zhang, J.; Wang, T.; Ye, L.; Zhang, X. MicroRNA-520b inhibits growth of hepatoma cells by targeting MEKK2 and cyclin D1. PLoS One 2012, 7, e31450. [Google Scholar]
- Park, S.G.; Chung, C.; Kang, H.; Kim, J.-Y.; Jung, G. Up-regulation of cyclin D1 by HBx is mediated by NF-kappaB2/BCL3 complex through kappaB site of cyclin D1 promoter. J. Biol. Chem. 2006, 281, 31770–31777. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tamakawa, S.; Yoshie, M.; Yaginuma, Y.; Ogawa, K. Neoplastic hepatocyte growth associated with cyclin D1 redistribution from the cytoplasm to the nucleus in mouse hepatocarcinogenesis. Mol. Carcinog. 2006, 45, 901–913. [Google Scholar]
- Diehl, J.A. Cycling to cancer with cyclin D1. Cancer Biol. Ther. 2002, 1, 226–231. [Google Scholar]
- Sato, Y.; Itoh, F.; Hareyama, M.; Satoh, M.; Hinoda, Y.; Seto, M.; Ueda, R.; Imai, K. Association of cyclin D1 expression with factors correlated with tumor progression in human hepatocellular carcinoma. J. Gastroenterol. 1999, 34, 486–493. [Google Scholar] [CrossRef]
- Peng, S.Y.; Chou, S.P.; Hsu, H.C. Association of downregulation of cyclin D1 and of overexpression of cyclin E with p53 mutation, high tumor grade and poor prognosis in hepatocellular carcinoma. J. Hepatol. 1998, 29, 281–289. [Google Scholar] [CrossRef]
- Ito, Y.; Matsuura, N.; Sakon, M.; Miyoshi, E.; Noda, K.; Takeda, T.; Umeshita, K.; Nagano, H.; Nakamori, S.; Dono, K.; et al. Expression and prognostic roles of the G1-S modulators in hepatocellular carcinoma: p27 independently predicts the recurrence. Hepatology 1999, 30, 90–99. [Google Scholar] [CrossRef]
- Deane, N.G.; Parker, M.A.; Aramandla, R.; Diehl, L.; Lee, W.J.; Washington, M.K.; Nanney, L.B.; Shyr, Y.; Beauchamp, R.D. Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 2001, 61, 5389–5395. [Google Scholar]
- Rennstam, K.; Baldetorp, B.; Kytölä, S.; Tanner, M.; Isola, J. Chromosomal rearrangements and oncogene amplification precede aneuploidization in the genetic evolution of breast cancer. Cancer Res. 2001, 61, 1214–1219. [Google Scholar]
- Ott, G.; Kalla, J.; Ott, M.M.; Schryen, B.; Katzenberger, T.; Müller, J.G.; Müller-Hermelink, H.K. Blastoid variants of mantle cell lymphoma: Frequent bcl-1 rearrangements at the major translocation cluster region and tetraploid chromosome clones. Blood 1997, 89, 1421–1429. [Google Scholar]
- Nelsen, C.J.; Kuriyama, R.; Hirsch, B.; Negron, V.C.; Lingle, W.L.; Goggin, M.M.; Stanley, M.W.; Albrecht, J.H. Short term cyclin D1 overexpression induces centrosome amplification, mitotic spindle abnormalities, and aneuploidy. J. Biol. Chem. 2005, 280, 768–776. [Google Scholar]
- Schraml, P.; Bucher, C.; Bissig, H.; Nocito, A.; Haas, P.; Wilber, K.; Seelig, S.; Kononen, J.; Mihatsch, M.J.; Dirnhofer, S.; et al. Cyclin E overexpression and amplification in human tumours. J. Pathol. 2003, 200, 375–382. [Google Scholar] [CrossRef]
- Jung, Y.J.; Lee, K.H.; Choi, D.W.; Han, C.J.; Jeong, S.H.; Kim, K.C.; Oh, J.W.; Park, T.K.; Kim, C.M. Reciprocal expressions of cyclin E and cyclin D1 in hepatocellular carcinoma. Cancer Lett. 2001, 168, 57–63. [Google Scholar] [CrossRef]
- Lv, Z.; Zhang, M.; Bi, J.; Xu, F.; Hu, S.; Wen, J. Promoter hypermethylation of a novel gene, ZHX2, in hepatocellular carcinoma. Am. J. Clin. Pathol. 2006, 125, 740–746. [Google Scholar] [CrossRef]
- Yue, X.; Zhang, Z.; Liang, X.; Gao, L.; Zhang, X.; Zhao, D.; Liu, X.; Ma, H.; Guo, M.; Spear, B.T.; et al. Zinc fingers and homeoboxes 2 inhibits hepatocellular carcinoma cell proliferation and represses expression of Cyclins A and E. Gastroenterology 2012, 142, 1559–1570.e2. [Google Scholar] [CrossRef]
- Tu, K.; Zheng, X.; Zan, X.; Han, S.; Yao, Y.; Liu, Q. Evaluation of Fbxw7 expression and its correlation with the expression of c-Myc, cyclin E and p53 in human hepatocellular carcinoma. Hepatol. Res. 2012, 42, 904–910. [Google Scholar] [CrossRef]
- Hagedorn, M.; Delugin, M.; Abraldes, I.; Allain, N.; Belaud-Rotureau, M.-A.; Turmo, M.; Prigent, C.; Loiseau, H.; Bikfalvi, A.; Javerzat, S. FBXW7/hCDC4 controls glioma cell proliferation in vitro and is a prognostic marker for survival in glioblastoma patients. Cell Div. 2007, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Akhoondi, S.; Sun, D.; von der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dafou, D.; Dofou, D.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef]
- Mao, J.-H.; Perez-losada, J.; Wu, D.; DelRosario, R.; Tsunematsu, R.; Nakayama, K.I.; Brown, K.; Bryson, S.; Balmain, A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004, 432, 775–779. [Google Scholar] [CrossRef]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef]
- Tu, K.; Zheng, X.; Zhou, Z.; Li, C.; Zhang, J.; Gao, J.; Yao, Y. Recombinant human adenovirus-p53 injection induced apoptosis in hepatocellular carcinoma cell lines mediated by p53-Fbxw7 pathway, which controls c-Myc and cyclin E. PLoS One 2013, 8, e68574. [Google Scholar]
- Li, K.; Lin, S.-Y.; Brunicardi, F.C.; Seu, P. Use of RNA interference to target cyclin E-overexpressing hepatocellular carcinoma. Cancer Res. 2003, 63, 3593–3597. [Google Scholar]
- Pok, S.; Wen, V.; Shackel, N.; Alsop, A.; Pyakurel, P.; Fahrer, A.; Farrell, G.C.; Teoh, N.C. Cyclin E facilitates dysplastic hepatocytes to bypass G 1/S checkpoint in hepatocarcinogenesis. J. Gastroenterol. Hepatol. 2013, 28, 1545–1554. [Google Scholar] [CrossRef]
- Erdal, E.; Ozturk, N.; Cagatay, T.; Eksioglu-Demiralp, E.; Ozturk, M. Lithium-mediated downregulation of PKB/Akt and cyclin E with growth inhibition in hepatocellular carcinoma cells. Int. J. Cancer 2005, 115, 903–910. [Google Scholar] [CrossRef]
- Sayan, A.E.; Sayan, B.S.; Findikli, N.; Ozturk, M. Acquired expression of transcriptionally active p73 in hepatocellular carcinoma cells. Oncogene 2001, 20, 5111–5117. [Google Scholar]
- Spruck, C.H.; Won, K.-A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef]
- Tane, S.; Chibazakura, T. Cyclin A overexpression induces chromosomal double-strand breaks in mammalian cells. Cell Cycle 2009, 8, 3900–3903. [Google Scholar] [CrossRef]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar]
- Chibazakura, T. Cyclin proteolysis and CDK inhibitors: Two redundant pathways to maintain genome stability in mammalian cells. Cell Cycle 2004, 3, 1243–1245. [Google Scholar]
- Wheeler, L.W.; Lents, N.H.; Baldassare, J.J. Cyclin A-CDK activity during G1 phase impairs MCM chromatin loading and inhibits DNA synthesis in mammalian cells. Cell Cycle 2008, 7, 2179–2188. [Google Scholar] [CrossRef]
- Wang, J.; Chenivesse, X.; Henglein, B.; Bréchot, C. Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature 1990, 343, 555–557. [Google Scholar] [CrossRef]
- Chao, Y.; Shih, Y.-L.; Chiu, J.-H.; Chau, G.-Y.; Lui, W.-Y.; Yang, W.K.; Lee, S.-D.; Huang, T.-S. Overexpression of cyclin A but not Skp 2 correlates with the tumor relapse of human hepatocellular carcinoma. Cancer Res. 1998, 58, 985–990. [Google Scholar]
- Bahnassy, A.A.; Zekri, A.-R.N.; Loutfy, S.A.; Mohamed, W.S.; Moneim, A.A.; Salem, S.E.; Sheta, M.M.; Omar, A.; Al-Zawahry, H. The role of cyclins and cyclin dependent kinases in development and progression of hepatitis C virus-genotype 4-associated hepatitis and hepatocellular carcinoma. Exp. Mol. Path. 2011, 91, 643–652. [Google Scholar] [CrossRef]
- Paterlini, P.; Flejou, J.-F.; de Mitri, M.S.; Pisi, E.; Franco, D.; Bréchot, C. Structure and expression of the cyclin A gene in human primary liver cancer. Correlation with flow cytometric parameters. J. Hepatol. 1995, 23, 47–52. [Google Scholar] [CrossRef]
- Nakajima, T.; Yasui, K.; Zen, K.; Inagaki, Y.; Fujii, H.; Minami, M.; Tanaka, S.; Taniwaki, M.; Itoh, Y.; Arii, S.; et al. Activation of B-Myb by E2F1 in hepatocellular carcinoma. Hepatol. Res. 2008, 38, 886–895. [Google Scholar]
- Park, T.J.; Kim, J.-Y.; Oh, S.P.; Kang, S.Y.; Kim, B.W.; Wang, H.J.; Song, K.Y.; Kim, H.C.; Lim, I.K. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology 2008, 47, 1533–1543. [Google Scholar] [CrossRef]
- Hui, A.-M.; Kanai, Y.; Sakamoto, M.; Tsuda, H.; Hirohashi, S. Reduced p21WAF1/CIP1 expression and p53 mutation in hepatocellular carcinomas. Hepatology 1997, 25, 575–579. [Google Scholar] [CrossRef]
- Qin, L.F.; Ng, I.O. Expression of p27KIP1 and p21WAF1/CIP1 in primary hepatocellular carcinoma: Clinicopathologic correlation and survival analysis. Hum. Pathol. 2001, 32, 778–784. [Google Scholar] [CrossRef]
- Kao, J.-T.; Chuah, S.-K.; Huang, C.-C.; Chen, C.-L.; Wang, C.-C.; Hung, C.-H.; Chen, C.-H.; Wang, J.-H.; Lu, S.-N.; Lee, C.-M.; et al. p21WAF1 is an independent survival prognostic factor for patients with hepatocellular carcinoma after resection. Liver Int. 2007, 27, 772–781. [Google Scholar] [CrossRef]
- Tannapfel, A.; Grund, D.; Katalinic, A.; Uhlmann, D.; Köckerling, F.; Haugwitz, U.; Wasner, M.; Hauss, J.; Engeland, K.; Wittekind, C. Decreased expression of p27 protein is associated with advanced tumor stage in hepatocellular carcinoma. Int. J. Cancer 2000, 89, 350–355. [Google Scholar] [CrossRef]
- Hui, A.-M.; Sun, L.; Kanai, Y.; Sakamoto, M.; Hirohashi, S. Reduced p27Kip1 expression in hepatocellular carcinomas. Cancer Lett. 1998, 132, 67–73. [Google Scholar] [CrossRef]
- Fiorentino, M.; Altimari, A.; D’Errico, A.; Cukor, B.; Barozzi, C.; Loda, M.; Grigioni, W.F. Acquired expression of p27 is a favorable prognostic indicator in patients with hepatocellular carcinoma. Clin. Cancer Res. 2000, 6, 3966–3972. [Google Scholar]
- Ray, R.B.; Steele, R.; Meyer, K.; Ray, R. Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene 1998, 208, 331–336. [Google Scholar] [CrossRef]
- Jung, E.Y.; Lee, M.N.; Yang, H.Y.; Yu, D.; Jang, K.L. The repressive activity of hepatitis C virus core protein on the transcription of p21Waf1 is regulated by protein kinase A-mediated phosphorylation. Virus Res. 2001, 79, 109–115. [Google Scholar] [CrossRef]
- Otsuka, M.; Kato, N.; Lan, K.; Yoshida, H.; Kato, J.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 2000, 275, 34122–34130. [Google Scholar]
- Lu, W.; Lo, S.Y.; Chen, M.; Wu, K.J.; Fung, Y.K.; Ou, J.H. Activation of p53 tumor suppressor by hepatitis C virus core protein. Virology 1999, 264, 134–141. [Google Scholar] [CrossRef]
- Yamanaka, T.; Kodama, T.; Doi, T. Subcellular localization of HCV core protein regulates its ability for p53 activation and p21 suppression. Biochem. Biophys. Res. Commun. 2002, 294, 528–534. [Google Scholar] [CrossRef]
- Shiu, T.-Y.; Huang, S.-M.; Shih, Y.-L.; Chu, H.-C.; Chang, W.-K.; Hsieh, T.-Y. Hepatitis C virus core protein down-regulates p21Waf1/Cip1 and inhibits curcumin-induced apoptosis through microRNA-345 targeting in human hepatoma cells. PLoS One 2013, 8, e61089. [Google Scholar]
- Nguyen, H.; Mudryj, M.; Guadalupe, M.; Dandekar, S. Hepatitis C virus core protein expression leads to biphasic regulation of the p21 cdk inhibitor and modulation of hepatocyte cell cycle. Virology 2003, 312, 245–253. [Google Scholar] [CrossRef]
- Qiao, L.; Leach, K.; McKinstry, R.; Gilfor, D.; Yacoub, A.; Park, J.S.; Grant, S.; Hylemon, P.B.; Fisher, P.B.; Dent, P. Hepatitis B virus X protein increases expression of p21Cip-1/WAF1/MDA6 and p27Kip-1 in primary mouse hepatocytes, leading to reduced cell cycle progression. Hepatology 2001, 34, 906–917. [Google Scholar] [CrossRef]
- Park, U.S.; Park, S.K.; Lee, Y.I.; Park, J.G. Hepatitis B virus-X protein upregulates the expression of p21waf1/cip1 and prolongs G1→S transition via a p53-independent pathway in human hepatoma cells. Oncogene 2000, 19, 3384. [Google Scholar] [CrossRef]
- Yano, M.; Ohkoshi, S.; Aoki, Y.-H.; Takahashi, H.; Kurita, S.; Yamazaki, K.; Suzuki, K.; Yamagiwa, S.; Sanpei, A.; Fujimaki, S.; et al. Hepatitis B virus X induces cell proliferation in the hepatocarcinogenesis via up-regulation of cytoplasmic p21 expression. Liver Int. 2013, 33, 1218–1229. [Google Scholar] [CrossRef]
- Han, H.J.; Jung, E.Y.; Lee, W.J.; Jang, K.L. Cooperative repression of cyclin-dependent kinase inhibitor p21 gene expression by hepatitis B virus X protein and hepatitis C virus core protein. FEBS Lett. 2002, 518, 169–172. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Chung, E.Y.; Kwun, H.J.; Jang, K.L. Transcriptional repression of p21Waf1 promoter by hepatitis B virus X protein via a p53-independent pathway. Gene 2001, 275, 163–168. [Google Scholar] [CrossRef]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Invest. 2008, 118, 3943–3953. [Google Scholar] [CrossRef]
- Buitrago-Molina, L.E.; Marhenke, S.; Longerich, T.; Sharma, A.D.; Boukouris, A.E.; Geffers, R.; Guigas, B.; Manns, M.P.; Vogel, A. The degree of liver injury determines the role of p21 in liver regeneration and hepatocarcinogenesis in mice. Hepatology 2013, 58, 1143–1152. [Google Scholar] [CrossRef]
- Willenbring, H.; Sharma, A.D.; Vogel, A.; Lee, A.Y.; Rothfuss, A.; Wang, Z.; Finegold, M.; Grompe, M. Loss of p21 permits carcinogenesis from chronically damaged liver and kidney epithelial cells despite unchecked apoptosis. Cancer Cell 2008, 14, 59–67. [Google Scholar] [CrossRef]
- Fero, M.L.; Rivkin, M.; Tasch, M.; Porter, P.; Carow, C.E.; Firpo, E.; Polyak, K.; Tsai, L.H.; Broudy, V.; Perlmutter, R.M.; et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell 1996, 85, 733–744. [Google Scholar] [CrossRef]
- Nakayama, K.; Ishida, N.; Shirane, M.; Inomata, A.; Inoue, T.; Shishido, N.; Horii, I.; Loh, D.Y.; Nakayama, K.-I. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 1996, 85, 707–720. [Google Scholar] [CrossRef]
- Kiyokawa, H.; Kineman, R.D.; Manova-Todorova, K.O.; Soares, V.C.; Hoffman, E.S.; Ono, M.; Khanam, D.; Hayday, A.C.; Frohman, L.A.; Koff, A. Enhanced growth of mice lacking the Cyclin-dependent kinase inhibitor function of p27Kip1. Cell 1996, 85, 721–732. [Google Scholar] [CrossRef]
- Sun, D.; Ren, H.; Oertel, M.; Sellers, R.S.; Zhu, L. Loss of p27Kip1 enhances tumor progression in chronic hepatocyte injury-induced liver tumorigenesis with widely ranging effects on Cdk2 or Cdc2 activation. Carcinogenesis 2007, 28, 1859–1866. [Google Scholar] [CrossRef]
- Matsuda, Y.; Wakai, T.; Kubota, M.; Takamura, M.; Yamagiwa, S.; Aoyagi, Y.; Osawa, M.; Fujimaki, S.; Sanpei, A.; Genda, T.; et al. Clinical significance of cell cycle inhibitors in hepatocellular carcinoma. Med. Mol. Morphol. 2013. [Google Scholar] [CrossRef]
- Lei, P.-P.; Zhang, Z.-J.; Shen, L.-J.; Li, J.-Y.; Zou, Q.; Zhang, H.-X. Expression and hypermethylation of p27kip1 in hepatocarcinogenesis. World J. Gastroenterol. 2005, 11, 4587–4591. [Google Scholar]
- Calvisi, D.F.; Ladu, S.; Pinna, F.; Frau, M.; Tomasi, M.L.; Sini, M.; Simile, M.M.; Bonelli, P.; Muroni, M.R.; Seddaiu, M.A.; et al. SKP2 and CKS1 promote degradation of cell cycle regulators and are associated with hepatocellular carcinoma prognosis. Gastroenterology 2009, 137, 1816–1826.e10. [Google Scholar] [CrossRef]
- Fornari, F.; Gramantieri, L.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Giovannini, C.; Croce, C.M.; Bolondi, L. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene 2008, 27, 5651–5661. [Google Scholar] [CrossRef]
- Fu, X.; Wang, Q.; Chen, J.; Huang, X.; Chen, X.; Cao, L.; Tan, H.; Li, W.; Zhang, L.; Bi, J.; et al. Clinical significance of miR-221 and its inverse correlation with p27Kip1 in hepatocellular carcinoma. Mol. Biol. Rep. 2011, 38, 3029–3035. [Google Scholar] [CrossRef]
- Rong, M.; Chen, G.; Dang, Y. Increased miR-221 expression in hepatocellular carcinoma tissues and its role in enhancing cell growth and inhibiting apoptosis in vitro. BMC Cancer 2013, 13, 21. [Google Scholar] [CrossRef]
- Lee, E.-K.; Kim, D.-G.; Kim, J.-S.; Yoon, Y. Cell-cycle regulator Cks1 promotes hepatocellular carcinoma by supporting NF-κB-dependent expression of interleukin-8. Cancer Res. 2011, 71, 6827–6835. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, Q.; Ni, R.; Huang, X.; Wang, Y.; Lu, C.; Shen, A.; Wang, Y.; Li, C.; Yuan, Q.; et al. Early mitotic inhibitor-1, an anaphase-promoting complex/cyclosome inhibitor, can control tumor cell proliferation in hepatocellular carcinoma: Correlation with Skp2 stability and degradation of p27Kip1. Hum. Pathol. 2013, 44, 365–373. [Google Scholar] [CrossRef]
- Vodermaier, H.C. APC/C and SCF: Controlling each other and the cell cycle. Curr. Biol. 2004, 14, R787–R796. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Chan, C.-H.; Lee, S.-W.; Wang, J.; Lin, H.-K. Regulation of Skp2 expression and activity and its role in cancer progression. TheScientificWorldJournal 2010, 10, 1001–1015. [Google Scholar] [CrossRef]
- He, S.; Lu, M.; Xue, W.; Wang, Y.; Zhao, Y.; Gao, S.; Ke, Q.; Liu, Y.; Li, P.; Cui, X.; et al. Phosphorylated p27Kip1 on Thr157 is an important prognosis in human hepatocellular carcinoma in vivo and in vitro. Med. Oncol. 2010, 28, 94–104. [Google Scholar]
- Lu, M.; Fei, M.; Cheng, C.; Wang, Y.; He, S.; Zhao, Y.; Gao, S.; Ke, Q.; Li, P.; Cui, X.; et al. Mutant p27Kip1 and its potential effect as hepatocellular gene therapy. Arch. Med. Res. 2008, 39, 573–581. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ichida, T.; Genda, T.; Yamagiwa, S.; Aoyagi, Y.; Asakura, H. Loss of p16 contributes to p27 sequestration by cyclin D1-cyclin-dependent kinase 4 complexes and poor prognosis in hepatocellular carcinoma. Clin. Cancer Res. 2003, 9, 3389–3396. [Google Scholar]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef]
- Rivadeneira, D.B.; Mayhew, C.N.; Thangavel, C.; Sotillo, E.; Reed, C.A.; Graña, X.; Knudsen, E.S. Proliferative suppression by CDK4/6 inhibition: Complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology 2010, 138, 1920–1930.e2. [Google Scholar]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.-K.; Perou, C.M.; Sharpless, N.E. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- Huang, X.; di Liberto, M.; Jayabalan, D.; Liang, J.; Ely, S.; Bretz, J.; Shaffer, A.L.; Louie, T.; Chen, I.; Randolph, S.; et al. Prolonged early G1 arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood 2012, 120, 1095–1106. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bisteau, X.; Caldez, M.J.; Kaldis, P. The Complex Relationship between Liver Cancer and the Cell Cycle: A Story of Multiple Regulations. Cancers 2014, 6, 79-111. https://doi.org/10.3390/cancers6010079
Bisteau X, Caldez MJ, Kaldis P. The Complex Relationship between Liver Cancer and the Cell Cycle: A Story of Multiple Regulations. Cancers. 2014; 6(1):79-111. https://doi.org/10.3390/cancers6010079
Chicago/Turabian StyleBisteau, Xavier, Matias J. Caldez, and Philipp Kaldis. 2014. "The Complex Relationship between Liver Cancer and the Cell Cycle: A Story of Multiple Regulations" Cancers 6, no. 1: 79-111. https://doi.org/10.3390/cancers6010079