Recent Advances in the Molecular Characterization of Circulating Tumor Cells

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Metastatic Disease and Circulating Tumor Cells
1.2. Clinical Applications for CTCs
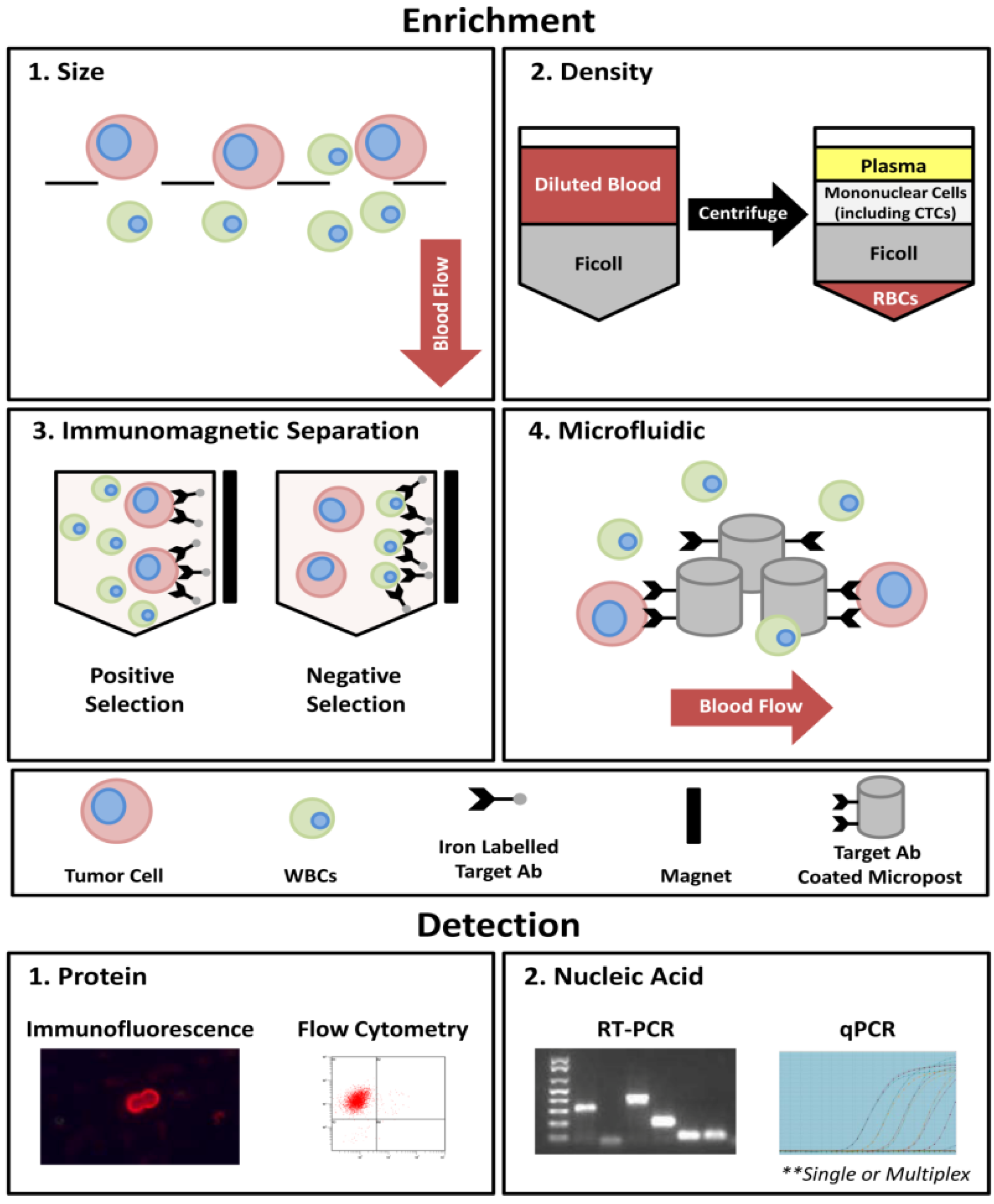
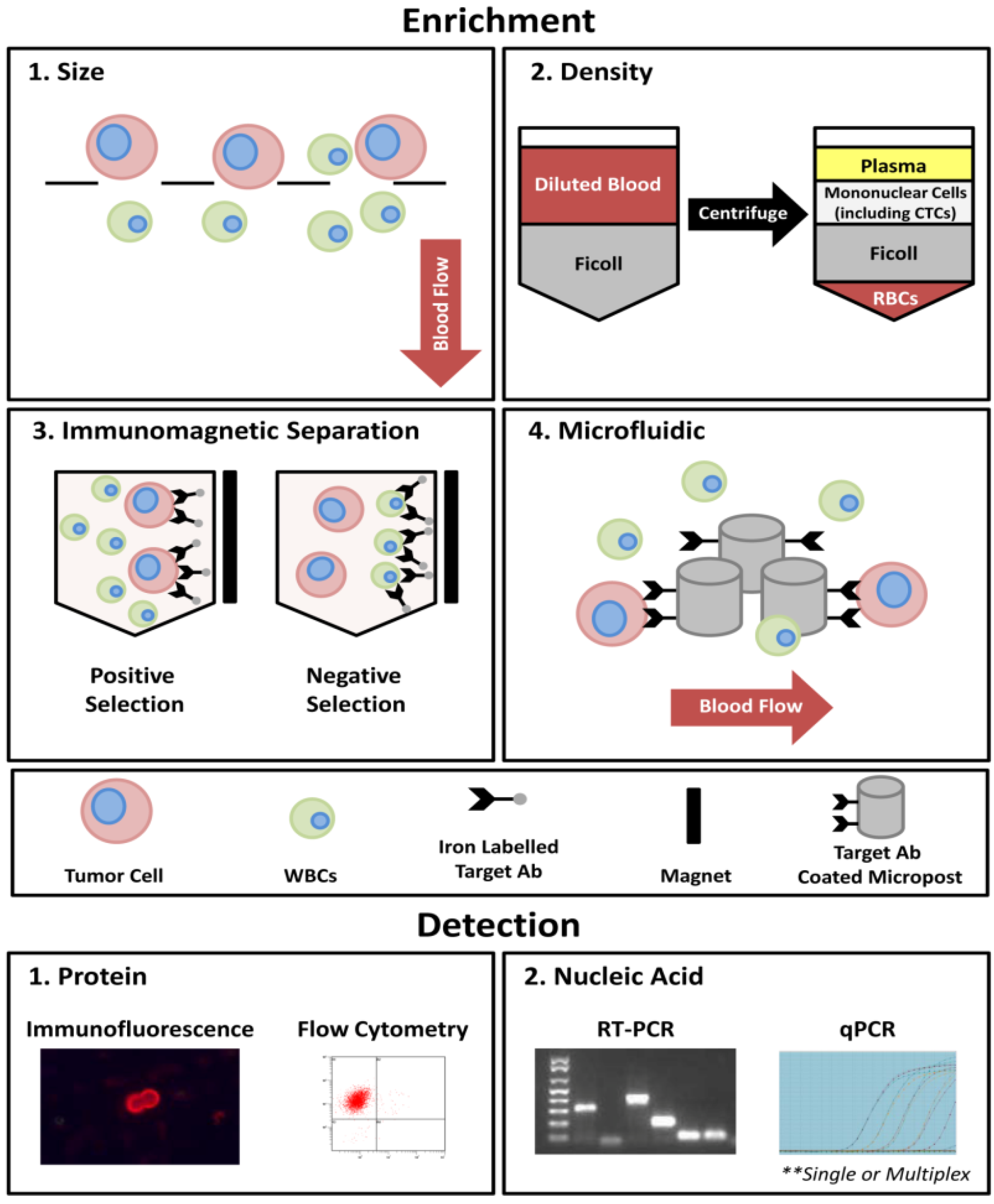
1.3. CTCs as Surrogate Biomarkers for Metastatic Biopsy
2. CTC Molecular Characterization Approaches
2.1. Protein-Based CTC Characterization Techniques
2.1.1. Immunofluorescence
2.1.2. Flow Cytometry
2.2. Nucleic Acid-Based CTC Characterization Approaches
2.2.1. Fluorescence in Situ Hybridization (FISH)
2.2.2. Reverse Transcription Polymerase Chain Reaction (RT-PCR) and Reverse Transcription Quantitative PCR (RT-qPCR)
2.2.3. Microarrays
2.2.4. Sequencing
2.3. General Considerations for CTC Characterization
3. Clinical Significance of CTC Molecular Characterization: HER2 as a Proof-of-Principle Marker
3.1. The Role of HER2 in Breast Cancer and Clinical Assessment
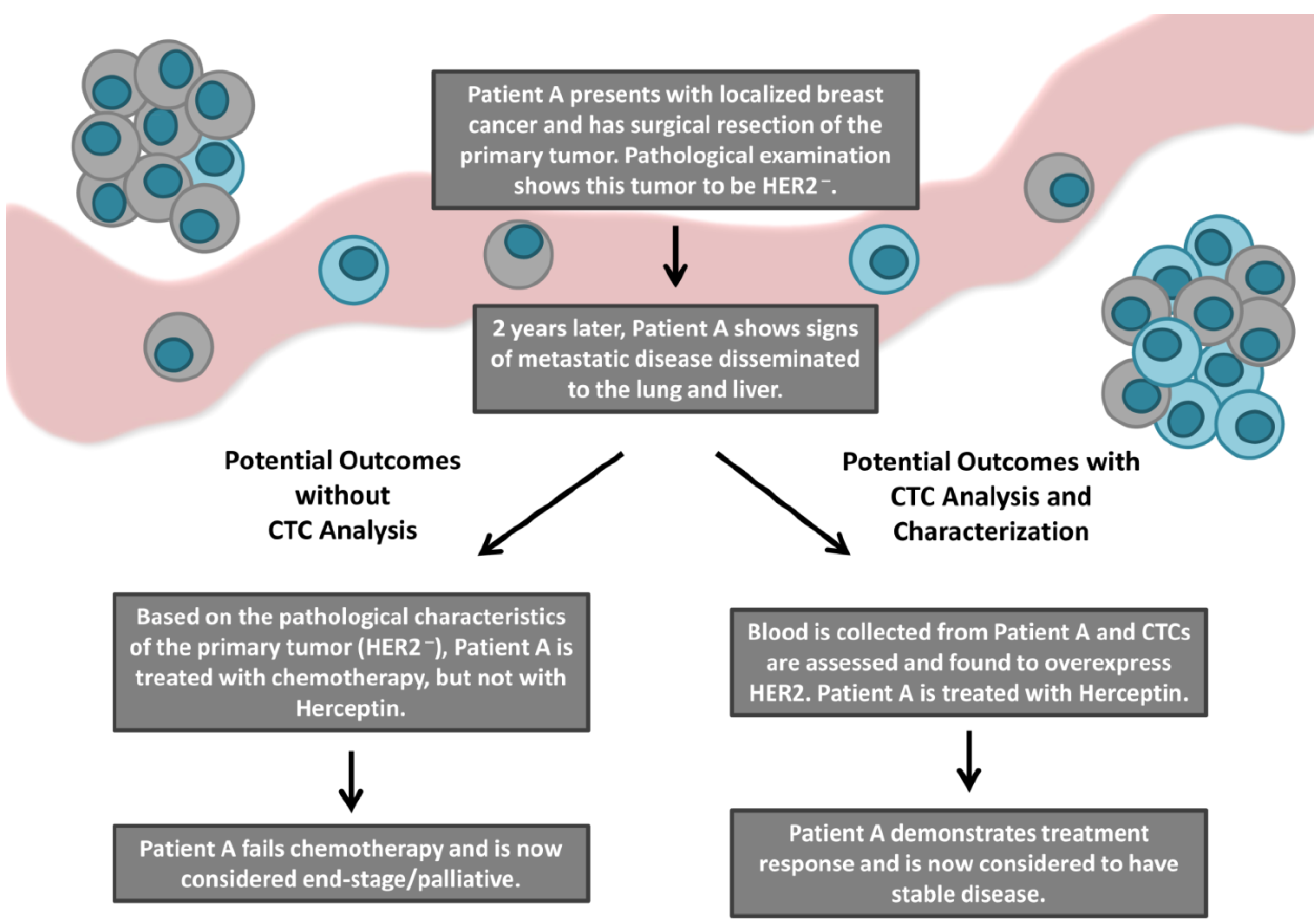
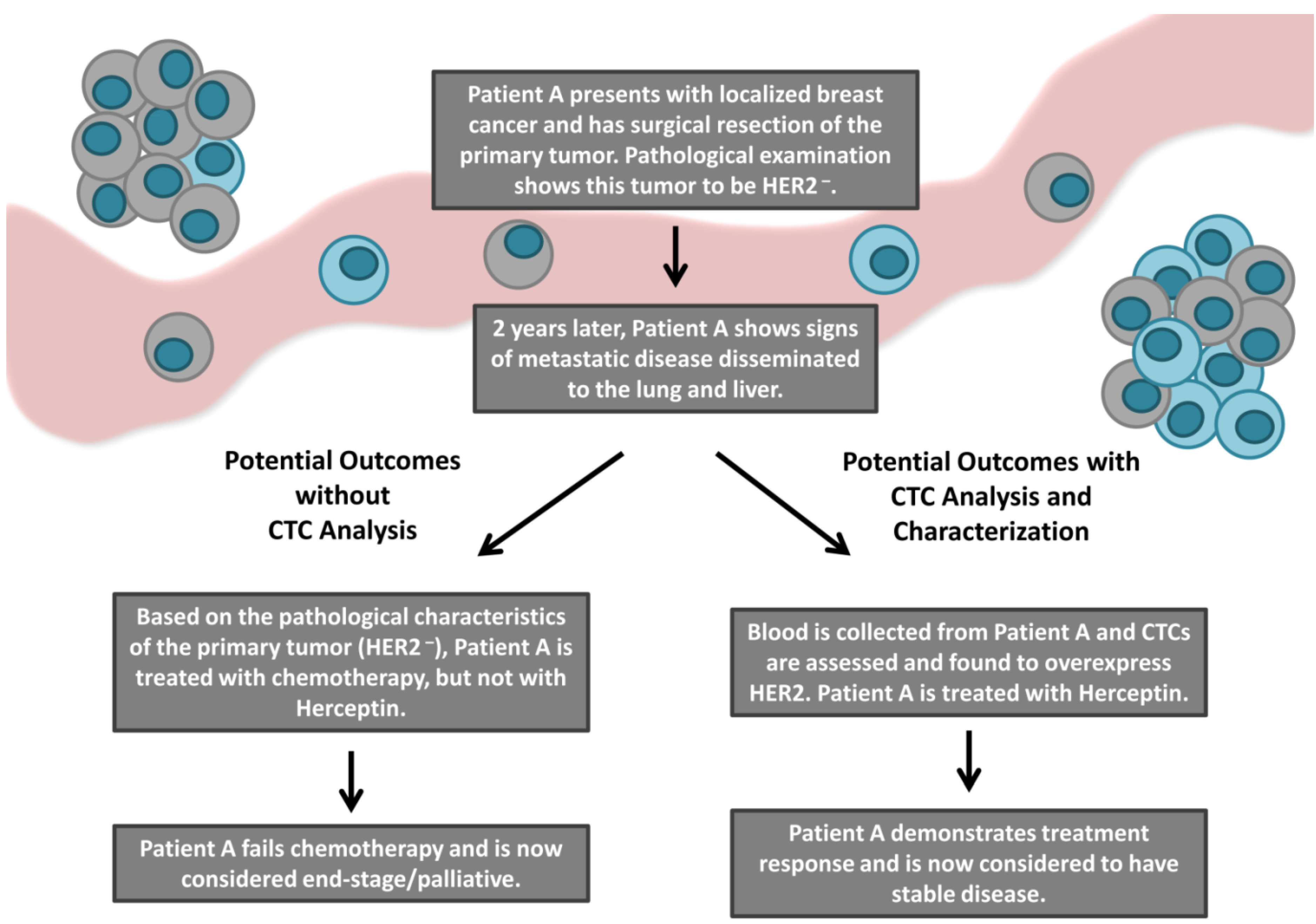
3.2. Discordance in HER2 Expression between Tumor Tissue and CTCs
3.3. Clinical Outcomes Based on HER2 Discrepancies
4. Conclusions: Current Limitations and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Sethi, N.; Kang, Y. Unravelling the complexity of metastasis—Molecular understanding and targeted therapies. Nat. Rev. Cancer 2011, 11, 735–748. [Google Scholar] [CrossRef]
- Aitken, S.J.; Thomas, J.S.; Langdon, S.P.; Harrison, D.J.; Faratian, D. Quantitative analysis of changes in ER, PR and HER2 expression in primary breast cancer and paired nodal metastases. Ann. Oncol. 2010, 21, 1254–1261. [Google Scholar] [CrossRef]
- Carlsson, J.; Nordgren, H.; Sjöström, J.; Wester, K.; Villman, K.; Bengtsson, N.O.; Ostenstad, B.; Lundqvist, H.; Blomqvist, C. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br. J. Cancer 2004, 90, 2344–2348. [Google Scholar]
- Regitnig, P.; Schippinger, W.; Lindbauer, M.; Samonigg, H.; Lax, S.F. Change of HER-2/neu status in a subset of distant metastases from breast carcinomas. J. Pathol. 2004, 203, 918–926. [Google Scholar] [CrossRef]
- Gancberg, D.; di Leo, A.; Cardoso, F.; Rouas, G.; Pedrocchi, M.; Paesmans, M.; Verhest, A.; Bernard-Marty, C.; Piccart, M.J.; Larsimont, D. Comparison of HER-2 status between primary breast cancer and corresponding distant metastatic sites. Ann. Oncol. 2002, 13, 1036–1043. [Google Scholar] [CrossRef]
- Vincent-Salomon, A.; Pierga, J.-Y.; Couturier, J.; d’Enghien, C.D.; Nos, C.; Sigal-Zafrani, B.; Lae, M.; Fréneaux, P.; Diéras, V.; Thiéry, J.-P.; et al. HER2 status of bone marrow micrometastasisand their corresponding primary tumours in a pilot study of 27 cases: A possible tool for anti-HER2 therapy management? Br. J. Cancer 2007, 96, 654–659. [Google Scholar] [CrossRef]
- Jensen, J.D.; Knoop, A.; Ewertz, M.; Laenkholm, A.V. ER, HER2, and TOP2A expression in primary tumor, synchronous axillary nodes, and asynchronous metastases in breast cancer. Breast Cancer Res. Treat. 2012, 132, 511–521. [Google Scholar] [CrossRef]
- Jabbour, M.N.; Massad, C.Y.; Boulos, F.I. Variability in hormone and growth factor receptor expression in primaryversus recurrent, metastatic, and post-neoadjuvant breast carcinoma. Breast Cancer Res. Treat. 2012, 135, 29–37. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H.; Brandt, B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer 2008, 8, 329–340. [Google Scholar] [CrossRef]
- Ashworth, T.R. A case of cancer in which cells similar to those in the tumors were seen in the blood after death. Aust. Med. J. 1869, 14, 146–147. [Google Scholar]
- Lowes, L.E.; Goodale, D.; Keeney, M.; Allan, A.L. Image cytometry analysis of circulating tumor cells. Methods Cell Biol. 2011, 102, 261–290. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef]
- Lianidou, E.S.; Markou, A. Circulating tumor cells in breast cancer: Detection systems, molecular characterization, and future challenges. Clin. Chem. 2011, 57, 1242–1255. [Google Scholar] [CrossRef]
- Yu, M.; Stott, S.; Toner, M.; Maheswaran, S.; Haber, D.A. Circulating tumor cells: Approaches to isolation and characterization. J. Cell Biol. 2011, 192, 373–382. [Google Scholar] [CrossRef]
- Mostert, B.; Sleijfer, S.; Foekens, J.A.; Gratama, J.W. Circulating tumor cells (CTCs): Detection methods and their clinical relevance in breast cancer. Cancer Treat. Rev. 2009, 35, 463–474. [Google Scholar] [CrossRef]
- Alunni-Fabbroni, M.; Sandri, M.T. Circulating tumour cells in clinical practice: Methods of detection and possible characterization. Methods 2010, 50, 289–297. [Google Scholar] [CrossRef]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef]
- Ozkumur, E.; Shah, A.M.; Ciciliano, J.C.; Emmink, B.L.; Miyamoto, D.T.; Brachtel, E.; Yu, M.; Chen, P.I.; Morgan, B.; Trautwein, J.; et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar]
- Peeters, D.J.; de Laere, B.; van den Eynden, G.G.; van Laere, S.J.; Rothé, F.; Ignatiadis, M.; Sieuwerts, A.M.; Lambrechts, D.; Rutten, A.; van Dam, P.A.; et al. Semiautomated isolation and molecular characterisation of single or highly purified tumour cells from CellSearch enriched blood samples using dielectrophoretic cell sorting. Br. J. Cancer 2013, 108, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabières, C.; Vendrell, J.P.; Slijper, M.; Pellé, O.; Barbotte, E.; Mercier, G.; Jacot, W.; Fabbro, M.; Pantel, K. Full-length cytokeratin-19 is released by human tumor cells: A potential role in metastatic progression of breast cancer. Breast Cancer Res. 2009, 11, R39. [Google Scholar] [CrossRef]
- Alix-Panabières, C. EPISPOT assay: Detection of viable DTCs/CTCs in solid tumor patients. Recent Res. Cancer 2012, 195, 69–76. [Google Scholar] [CrossRef]
- Denève, E.; Riethdorf, S.; Ramos, J.; Nocca, D.; Coffy, A.; Daurès, J.P.; Maudelonde, T.; Fabre, J.M.; Pantel, K.; Alix-Panabières, C. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin. Chem. 2013, 59, 1384–1392. [Google Scholar] [CrossRef]
- Tibbe, A.G.J.; Miller, M.C.; Terstappen, L.W. Statistical considerations for enumeration of circulating tumor cells. Cytometry A 2007, 71, 154–162. [Google Scholar]
- Ross, A.A.; Cooper, B.W.; Lazarus, H.M.; Mackey, W.; Moss, T.J.; Ciobanu, N.; Tallman, M.S.; Kennedy, M.J.; Davidson, N.E.; Sweet, D.; et al. Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood 1993, 82, 2605–2610. [Google Scholar]
- Sleijfer, S.; Gratama, J.-W.; Sieuwerts, A.M.; Kraan, J.; Martens, J.W.M.; Foekens, J.A. Circulating tumour cell detection on its way to routine diagnostic implementation? Eur. J. Cancer 2007, 43, 2645–2650. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V; Allard, W.J.; Terstappen, L.W.M.M.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V; Terstappen, L.W.W.M.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W.W.M. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res. 2006, 12, 4218–4224. [Google Scholar] [CrossRef]
- Budd, G.T.; Cristofanilli, M.; Ellis, M.J.; Stopeck, A.; Borden, E.; Miller, M.C.; Matera, J.; Repollet, M.; Doyle, G.V.; Terstappen, L.W.M.M.; et al. Circulating tumor cells versus imaging—Predicting overall survival in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 6403–6409. [Google Scholar] [CrossRef]
- Olmos, D.; Arkenau, H.-T.; Ang, J.E.; Ledaki, I.; Attard, G.; Carden, C.P.; Reid, A.H.M.; A’Hern, R.; Fong, P.C.; Oomen, N.B.; et al. Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): A single-centre experience. Ann. Oncol. 2009, 20, 27–33. [Google Scholar]
- Punnoose, E.A.; Atwal, S.K.; Spoerke, J.M.; Savage, H.; Pandita, A.; Yeh, R.-F.; Pirzkall, A.; Fine, B.M.; Amler, L.C.; Chen, D.S.; et al. Molecular biomarker analyses using circulating tumor cells. PLoS One 2010, 5, e12517. [Google Scholar] [CrossRef]
- Pestrin, M.; Bessi, S.; Galardi, F.; Truglia, M.; Biggeri, A.; Biagioni, C.; Cappadona, S.; Biganzoli, L.; Giannini, A.; di Leo, A. Correlation of HER2 status between primary tumors and corresponding circulating tumor cells in advanced breast cancer patients. Breast Cancer Res. Treat. 2009, 118, 523–530. [Google Scholar] [CrossRef]
- Shaffer, D.R.; Leversha, M.A.; Danila, D.C.; Lin, O.; Gonzalez-Espinoza, R.; Gu, B.; Anand, A.; Smith, K.; Maslak, P.; Doyle, G.V.; et al. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clin. Cancer Res. 2007, 13, 2023–2029. [Google Scholar] [CrossRef]
- Meng, S.; Tripathy, D.; Shete, S.; Ashfaq, R.; Haley, B.; Perkins, S.; Beitsch, P.; Khan, A.; Euhus, D.; Osborne, C.; et al. HER-2 gene amplification can be acquired as breast cancer progresses. Proc. Natl. Acad. Sci. USA 2004, 101, 9393–9398. [Google Scholar] [CrossRef]
- Meng, S.; Tripathy, D.; Shete, S.; Ashfaq, R.; Saboorian, H.; Haley, B.; Frenkel, E.; Euhus, D.; Leitch, M.; Osborne, C.; et al. uPAR and HER-2 gene status in individual breast cancer cells from blood and tissues. Proc. Natl. Acad. Sci. USA 2006, 103, 17361–17365. [Google Scholar] [CrossRef]
- Fehm, T.; Müller, V.; Aktas, B.; Janni, W.; Schneeweiss, A.; Stickeler, E.; Lattrich, C.; Löhberg, C.R.; Solomayer, E.; Rack, B.; et al. HER2 status of circulating tumor cells in patients with metastatic breast cancer: A prospective, multicenter trial. Breast Cancer Res. Treat. 2010, 124, 403–412. [Google Scholar] [CrossRef]
- Munzone, E.; Nolé, F.; Goldhirsch, A.; Botteri, E.; Esposito, A.; Zorzino, L.; Curigliano, G.; Minchella, I.; Adamoli, L.; Cassatella, M.C.; et al. Changes of HER2 status in circulating tumor cells compared with the primary tumor during treatment for advanced breast cancer. Clin. Breast Cancer 2010, 10, 392–397. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Rothé, F.; Chaboteaux, C.; Durbecq, V.; Rouas, G.; Criscitiello, C.; Metallo, J.; Kheddoumi, N.; Singhal, S.K.; Michiels, S.; et al. HER2-positive circulating tumor cells in breast cancer. PLoS One 2011, 6, e15624. [Google Scholar] [CrossRef]
- Somlo, G.; Lau, S.K.; Frankel, P.; Hsieh, H.B.; Liu, X.; Yang, L.; Krivacic, R.; Bruce, R.H. Multiple biomarker expression on circulating tumor cells in comparison to tumor tissues from primary and metastatic sites in patients with locally advanced/inflammatory, and stage IV breast cancer, using a novel detection technology. Breast Cancer Res. Treat. 2011, 128, 155–163. [Google Scholar] [CrossRef]
- Hayashi, N.; Nakamura, S.; Tokuda, Y.; Shimoda, Y.; Yagata, H.; Yoshida, A.; Ota, H.; Hortobagyi, G.N.; Cristofanilli, M.; Ueno, N.T. Prognostic value of HER2-positive circulating tumor cells in patients with metastatic breast cancer. Int. J. Clin. Oncol. 2012, 17, 96–104. [Google Scholar] [CrossRef]
- Pestrin, M.; Bessi, S.; Puglisi, F.; Minisini, A.M.; Masci, G.; Battelli, N.; Ravaioli, A.; Gianni, L.; di Marsico, R.; Tondini, C.; et al. Final results of a multicenter phase II clinical trial evaluating the activity of single-agent lapatinib in patients with HER2-negative metastatic breast cancer and HER2-positive circulating tumor cells. A proof-of-concept study. Breast Cancer Res. Treat. 2012, 134, 283–289. [Google Scholar] [CrossRef]
- Hayes, D.F.; Walker, T.M.; Singh, B.; Vitetta, E.S.; Uhr, J.W.; Gross, S.; Rao, C.; Doyle, G.V.; Terstappen, L.W.M.M. Monitoring expression of HER-2 on circulating epithelial cells in patients with advanced breast cancer. Int. J. Oncol. 2002, 21, 1111–1117. [Google Scholar]
- Riethdorf, S.; Müller, V.; Zhang, L.; Rau, T.; Loibl, S.; Komor, M.; Roller, M.; Huober, J.; Fehm, T.; Schrader, I.; et al. Detection and HER2 expression of circulating tumor cells: Prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin. Cancer Res. 2010, 16, 2634–2645. [Google Scholar] [CrossRef]
- Cao, S.; Li, Y.; Li, J.; Li, C.; Zhang, W.; Yang, Z.-Q.; Meng, S.-D. Quantitative determination of HER2 expression by confocal microscopy assay in CTCs of breast cancer. Oncol. Rep. 2010, 23, 423–428. [Google Scholar]
- Fehm, T.; Becker, S.; Duerr-Stoerzer, S.; Sotlar, K.; Mueller, V.; Wallwiener, D.; Lane, N.; Solomayer, E.; Uhr, J. Determination of HER2 status using both serum HER2 levels and circulating tumor cells in patients with recurrent breast cancer whose primary tumor was HER2 negative or of unknown HER2 status. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef]
- Nadal, R.; Fernandez, A.; Sanchez-Rovira, P.; Salido, M.; Rodríguez, M.; García-Puche, J.L.; Macià, M.; Corominas, J.M.; Delgado-Rodriguez, M.; Gonzalez, L.; et al. Biomarkers characterization of circulating tumour cells in breast cancer patients. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef]
- Stebbing, J.; Payne, R.; Reise, J.; Frampton, A.E.; Avery, M.; Woodley, L.; di Leo, A.; Pestrin, M.; Krell, J.; Coombes, R.C. The efficacy of lapatinib in metastatic breast cancer with HER2 non-amplified primary tumors and EGFR positive circulating tumor cells: A proof-of-concept study. PLoS One 2013, 8, e62543. [Google Scholar] [CrossRef]
- Miyamoto, D.T.; Lee, R.J.; Stott, S.L.; Ting, D.T.; Wittner, B.S.; Ulman, M.; Smas, M.E.; Lord, J.B.; Brannigan, B.W.; Trautwein, J.; et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov. 2012, 2, 995–1003. [Google Scholar] [CrossRef]
- Lazar, D.C.; Cho, E.H.; Luttgen, M.S.; Metzner, T.J.; Uson, M.L.; Torrey, M.; Gross, M.E.; Kuhn, P. Cytometric comparisons between circulating tumor cells from prostate cancer patients and the prostate-tumor-derived LNCaP cell line. Phys. Biol. 2012, 9. [Google Scholar] [CrossRef]
- Lin, H.K.; Zheng, S.; Williams, A.J.; Balic, M.; Groshen, S.; Scher, H.I.; Fleisher, M.; Stadler, W.; Datar, R.H.; Tai, Y.-C.; et al. Portable filter-based microdevice for detection and characterization of circulating tumor cells. Clin. Cancer Res. 2010, 16, 5011–5018. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Mittag, A.; Tarnok, A. Recent advances in cytometry applications: Preclinical, clinical, and cell biology. Methods Cell Biol. 2011, 103, 1–20. [Google Scholar] [CrossRef]
- Jaye, D.L.; Bray, R.A.; Gebel, H.M.; Harris, W.A.C.; Waller, E.K. Translational applications of flow cytometry in clinical practice. J. Immunol. 2012, 188, 4715–4719. [Google Scholar] [CrossRef]
- Tinhofer, I.; Hristozova, T.; Stromberger, C.; Keilhoiz, U.; Budach, V. Monitoring of circulating tumor cells and their expression of EGFR/phospho-EGFR during combined radiotherapy regimens in locally advanced squamous cell carcinoma of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, e685–e690. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Hristozova, T.; Konschak, R.; Budach, V.; Tinhofer, I. A simple multicolor flow cytometry protocol for detection and molecular characterization of circulating tumor cells in epithelial cancers. Cytometry A 2012, 81, 489–495. [Google Scholar]
- Wang, L.; Wang, Y.; Liu, Y.; Cheng, M.; Wu, X.; Wei, H. Flow cytometric analysis of CK19 expression in the peripheral blood of breast carcinoma patients: Relevance for circulating tumor cell detection. J. Exp. Clin. Cancer Res. 2009, 28. [Google Scholar] [CrossRef]
- Hu, Y.; Fan, L.; Zheng, J.; Cui, R.; Liu, W.; He, Y.; Li, X.; Huang, S. Detection of circulating tumor cells in breast cancer patients utilizing multiparameter flow cytometry and assessment of the prognosis of patients in different CTCs levels. Cytometry A 2010, 77, 213–219. [Google Scholar]
- Pailler, E.; Adam, J.; Barthélémy, A.; Oulhen, M.; Auger, N.; Valent, A.; Borget, I.; Planchard, D.; Taylor, M.; André, F.; et al. Detection of circulating tumor cells harboring a unique ALK rearrangement in ALK-positive non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 2273–2281. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Bischoff, F.; Ann Mayer, J.; Wong, K.; Pham, T.; Kuerer, H.; Lodhi, A.; Bhattacharyya, A.; Hall, C.; Lucci, A. Discordance in HER2 gene amplification in circulating and disseminated tumor cells in patients with operable breast cancer. Cancer Med. 2013, 2, 226–233. [Google Scholar] [CrossRef]
- Ilie, M.; Long, E.; Butori, C.; Hofman, V.; Coelle, C.; Mauro, V.; Zahaf, K.; Marquette, C.H.; Mouroux, J.; Paterlini-Bréchot, P.; et al. ALK-gene rearrangement: A comparative analysis on circulating tumour cells and tumour tissue from patients with lung adenocarcinoma. Ann. Oncol. 2012, 23, 2907–2913. [Google Scholar] [CrossRef]
- Flores, L.M.; Kindelberger, D.W.; Ligon, A.H.; Capelletti, M.; Fiorentino, M.; Loda, M.; Cibas, E.S.; Jänne, P.A.; Krop, I.E. Improving the yield of circulating tumour cells facilitates molecular characterisation and recognition of discordant HER2 amplification in breast cancer. Br. J. Cancer 2010, 102, 1495–1502. [Google Scholar] [CrossRef]
- Leversha, M.A.; Han, J.; Asgari, Z.; Danila, D.C.; Lin, O.; Anand, A.; Lilja, H.; Heller, G.; Fleisher, M.; Scher, H.I. Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin. Cancer Res. 2009, 15, 2091–2097. [Google Scholar] [CrossRef]
- Mao, X.; Shaw, G.; James, S.Y.; Purkis, P.; Kudahetti, S.C.; Tsigani, T.; Kia, S.; Young, B.D.; Oliver, R.T.D.; Berney, D.; et al. Detection of TMPRSS2:ERG fusion gene in circulating prostate cancer cells. Asian J. Androl. 2008, 10, 467–473. [Google Scholar] [CrossRef]
- Attard, G.; Swennenhuis, J.F.; Olmos, D.; Reid, A.H.M.; Vickers, E.; A’Hern, R.; Levink, R.; Coumans, F.; Moreira, J.; Riisnaes, R.; et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009, 69, 2912–2918. [Google Scholar] [CrossRef]
- Punnoose, E.; Tucker, E.; Marrinucci, D.; Amler, L.C.; Koeppen, H.; Patel, P.H.; Yan, Y.; Riisnaes, R.; Attard, G.; de Bono, J.S. Evaluation of PTEN status in circulating tumor cells (CTCs) and matched tumor tissue from patients with castrate-resistant prostate cancer (CRPC). In Proceedings of the 2013 ASCO Genitourinary Cancers Symposium, Orlando, FL, USA, 14–16 Febuary 2013.
- Mayer, J.A.; Pham, T.; Wong, K.L.; Scoggin, J.; Sales, E.V.; Clarin, T.; Pircher, T.J.; Mikolajczyk, S.D.; Cotter, P.D.; Bischoff, F.Z. FISH-based determination of HER2 status in circulating tumor cells isolated with the microfluidic CEE™ platform. Cancer Genet. 2011, 204, 589–595. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 2007, 25, 118–145. [Google Scholar]
- Koksal, I.T.; Dirice, E.; Yasar, D.; Sanlioglu, A.D.; Ciftcioglu, A.; Gulkesen, K.H.; Ozes, N.O.; Baykara, M.; Luleci, G.; Sanlioglu, S. The assessment of PTEN tumor suppressor gene in combination with Gleason scoring and serum PSA to evaluate progression of prostate carcinoma. Urol. Oncol. 2004, 22, 307–312. [Google Scholar] [CrossRef]
- Besson, A.; Robbins, S.M.; Yong, V.W. PTEN/MMAC1/TEP1 in signal transduction and tumorigenesis. Eur. J. Biochem. 1999, 263, 605–611. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Cunha, I.W.; Coudry, R.A.; Fonseca, F.P.; Torres, C.H.; Soares, F.A.; Squire, J.A. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br. J. Cancer 2007, 97, 678–685. [Google Scholar] [CrossRef]
- Fehm, T.; Hoffmann, O.; Aktas, B.; Becker, S.; Solomayer, E.F.; Wallwiener, D.; Kimmig, R.; Kasimir-Bauer, S. Detection and characterization of circulating tumor cells in blood of primary breast cancer patients by RT-PCR and comparison to status of bone marrow disseminated cells. Breast Cancer Res. 2009, 11. [Google Scholar] [CrossRef]
- Aktas, B.; Müller, V.; Tewes, M.; Zeitz, J.; Kasimir-Bauer, S.; Loehberg, C.R.; Rack, B.; Schneeweiss, A.; Fehm, T. Comparison of estrogen and progesterone receptor status of circulating tumor cells and the primary tumor in metastatic breast cancer patients. Gynecol. Oncol. 2011, 122, 356–360. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Kraan, J.; Bolt-de Vries, J.; van der Spoel, P.; Mostert, B.; Martens, J.W.M.; Gratama, J.-W.; Sleijfer, S.; Foekens, J.A. Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex Real-Time PCR. Breast Cancer Res. Treat. 2009, 118, 455–468. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Mostert, B.; Bolt-de Vries, J.; Peeters, D.; de Jongh, F.E.; Stouthard, J.M.L.; Dirix, L.Y.; van Dam, P.A.; van Galen, A.; de Weerd, V.; et al. mRNA and microRNA expression profiles in circulating tumor cells and primary tumors of metastatic breast cancer patients. Clin. Cancer Res. 2011, 17, 3600–3618. [Google Scholar] [CrossRef]
- Markou, A.; Strati, A.; Malamos, N.; Georgoulias, V.; Lianidou, E.S. Molecular characterization of circulating tumor cells in breast cancer by a liquid bead array hybridization assay. Clin. Chem. 2011, 57, 421–430. [Google Scholar] [CrossRef]
- De Albuquerque, A.; Kaul, S.; Breier, G.; Krabisch, P.; Fersis, N. Multimarker analysis of circulating tumor cells in peripheral blood of metastatic breast cancer patients: A step forward in personalized medicine. Breast Care 2012, 7, 7–12. [Google Scholar] [CrossRef]
- Chimonidou, M.; Strati, A.; Tzitzira, A.; Sotiropoulou, G.; Malamos, N.; Georgoulias, V.; Lianidou, E.S. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin. Chem. 2011, 57, 1169–1177. [Google Scholar] [CrossRef]
- Mostert, B.; Jiang, Y.; Sieuwerts, A.M.; Wang, H.; Bolt-de Vries, J.; Biermann, K.; Kraan, J.; Lalmahomed, Z.; van Galen, A.; de Weerd, V.; et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int. J. Cancer 2013, 133, 130–141. [Google Scholar] [CrossRef]
- Lowes, L.E.; Lock, M.; Rodrigues, G.; D’Souza, D.; Bauman, G.; Ahmad, B.; Venkatesan, V.; Allan, A.L.; Sexton, T. Circulating tumour cells in prostate cancer patients receiving salvage radiotherapy. Clin. Trans. Oncol. 2012, 14, 150–156. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Sosa, E.V.; Roy, R.; Eisenbud, L.E.; Scott, J.H.; Olshen, A.; Pinkel, D.; Rugo, H.S.; Park, J.W. Genomic profiling of isolated circulating tumor cells from metastatic breast cancer patients. Cancer Res. 2013, 73, 30–40. [Google Scholar] [CrossRef]
- Barbazán, J.; Alonso-Alconada, L.; Muinelo-Romay, L.; Vieito, M.; Abalo, A.; Alonso-Nocelo, M.; Candamio, S.; Gallardo, E.; Fernández, B.; Abdulkader, I.; et al. Molecular characterization of circulating tumor cells in human metastatic colorectal cancer. PLoS One 2012, 7, e40476. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Sosa, E.V.; Scott, J.H.; Simko, J.; Collins, C.; Pinkel, D.; Ryan, C.J.; Park, J.W. Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Sergeant, G.; van Eijsden, R.; Roskams, T.; van Duppen, V.; Topal, B. Pancreatic cancer circulating tumour cells express a cell motility gene signature that predicts survival after surgery. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Paris, P.L.; Kobayashi, Y.; Zhao, Q.; Zeng, W.; Sridharan, S.; Fan, T.; Adler, H.L.; Yera, E.R.; Zarrabi, M.H.; Zucker, S.; et al. Functional phenotyping and genotyping of circulating tumor cells from patients with castration resistant prostate cancer. Cancer Lett. 2009, 277, 164–173. [Google Scholar] [CrossRef]
- Buyse, M.; Loi, S.; van’t Veer, L.; Viale, G.; Delorenzi, M.; Glas, A.M.; d’Assignies, M.S.; Bergh, J.; Lidereau, R.; Ellis, P.; et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J. Natl. Cancer Inst. 2006, 98, 1183–1192. [Google Scholar] [CrossRef]
- Li, X.; Quigg, R.J.; Zhou, J.; Gu, W.; Nagesh Rao, P.; Reed, E.F. Clinical utility of microarrays: Current status, existing challenges and future outlook. Curr. Genomics 2008, 9, 466–474. [Google Scholar] [CrossRef]
- Dancey, J.E.; Bedard, P.L.; Onetto, N.; Hudson, T.J. The genetic basis for cancer treatment decisions. Cell 2012, 148, 409–420. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Ross, J.S.; Cronin, M. Whole cancer genome sequencing by next-generation methods. Am. J. Clin. Pathol. 2011, 136, 527–539. [Google Scholar] [CrossRef]
- Cronin, M.; Ross, J.S. Comprehensive next-generation cancer genome sequencing in the era of targeted therapy and personalized oncology. Biomark. Med. 2011, 5, 293–305. [Google Scholar] [CrossRef]
- Shendure, J.; Lieberman Aiden, E. The expanding scope of DNA sequencing. Nat. Biotechnol. 2012, 30, 1084–1094. [Google Scholar] [CrossRef]
- Shendure, J.A.; Porreca, G.J.; Church, G.M. Overview of DNA sequencing strategies. Curr. Protoc. Cell Biol. 2008. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef]
- Ran, R.; Li, L.; Wang, M.; Wang, S.; Zheng, Z.; Lin, P.P. Determination of EGFR mutations in single cells microdissected from enriched lung tumor cells in peripheral blood. Anal. Bioanal. Chem. 2013, 405, 7377–7382. [Google Scholar] [CrossRef]
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.R.; Pantel, K.; Riethdorf, S. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef]
- Khan, Z.A.; Jonas, S.K.; Le-Marer, N.; Patel, H.; Wharton, R.Q.; Tarragona, A.; Ivison, A.; Allen-Mersh, T.G. P53 mutations in primary and metastatic tumors and circulating tumor cells from colorectal carcinoma patients. Clin. Cancer Res. 2000, 6, 3499–3504. [Google Scholar]
- Jiang, Y.; Palma, J.F.; Agus, D.B.; Wang, Y.; Gross, M.E. Detection of androgen receptor mutations in circulating tumor cells in castration-resistant prostate cancer. Clin. Chem. 2010, 56, 1492–1495. [Google Scholar] [CrossRef]
- Ashida, S.; Okuda, H.; Chikazawa, M.; Tanimura, M.; Sugita, O.; Yamamoto, Y.; Nakamura, S.; Moriyama, M.; Shuin, T. Detection of circulating cancer cells with von hippel-lindau gene mutation in peripheral blood of patients with renal cell carcinoma. Clin. Cancer Res. 2000, 6, 3817–3822. [Google Scholar]
- Kirby, B.J.; Jodari, M.; Loftus, M.S.; Gakhar, G.; Pratt, E.D.; Chanel-Vos, C.; Gleghorn, J.P.; Santana, S.M.; Liu, H.; Smith, J.P.; et al. Functional characterization of circulating tumor cells with a prostate-cancer-specific microfluidic device. PLoS One 2012, 7, e35976. [Google Scholar] [CrossRef]
- Maheswaran, S.; Sequist, L.V; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederichs, S.; Iafrate, A.J.; Bell, D.W.; et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 2008, 359, 366–377. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bang, Y.-J.; van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Bookman, M.A. Evaluation of Monoclonal Humanized Anti-HER2 Antibody, Trastuzumab, in Patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: A phase II trial of the gynecologic oncology group. J. Clin. Oncol. 2003, 21, 283–290. [Google Scholar] [CrossRef]
- Fleming, G.F.; Sill, M.W.; Darcy, K.M.; McMeekin, D.S.; Thigpen, J.T.; Adler, L.M.; Berek, J.S.; Chapman, J.A.; DiSilvestro, P.A.; Horowitz, I.R.; et al. Phase II trial of trastuzumab in women with advanced or recurrent, HER2-positive endometrial carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2010, 116, 15–20. [Google Scholar] [CrossRef]
- Roukos, D.H. Next-generation, genome sequencing-based biomarkers: Concerns and challenges for medical practice. Biomark. Med. 2010, 4, 583–586. [Google Scholar] [CrossRef]
- Schrijver, I.; Aziz, N.; Farkas, D.H.; Furtado, M.; Gonzalez, A.F.; Greiner, T.C.; Grody, W.W.; Hambuch, T.; Kalman, L.; Kant, J.A.; et al. Opportunities and challenges associated with clinical diagnostic genome sequencing: A report of the association for molecular pathology. J. Mol. Diagn. 2012, 14, 525–540. [Google Scholar] [CrossRef]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch. Pathol. Lab. Med. 2010, 134, e48–e72. [Google Scholar]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef]
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Trapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997, 57, 314–319. [Google Scholar]
- Rakha, E.A.; El-Sheikh, S.E.; Kandil, M.A.; El-Sayed, M.E.; Green, A.R.; Ellis, I.O. Expression of BRCA1 protein in breast cancer and its prognostic significance. Hum. Pathol. 2008, 39, 857–865. [Google Scholar] [CrossRef]
- Cicenas, J. The potential role of Akt phosphorylation in human cancers. Int. J. Biol. Markers 2008, 23, 1–9. [Google Scholar]
- Kallergi, G.; Agelaki, S.; Kalykaki, A.; Stournaras, C.; Mavroudis, D.; Georgoulias, V. Phosphorylated EGFR and PI3K/Akt signaling kinases are expressed in circulating tumor cells of breast cancer patients. Breast Cancer Res. 2008, 10. [Google Scholar] [CrossRef]
- Parkinson, D.R.; Dracopoli, N.; Petty, B.G.; Compton, C.; Cristofanilli, M.; Deisseroth, A.; Hayes, D.F.; Kapke, G.; Kumar, P.; Lee, J.S.; et al. Considerations in the development of circulating tumor cell technology for clinical use. J. Transl. Med. 2012, 10. [Google Scholar] [CrossRef]
- Danila, D.C.; Pantel, K.; Fleisher, M.; Scher, H.I. Circulating tumors cells as biomarkers: Progress toward biomarker qualification. Cancer J. 2011, 17, 438–450. [Google Scholar] [CrossRef]
- Devriese, L.A.; Voest, E.E.; Beijnen, J.H.; Schellens, J.H. Circulating tumor cells as pharmacodynamic biomarker in early clinical oncological trials. Cancer Treat. Rev. 2011, 37, 579–589. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar]
- Ross, J.S.; Gray, G.S. Targeted therapy for cancer: The HER-2/neu and Herceptin story. Clin. Leadersh Manag. Rev. 2003, 17, 333–340. [Google Scholar]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Vogel, C.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. First-line, single-agent Herceptin(R) (trastuzumab) in metastatic breast cancer. A preliminary report. Eur. J. Cancer 2001, 37, 25–29. [Google Scholar]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.S.; Sledge, G.; Aktan, G.; Ellis, C.; Florance, A.; Vukelja, S.; Bischoff, J.; et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: Final results from the EGF104900 Study. J. Clin. Oncol. 2012, 30, 2585–2592. [Google Scholar] [CrossRef]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.; Sledge, G.; Koehler, M.; Ellis, C.; Casey, M.; Vukelja, S.; Bischoff, J.; et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J. Clin. Oncol. 2010, 28, 1124–1130. [Google Scholar] [CrossRef]
- Baselga, J.; Cortés, J.; Kim, S.-B.; Im, S.-A.; Hegg, R.; Im, Y.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 2012, 366, 109–119. [Google Scholar] [CrossRef]
- Zidan, J.; Dashkovsky, I.; Stayerman, C.; Basher, W.; Cozacov, C. Hadary, a Comparison of HER-2 overexpression in primary breast cancer and metastatic sites and its effect on biological targeting therapy of metastatic disease. Br. J. Cancer 2005, 93, 552–556. [Google Scholar] [CrossRef]
- Curigliano, G.; Bagnardi, V.; Viale, G.; Fumagalli, L.; Rotmensz, N.; Aurilio, G.; Locatelli, M.; Pruneri, G.; Giudici, S.; Bellomi, M.; et al. Should liver metastases of breast cancer be biopsied to improve treatment choice? Ann. Oncol. 2011, 22, 2227–2233. [Google Scholar] [CrossRef]
- Amir, E.; Clemons, M.; Purdie, C.A.; Miller, N.; Quinlan, P.; Geddie, W.; Coleman, R.E.; Freedman, O.C.; Jordan, L.B.; Thompson, A.M. Tissue confirmation of disease recurrence in breast cancer patients: Pooled analysis of multi-centre, multi-disciplinary prospective studies. Cancer Treat. Rev. 2012, 38, 708–714. [Google Scholar] [CrossRef]
- Amir, E.; Miller, N.; Geddie, W.; Freedman, O.; Kassam, F.; Simmons, C.; Oldfield, M.; Dranitsaris, G.; Tomlinson, G.; Laupacis, A.; et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol. 2012, 30, 587–592. [Google Scholar] [CrossRef]
- Riethdorf, S.; Pantel, K. Advancing personalized cancer therapy by detection and characterization of circulating carcinoma cells. Ann. N. Y. Acad. Sci. 2010, 1210, 66–77. [Google Scholar]
- Georgoulias, V.; Bozionelou, V.; Agelaki, S.; Perraki, M.; Apostolaki, S.; Kallergi, G.; Kalbakis, K.; Xyrafas, A.; Mavroudis, D. Trastuzumab decreases the incidence of clinical relapses in patients with early breast cancer presenting chemotherapy-resistant CK-19mRNA-positive circulating tumor cells: Results of a randomized phase II study. Ann. Oncol. 2012, 23, 1744–1750. [Google Scholar] [CrossRef]
- Efficacy study of herceptin to treat HER2-negative CTC breast cancer (TREAT-CTC). Available online: http://clinicaltrials.gov/ct2/show/NCT01548677?term=CTC+Treat&rank=1 (accessed on 1 August 2013).
- DETECT III—A multicenter, phase III study to compare standard therapy +/− lapatinib in HER2-ve MBC-patients with HER2+ve CTCs. Available online: http://clinicaltrials.gov/ct2/show/NCT01619111?term=DETECT+III&rank=1 (accessed on 1 August 2013).
- Circulating tumor cells to guide chemotherapy for metastatic breast cancer (CirCé01). Available online: http://clinicaltrials.gov/ct2/show/NCT01349842?term=Circe&rank=2 (accessed on 1 August 2013).
- Characterization of circulating tumor cells (CTC) from patients with metastatic breast cancer using the CTC-endocrine therapy index (COMETI P2). Available online: http://clinicaltrials.gov/ct2/show/NCT01701050?term=COMETI+P2&rank=1 (accessed on 1 August 2013).
- Bidard, F.-C.; Fehm, T.; Ignatiadis, M.; Smerage, J.B.; Alix-Panabières, C.; Janni, W.; Messina, C.; Paoletti, C.; Müller, V.; Hayes, D.F.; et al. Clinical application of circulating tumor cells in breast cancer: Overview of the current interventional trials. Cancer Metastasis Rev. 2013, 32, 179–188. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.J.; Uhr, J.W.; Terstappen, L.W.M.M. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Gradilone, A.; Raimondi, C.; Nicolazzo, C.; Petracca, A.; Gandini, O.; Vincenzi, B.; Naso, G.; Aglianò, A.M.; Cortesi, E.; Gazzaniga, P. Circulating tumour cells lacking cytokeratin in breast cancer: The importance of being mesenchymal. J. Cell Mol. Med. 2011, 15, 1066–1070. [Google Scholar] [CrossRef]
- Königsberg, R.; Obermayr, E.; Bises, G.; Pfeiler, G.; Gneist, M.; Wrba, F.; DE Santis, M.; Zeillinger, R.; Hudec, M.; Dittrich, C. Detection of EpCAM positive and negative circulating tumor cells in metastatic breast cancer patients. Acta Oncol. 2011, 50, 700–710. [Google Scholar] [CrossRef]
- Kasimir-Bauer, S.; Hoffmann, O.; Wallwiener, D.; Kimmig, R.; Fehm, T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012, 14, R15. [Google Scholar] [CrossRef]
- Mego, M.; Mani, S.A.; Lee, B.-N.; Li, C.; Evans, K.W.; Cohen, E.N.; Gao, H.; Jackson, S.A.; Giordano, A.; Hortobagyi, G.N.; et al. Expression of epithelial-mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int. J. Cancer 2012, 130, 808–816. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Gorges, T.M.; Tinhofer, I.; Drosch, M.; Röse, L.; Zollner, T.M.; Krahn, T.; von Ahsen, O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar]
- Kim, P.; Liu, X.; Lee, T.; Liu, L.; Barham, R.; Kirkland, R.; Leesman, G.; Kuller, A.; Ybarrondo, B.; Ng, S.-C.; et al. Highly sensitive proximity mediated immunoassay reveals HER2 status conversion in the circulating tumor cells of metastatic breast cancer patients. Proteome Sci. 2011, 9. [Google Scholar] [CrossRef]
- Powell, A.A.; Talasaz, A.H.; Zhang, H.; Coram, M.A.; Reddy, A.; Deng, G.; Telli, M.L.; Advani, R.H.; Carlson, R.W.; Mollick, J.A.; et al. Single cell profiling of circulating tumor cells: Transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One 2012, 7, e33788. [Google Scholar] [CrossRef]
- Gallant, J.-N.; Matthew, E.M.; Cheng, H.; Harouaka, R.; Lamparella, N.E.; Kunkel, M.; Yang, Z.; Harvey, H.A.; Cream, L.V.; Kumar, S.M.; et al. Predicting therapy response in live tumor cells isolated with the flexible micro spring array device. Cell Cycle 2013, 12, 2132–2143. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lowes, L.E.; Allan, A.L. Recent Advances in the Molecular Characterization of Circulating Tumor Cells. Cancers 2014, 6, 595-624. https://doi.org/10.3390/cancers6010595
Lowes LE, Allan AL. Recent Advances in the Molecular Characterization of Circulating Tumor Cells. Cancers. 2014; 6(1):595-624. https://doi.org/10.3390/cancers6010595
Chicago/Turabian StyleLowes, Lori E., and Alison L. Allan. 2014. "Recent Advances in the Molecular Characterization of Circulating Tumor Cells" Cancers 6, no. 1: 595-624. https://doi.org/10.3390/cancers6010595