Chronic Inflammation: Synergistic Interactions of Recruiting Macrophages (TAMs) and Eosinophils (Eos) with Host Mast Cells (MCs) and Tumorigenesis in CALTs. M-CSF, Suitable Biomarker for Cancer Diagnosis!

Abstract
:1. Introduction
- (a)
- A direct evidence for an association between inflammation-induced immune alterations that would lead to tumorigenesis and angiogenesis;
- (b)
- Identification of cellular composition of host/target tissue resident or recruited immune and non-immune cells and the nature of interactions between mediators, under a wide range of stimuli-induced immune response profiles that would result in cellular growth and site-specific cancers;
- (c)
- Time-course kinetics of developmental phases of immune response alterations that are identified during early stages of tumorigenesis and angiogenesis which potentially are preventable, reversible or correctable.
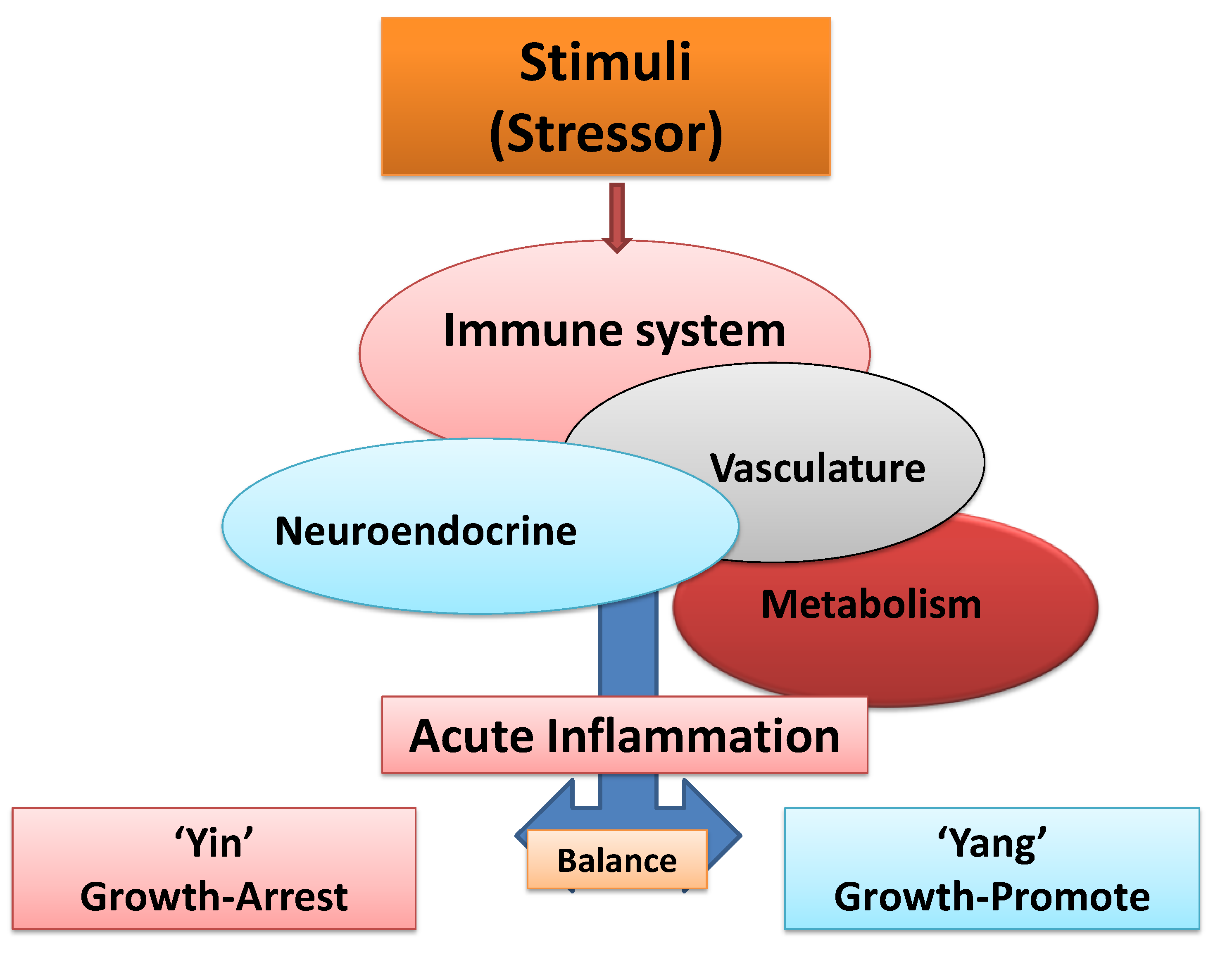
2. Acute Inflammation: Protective, Self-Terminating Property of Immune Surveillance
- (a)
- Encounter, sense/recognize, destroy/defeat and eliminate foreign elements and the injured host tissue during “Yin” (tumoricidal) responses;
- (b)
- b) Terminate, neutralize or resolve inflammatory responses and repair or remodel the host tissue during “Yang” (tumorigenic) responses.
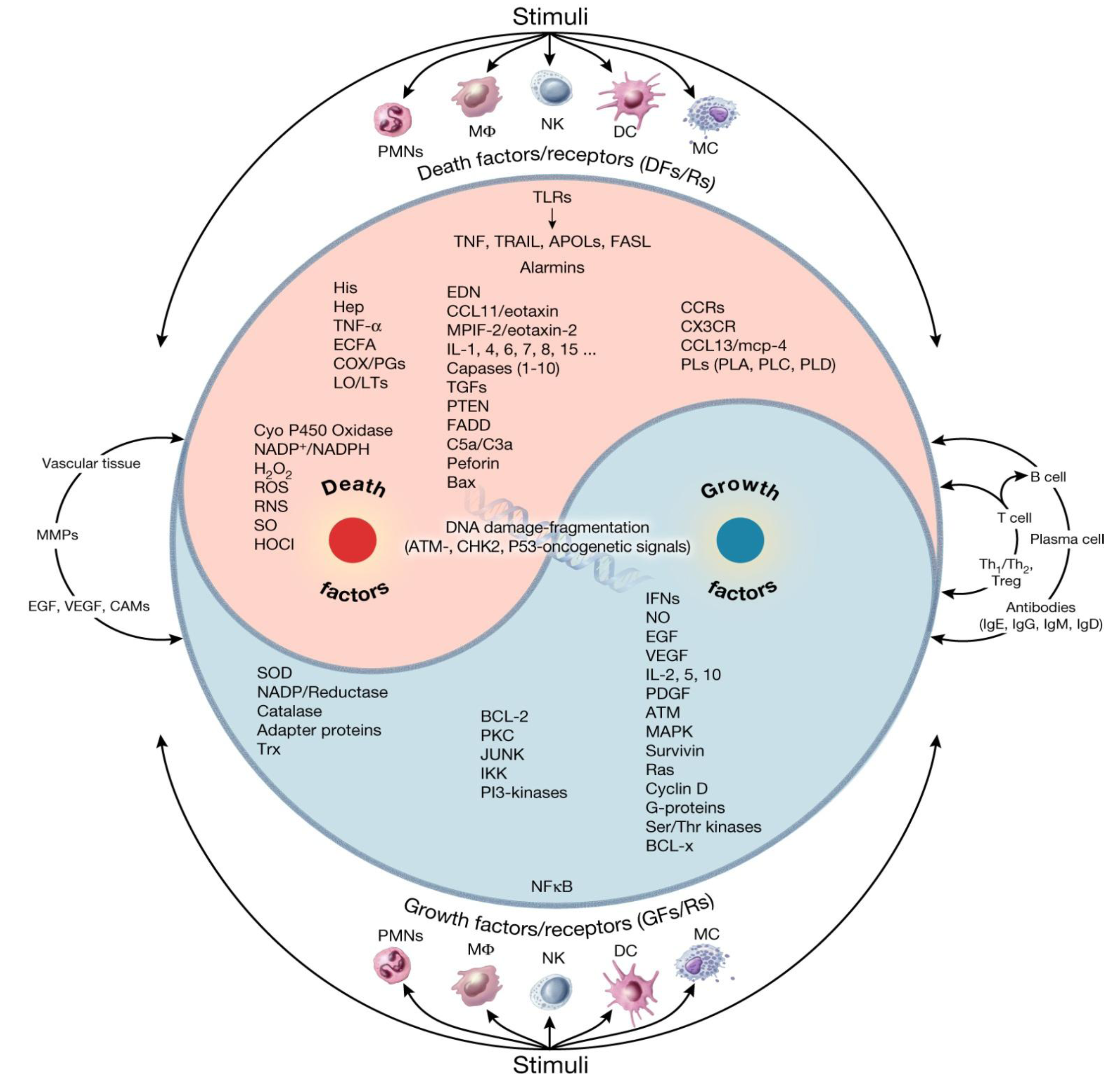
3. Specialized, Shared and Synergistic Features of APCs
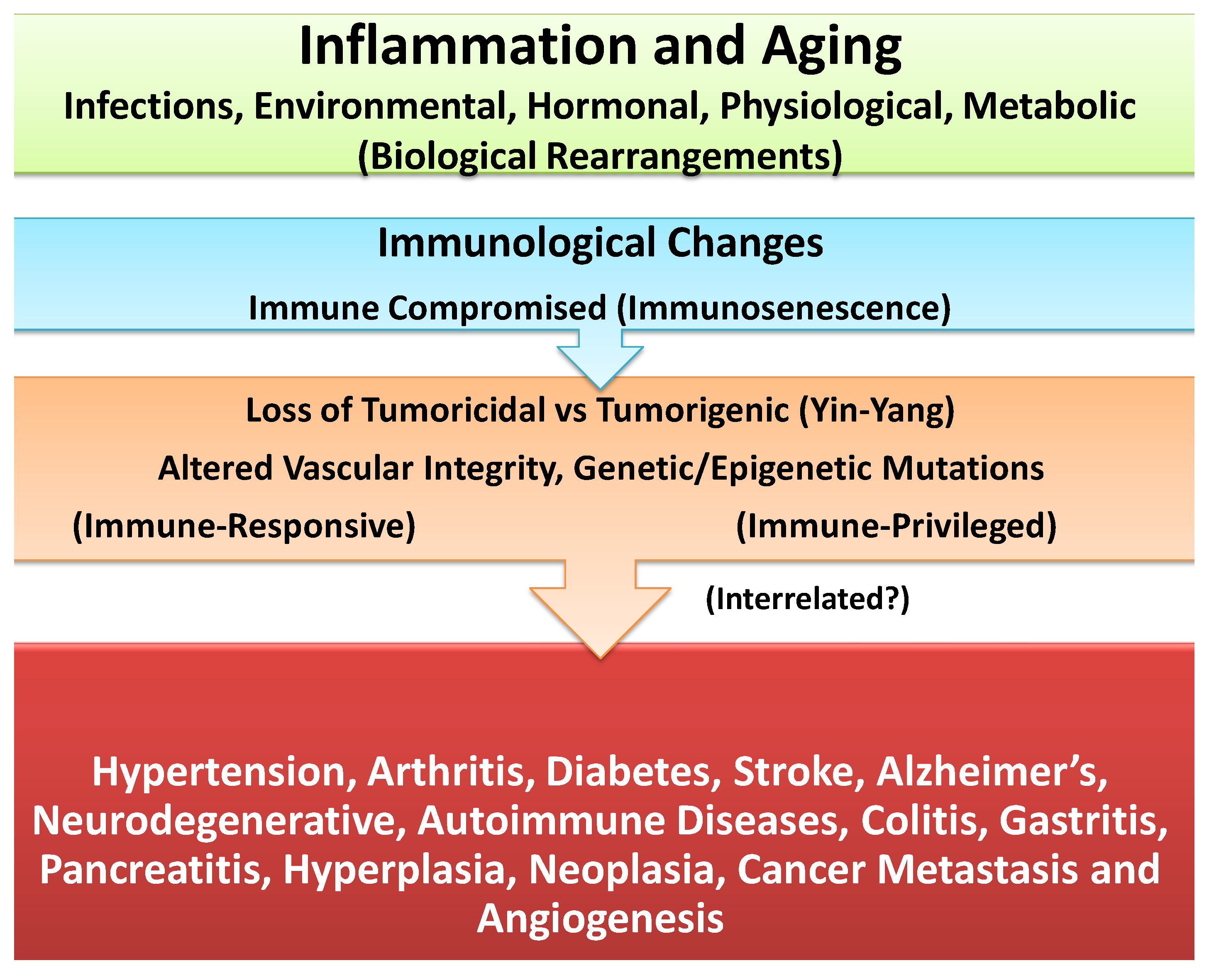
4. Unresolved Inflammation: Common Denominator in Induction of Age-Associated Chronic Diseases or Carcinogenesis
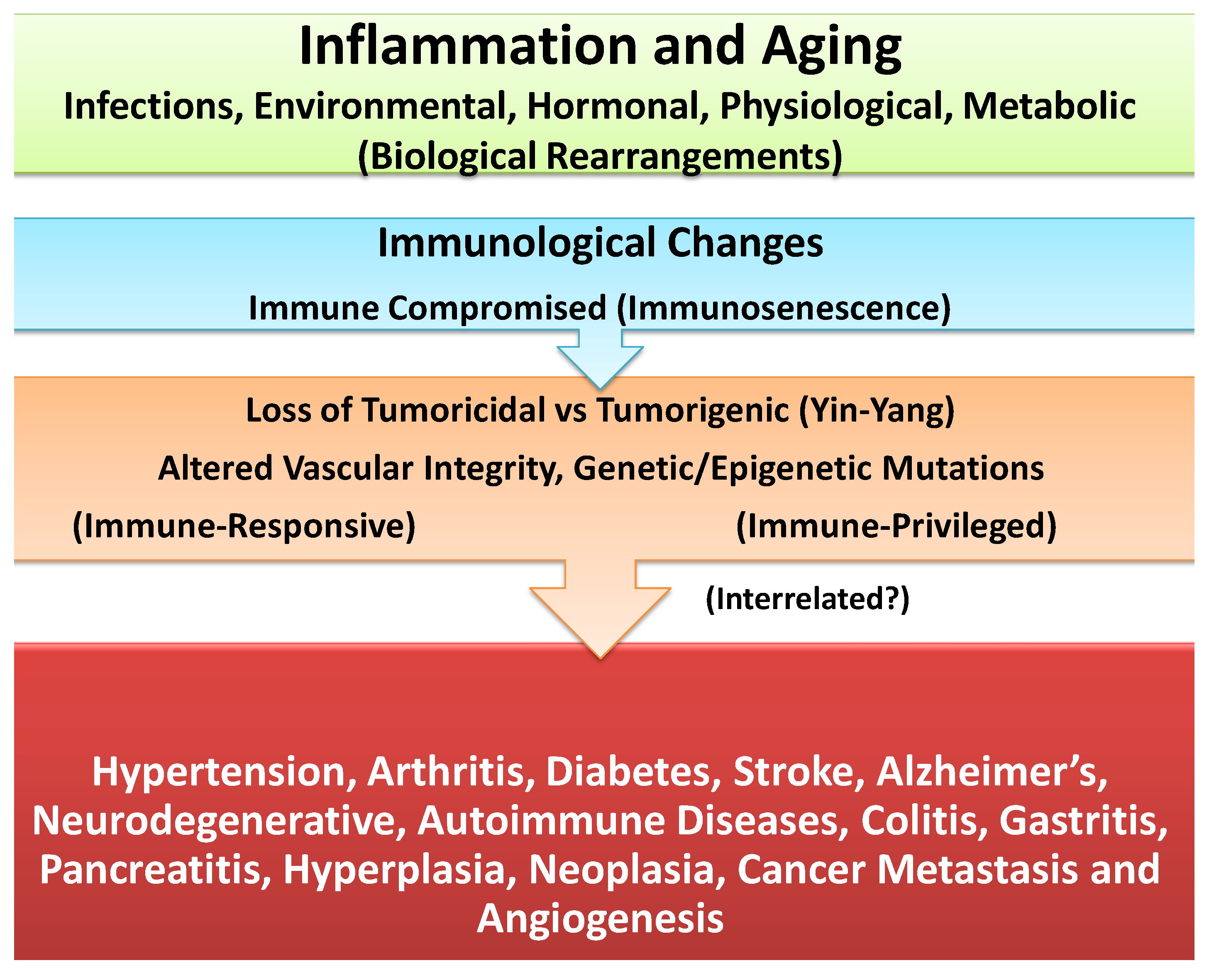
5. Differential Impact of Inflammation in Immune-Responsive and Immune-Privileged Tissues and Chronic Diseases or Cancer
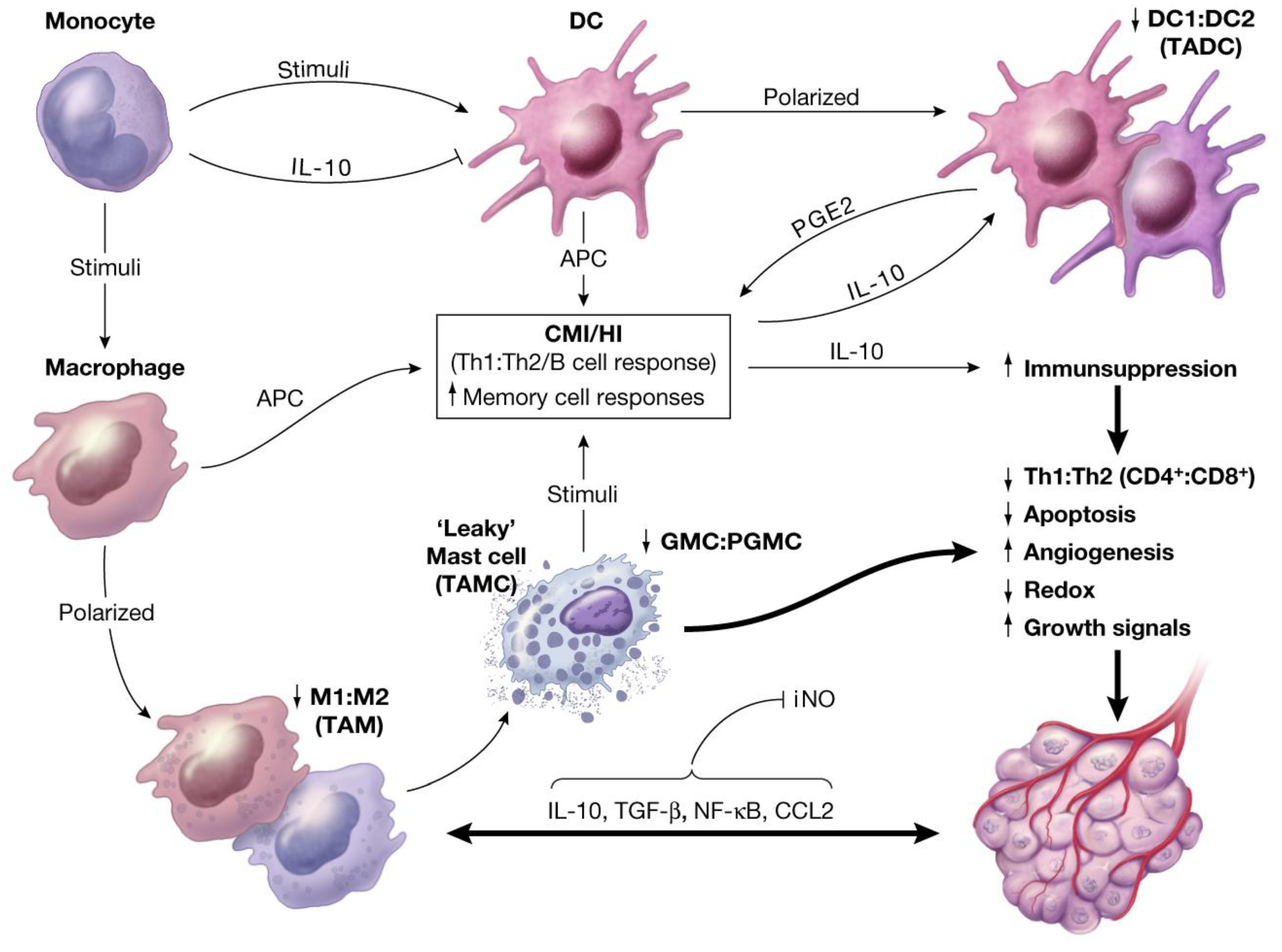
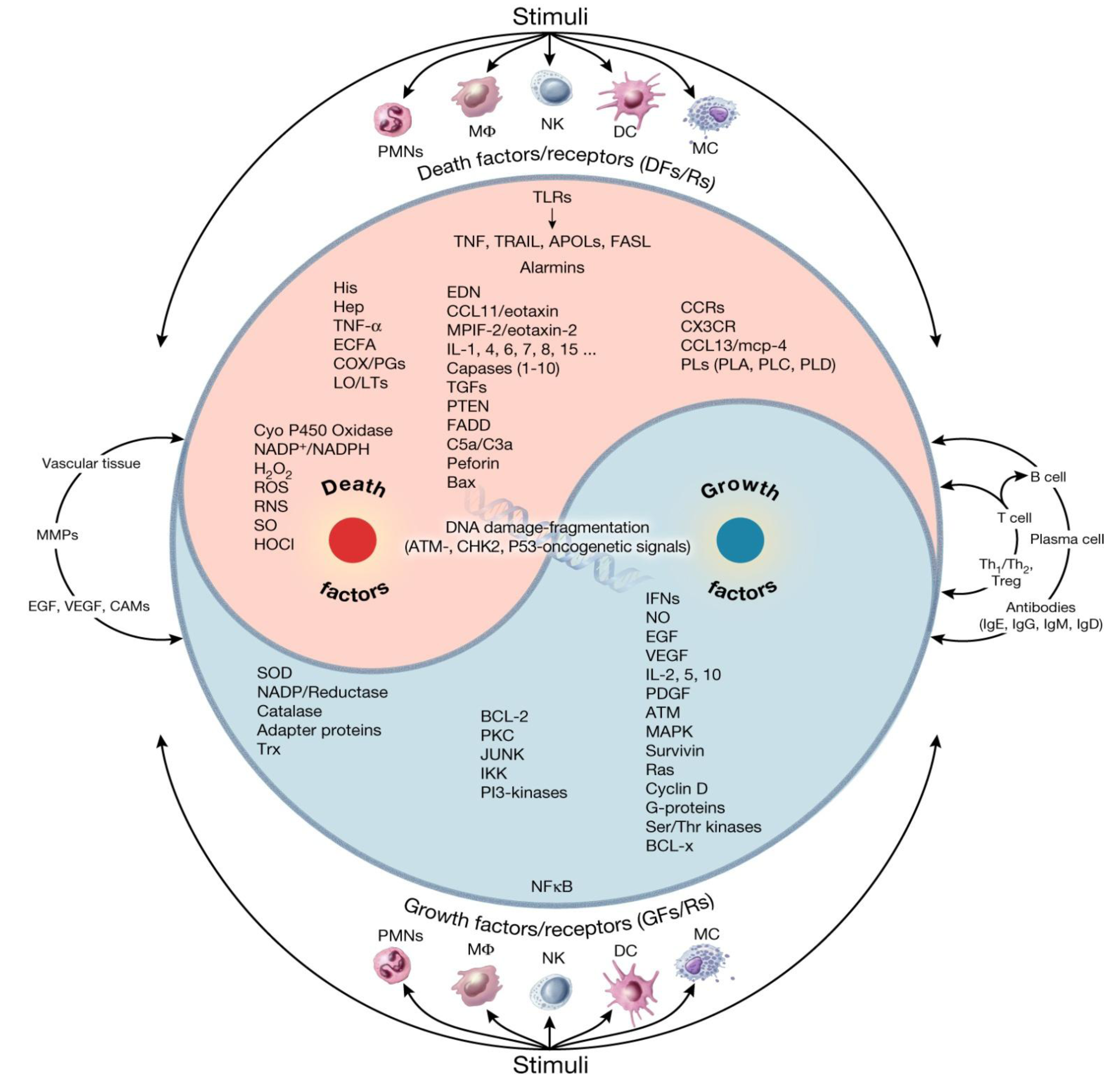
6. Interactions and Synergies between Resident and Recruited APCs in Host Tissue
7. Circumstantial Evidence for Association between Inflammation and Cancer

| Tumor (Cancer) Site | Inflammatory Disease |
|---|---|
| Bowel/Colon | Ulcerative Colitis (Crohn’s) |
| Urinary Bladder | Schistomiasis, Stones, Catheters |
| Prostate | Prostatitis---PIA-PIN |
| Breast | Inflammatory Conditions |
| Cervical | Pericarditis? |
| Esophagus | Barretts’ |
| Pancreas | Pancreatitis |
| Stomach | Gastric Infection (e.g., H Pylori) |
| Ulcers/Gastritis | |
| Lung | Asthma, Emphysema, Smoking |
| Liver | Hepatitis B, C |
| Thyroid | Thyroiditis |
| Conjunctiva | Conjunctivitis/CALTs (?) |
| Skin | Melanomas Burns/Radiations (?) |
| Uveal | Melanomas Uveitis (?) |
| Others | Cystitis (?) |
| Hyperplasia of GALTs (?) |
8. Direct Association between Inflammation and Tumorigenesis and Angiogenesis: Interactions between Resident and Recruited APCs in Experimental Models of Acute and Chronic Ocular Inflammatory Diseases
- (a)
- Acute-phase response: Initiated 9 days after topical sensitization and challenge with FLOA. Clinical and histopathological findings included a combination of strong or weak acute (type 1, hypersensitivity) reactions, tearing, scratching and conjunctival edema, milky secretions, IgE-dependent mast cells (MCs) degranulation, release of histamine and PGs, vascular hyperpermeability and coagulation of lipid-protein complex exudates. Time-course kinetics of release of histamine and PGs (6-keto-PGF-1α, a stable product of PGI2) into tears suggested that histamine (a potent and preformed vasoactive agent) was a primary mediator that activated membrane arachidonic acid metabolic pathways, e.g., activation of cyclooxygenase and lipooxygenase and the synthesis and release of prostanoids (PGs). No correlation was found between circulating homocytotropic-IgE and the degree of clinical reactions. Further studies with untreated eyes, or lung tissues and ocular challenges of new-born babies from sensitized animals suggested high affinity binding of IgE-MCs-Fc-ε receptors and removal of circulating IgE by MCs throughout the body [3,35,37,89].
- (b)
- Intermediate-phase responses; induction of down-regulation phenomenon: Occurring within 2 months of continuous sensitization and challenge. Findings included minimal tearing or tissue edema, loss (exhaustion) of number of functional or tumoricidal (mature) MCs, extensive infiltration of eosinophils into subepithelium and mucus-secreting GCs, tissue hypertrophy and neovascularization (Figure 5). Topical application of compound 48/80, a mast cell degranulating agent, to desensitized eyes produced little reactions. In contrast, histamine applied to desensitized eyes produced strong type 1 reactions. These observations further demonstrated that mast cells were exhausted (non-functional) while histamine receptors were not affected by repeated challenges at this stage of sensitization [3,37,87].
- (c)
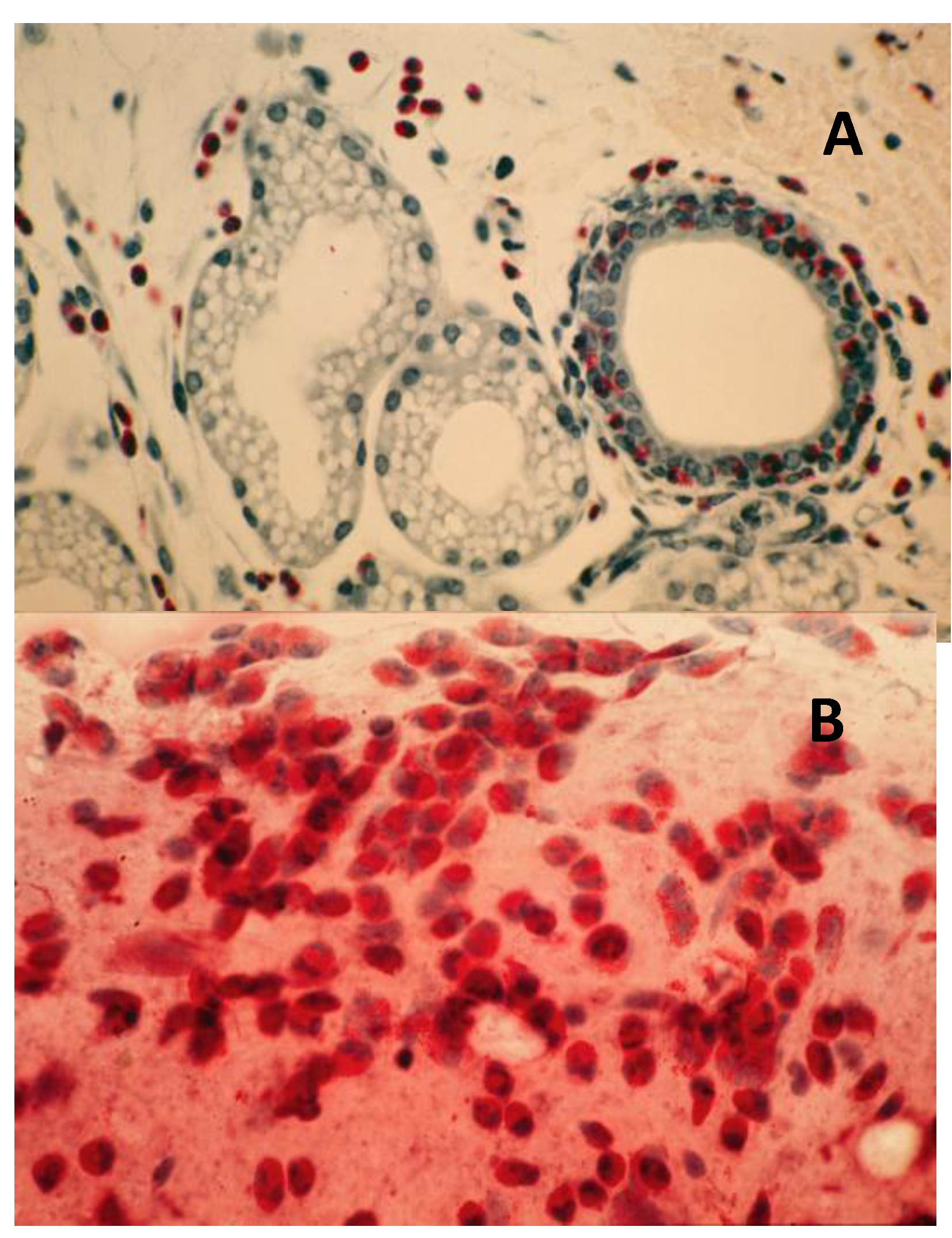
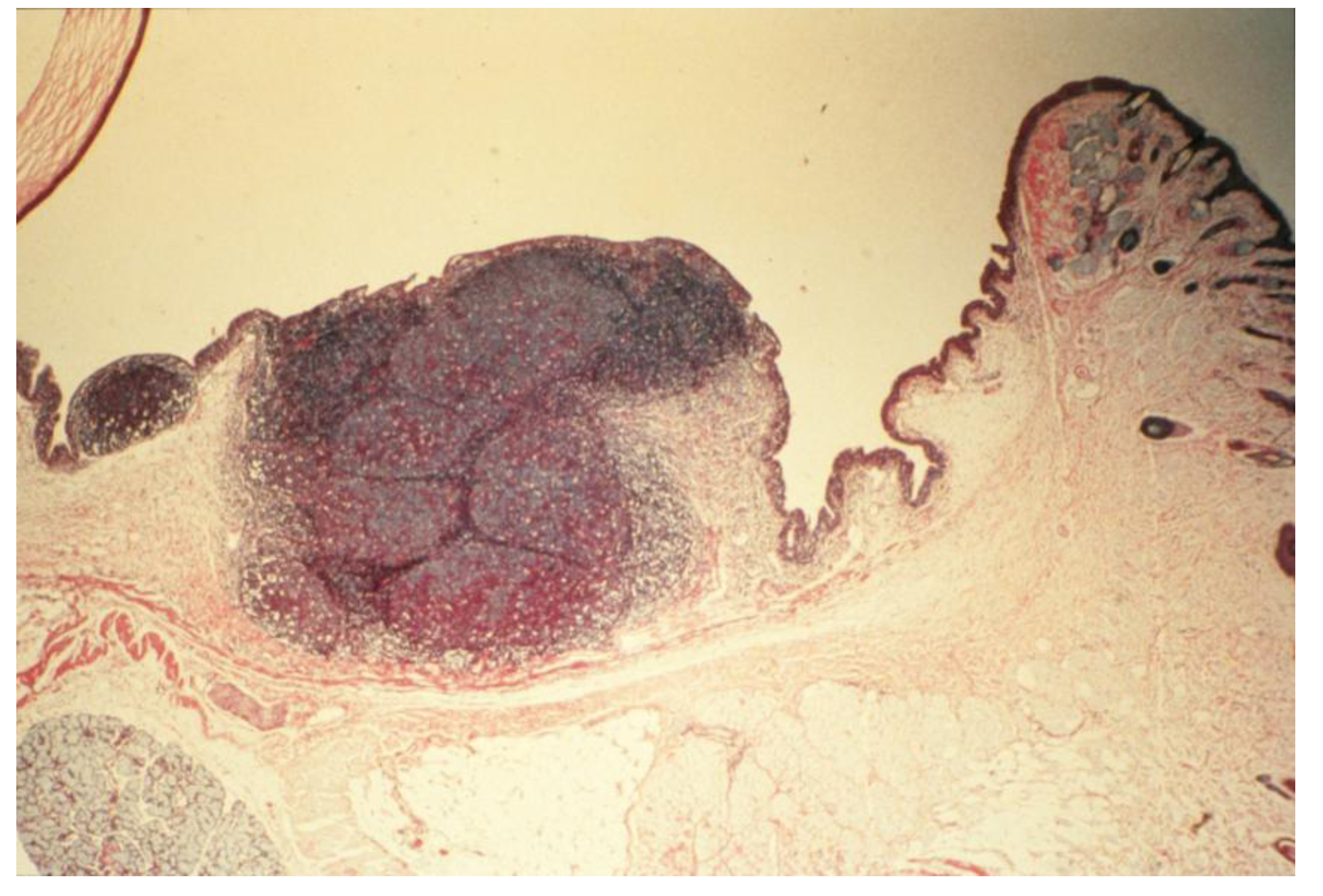
- Chronic-phase response, induction of tumorigenesis and angiogenesis: Occurring between 12 to 30 months of repeated tissue stimulation with FLOA. Findings included induction of tumor-like lesions in conjunctival tissues, angiogenesis, massive lymphoid hyperplasia, follicular formation with germinal centers, activated MFs, presence of histiocytes, loss of lymphocyte capsular membrane and extension of various sized B lymphocytes into surrounding epithelial tissues, increased swollen GCs, increased degranulated or partially granulated (“leaky”) MCs, involvement of lymphatic channels, extensive epithelial thickening (growth) and/or thinning (necrosis) often noted in the same tissue sections. Cross-sectional areas of massive hyperplastic lymphoid nodules from animals that were continuously challenged with antigen were at least five times larger than lymphoid tissues in normal-untreated animals (Figure 6) [3,36,37,39,45].Figure 5. Induction of down regulation in CALTs and eosinophil infiltration. Panel A; heavy eosinophils infiltration into GCs. Panel B; heavy eosinophils infiltration in conjunctival secretions from animals that were repeatedly challenged with topical application of FLOA and systemically immunized with A. Suum. Reproduced from Khatami et al. [35], @1984 American Medical Association, all rights reserved.Figure 5. Induction of down regulation in CALTs and eosinophil infiltration. Panel A; heavy eosinophils infiltration into GCs. Panel B; heavy eosinophils infiltration in conjunctival secretions from animals that were repeatedly challenged with topical application of FLOA and systemically immunized with A. Suum. Reproduced from Khatami et al. [35], @1984 American Medical Association, all rights reserved.
![Cancers 06 00297 g005]()
- (d)
- Induction of tumorigenesis with mixture of antigen and TPAs: Animals that were topically treated with a mixture of FLOA and TPAs developed tumor-like lesions within 6 months after commencement of sensitization. These preliminary observations were suggestive of additive impact of TPAs that shifted the kinetics of altered immune responses and tumorigenesis to earlier events, perhaps through activation of protein kinase C (PKC) and/or other tumor growth pathways [3,36,37].
- (e)
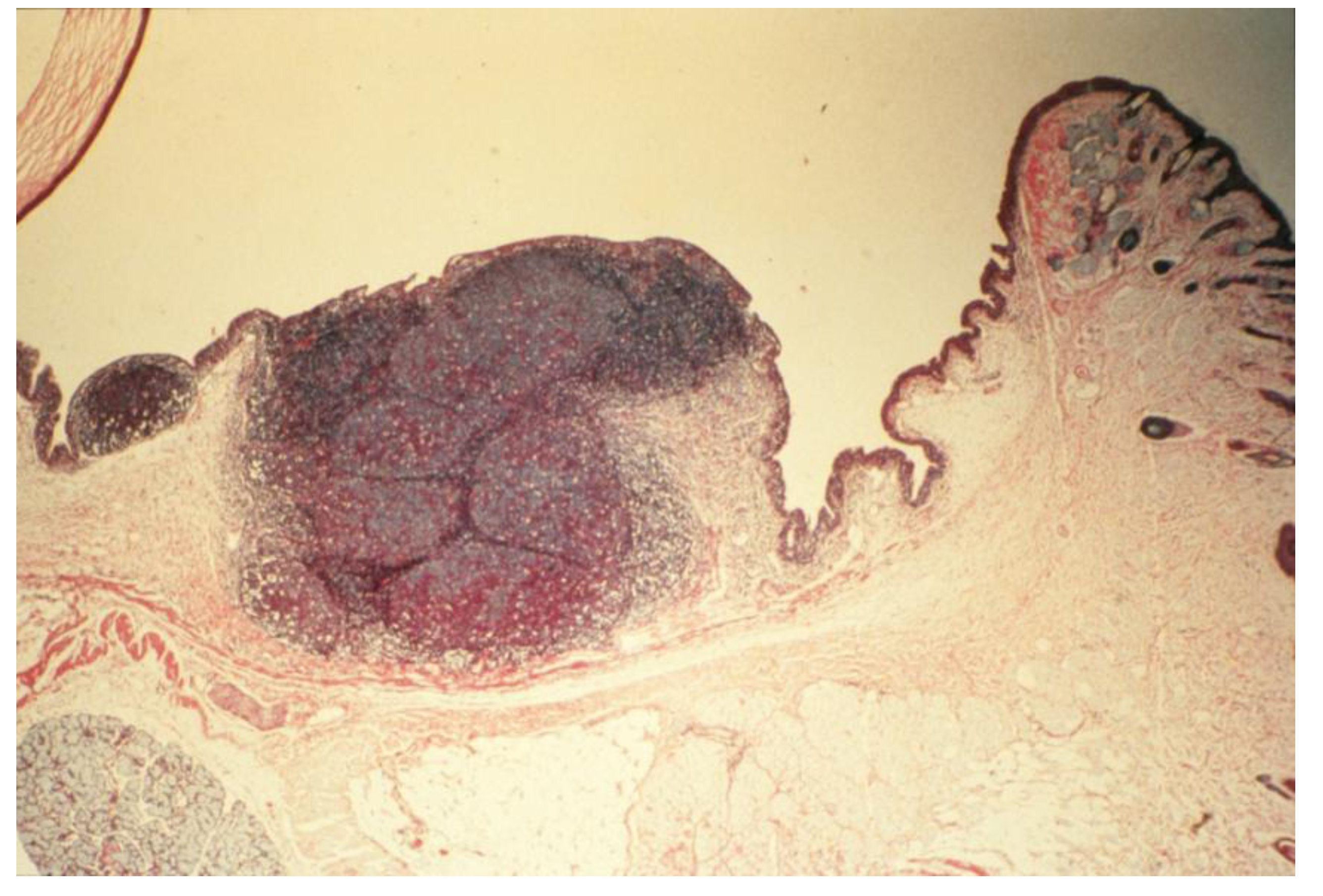
- Antibody profiles (humoral immunity, HI): Repeated stimulation of tissues and the induction of tumorigenesis produced significant increase in the expression of immunoglobulin isotypes (e.g., IgG1/IgG2 ratios) in culture media of massive hyperplastic CALTs, suggesting that frequent exposure to large dosage of antigen into substantia propria, or sub-epithelial tissues altered antibody profiles [3,37,88]. Indirect support for these observations came from the studies when guinea pigs were injected sub-conjunctivally with low dosage (or less frequent exposure) of nematode Onchocerca lienalis microfilaria; where no significant changes in biosynthesis of local IgG1 to IgG2 antibodies were observed in the cultures [3,90]. Others demonstrated diversities in the expression of cytokines and antibodies in B lymphocytes in humans and transgenic CCL2-deficient mouse models in the induction of inflammatory diseases or carcinogenesis [27,32,37]. The B lymphocytes in CCL2-deficient mice were shown to be unable to synthesize normal profiles of subclasses of antibodies, and continued synthesis of high levels of IgG2a and IgG2b, and low levels of IgG1, after immunization [3,27,28,32,37].Figure 6. Histopathologic section of eyelid of repeatedly topically immunized and challenged guinea pig showing massive hyperplasia of CALTs. Reproduced from Khatami et al. [36], @1989 American Medical Association, all rights reserved.Figure 6. Histopathologic section of eyelid of repeatedly topically immunized and challenged guinea pig showing massive hyperplasia of CALTs. Reproduced from Khatami et al. [36], @1989 American Medical Association, all rights reserved.
![Cancers 06 00297 g006]()
- (f)
- Statistics and data analyses: From a total of 400 eyes that were examined, 12/40 (30%) of the eyes from animals that were not sacrificed during earlier immunization periods developed tumor-like lesions or hyperplasia of CALTs. These preliminary data that suggested that tumor developed primarily in animals that initially produced minimal early type 1 hypersensitivity reactions, deserve further investigations [3,36,37].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
9. Assessment of Cancer Claimed “Targeted” Therapies or “Personalized” Medicine: False Foundation and Failed Outcomes!
10. Questions, Major Knowledge Gaps and Future Research: Systematic Studies on Biology of Multistep Carcinogenesis toward Prevention and Therapy
- (a)
- Antigen clearing effects: Understanding the basis for heterogeneities in immune response profiles (strong or weak acute inflammatory reactions) that were observed in acute phase in CALTs toward antigen challenges is important, since the extent of antigen permeability and access to inter-epithelial and sub-epithelial cells may produce significantly different outcomes. A strong type 1 reaction may restrict the penetration/exposure of antigen to trans-epithelial surface by an outward flow of fluids (e.g., mucus secretion and/or tearing) or copious leakage of vascular plasma from hyperpermeable vasculature. The increased tearing after a strong type 1 reaction also would wash away (dilute) or expel the antigen and protect the host during initial responses. However, increased or repeated exposures to stimuli and cumulative permeability of low levels of antigens may predispose the tissue toward antigen penetration. In contrast, a weak initial type 1 response, due to impaired function of MCs and/or B/plasma cells or GCs, may result in a greater net promotion of antigen penetration and/or increased epithelial exposure to higher doses of antigen or environmental toxins [3,37].
- (b)
- Role of mucus-secreting goblet cells (GCs): Although little is known about the roles played by IgA-mucus-secreting GCs in the genesis of tumors, increased presence of these cells in the epithelial tissues of conjunctiva, and other epithelial tissues should be considered potentially important in acute inflammation, as well as, in the genesis and progression of chronic diseases or many cancers [3,37]. Involvement of GCs in diseases such as microglandular GCs, carcinoma or adenocarcinoid, crypt cell carcinoma, or mucinous carcinoid in tumors of the appendix, stomach and intestinal cancers, mucosal-associated diseases, colonic carcinomas, Barrett’s esophagus, conjunctival-associated lymphoma, lung cancer or neoplasia and tumors of endocrine systems have been documented [3,37,39,54,79,92]. In addition to the infiltration of eosinophils into GCs that we observed during the intermediate phase of inflammatory responses in CALTs [3,37,39], mature GCs tumors have been reported to be composed of infiltrating virus, or the glandular cords appeared to contain mucin-containing cuboidal cells with small eccentric nuclei [3,79].
- (c)
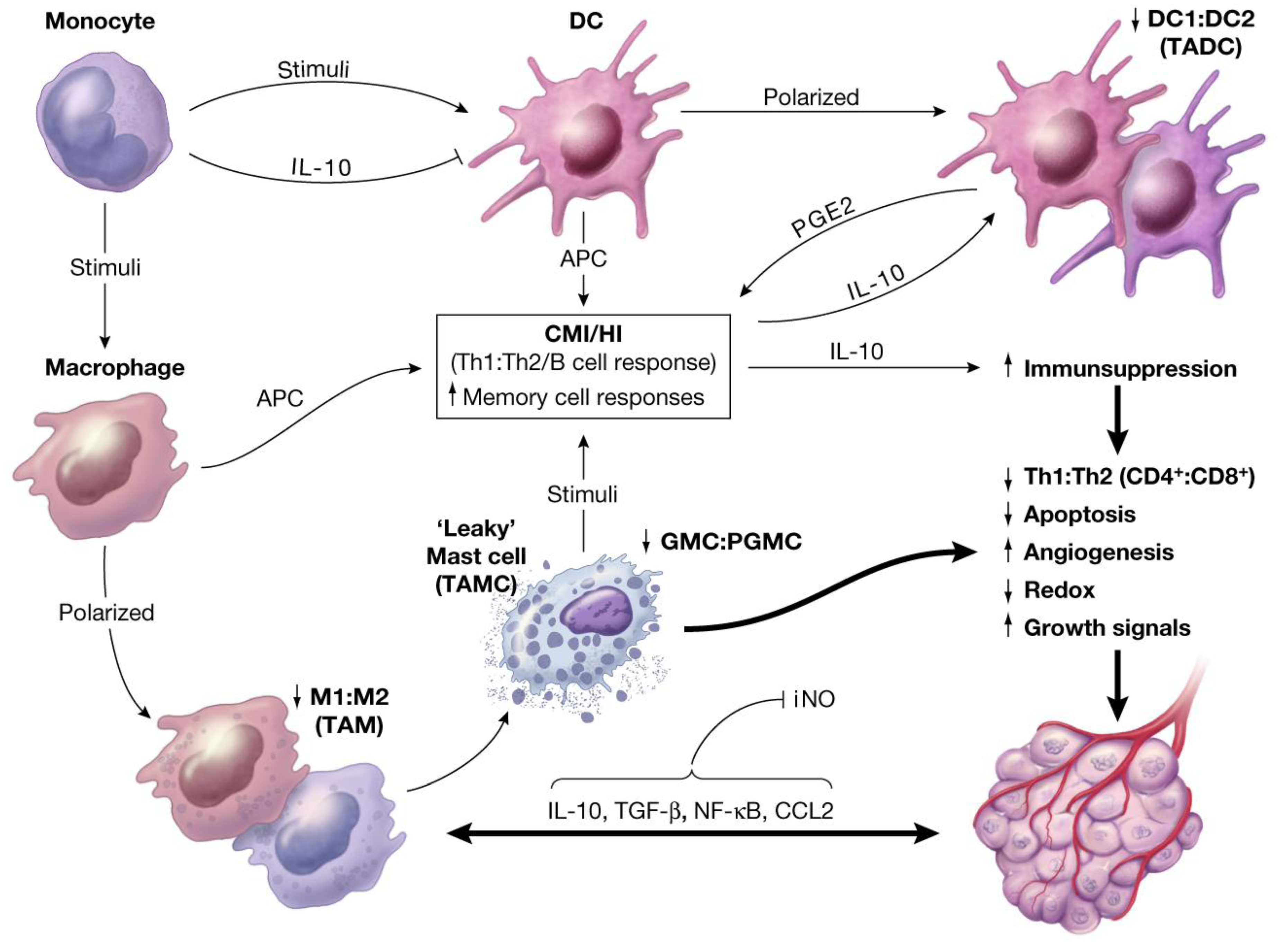
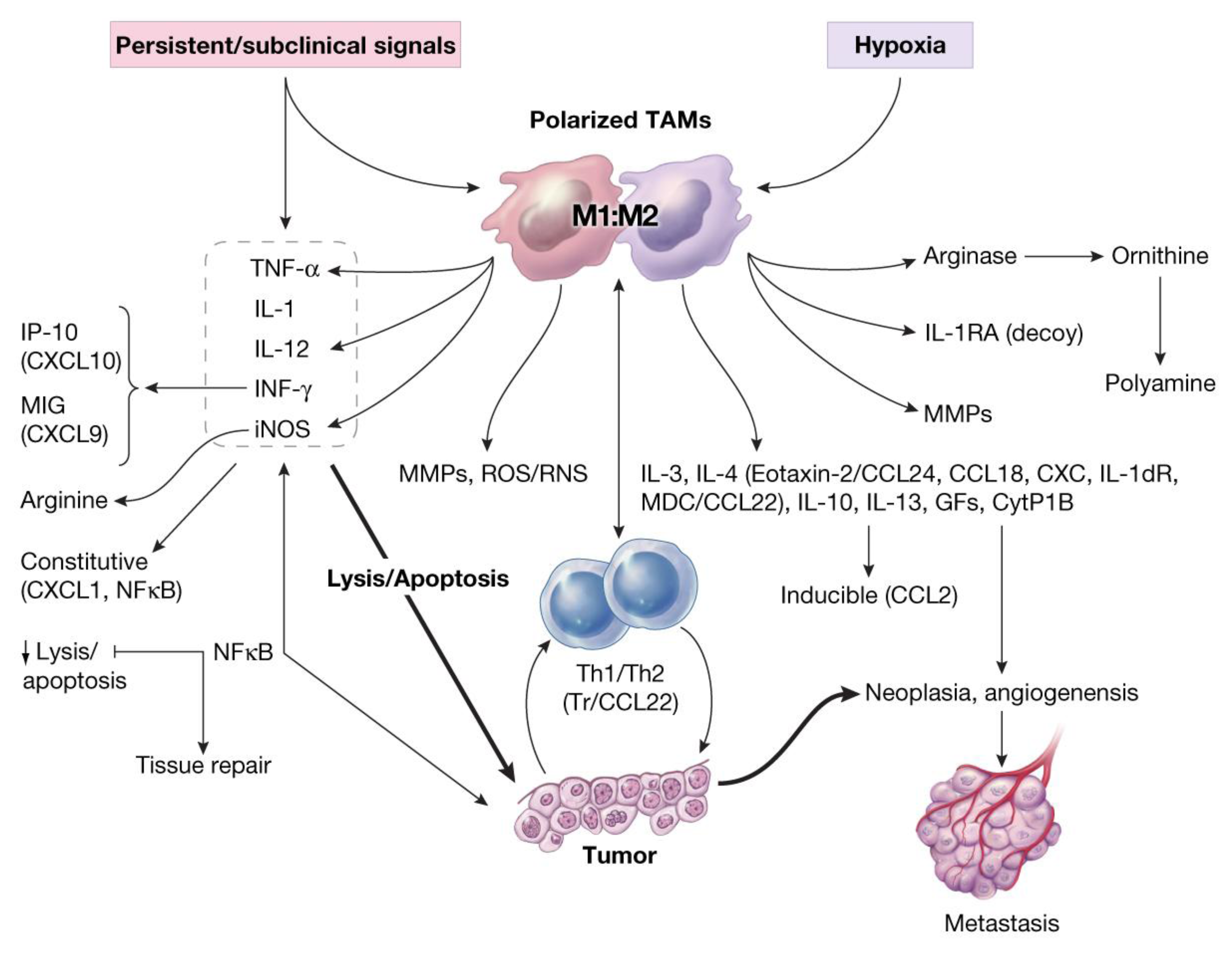
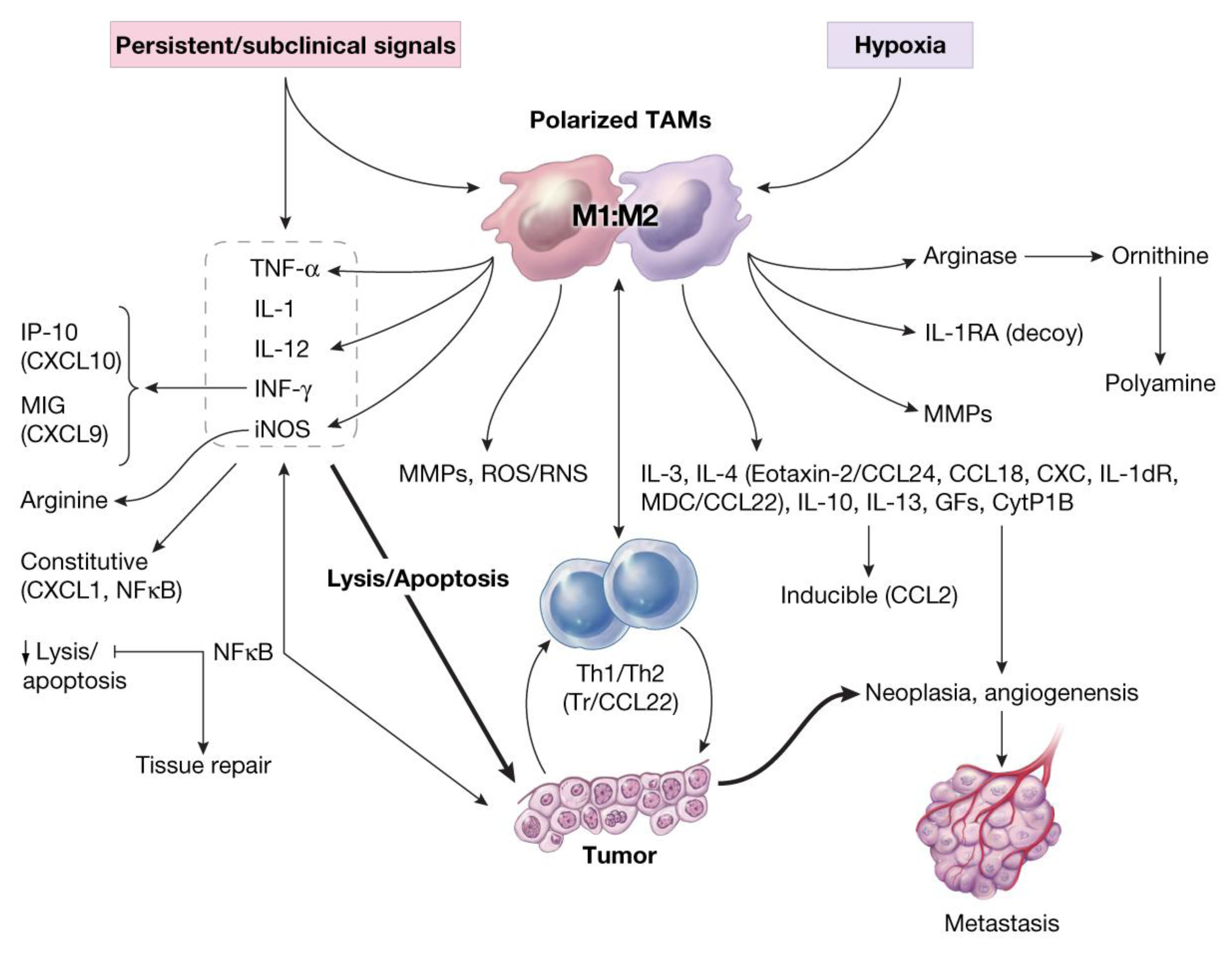
- Recruited macrophages: Macrophages recruited into the CALTs during the chronic phase of immune responses, in all likelihoods, play crucial roles in their M2 phenotype (TAMs, wound healing or tumorigenic) to facilitate the induction of tumorigenesis. Synergistic expression of immune suppressive mediators such as histamine and/or PGE2 from “leaky” MCs combined with expression of suppressive mediators from TAMs into CALTs could further facilitate the growth properties of host tissue [3].
11. Oxidative Stress and Induction of Tumor-Associated MFs (TAMs) in Carcinogenesis
12. Foundation of a Cancer Biomarkers Database: M-CSF as Prototype; Suitable Marker
- (a)
- high specificity; marker will not be detected in healthy individual;
- (b)
- high sensitivity; marker be detected at earliest stage of tissue growth or dysfunction (prior to cancer formation or when few cancerous cells are formed);
- (c)
- marker possess predictive values; its levels in patient samples correlate with tumor stage;
- (d)
- marker possess superior specificity and sensitivity compared with traditional markers;
13. Concluding Remarks: Promoting Immune Surveillance for Healthy Aging, Effective Cancer Prevention, Risk Assessment Formulation and Therapeutic Approaches
Acknowledgements
Conflicts of Interest
References
- Ehrlich, P. Uber den jetzigen Stand der Karzinomforschung. Ned. Tijdschr. Geneeskd. 1909, 5, 273–290. [Google Scholar]
- Burnet, M. Cancer; a biologic approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef]
- Khatami, M. Inflammation, Aging and Cancer: Friend or For? In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 3–30. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Aprahamian, T. Autoimmunity, atherosclerosis and apoptic cell clearance. In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 75–96. [Google Scholar]
- Fischetti, F.; Tedesco, F. Cross-talk between the complement system and endothelial cells in physiologic conditions and vascular diseases. Autoimmunity 2006, 39, 417–428. [Google Scholar] [CrossRef]
- Culmsee, C.; Landshamer, S. Molecular insights into mechanisms of the cell death program: role in the progression of neurodegenerative disorders. Curr. Alzheimer Res. 2006, 3, 269–283. [Google Scholar] [CrossRef]
- Keibel, A.; Singh, V.; Sharma, M.C. Inflammation, microenvironment, and the immune system in cancer progression. Curr. Pharm. Des. 2009, 15, 1949–1955. [Google Scholar] [CrossRef]
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef]
- Hanson, A.; Gosemann, M.; Pruss, A.; Reiter, K.; Ruzickova, S.; Lipsky, P.E.; Domer, T. Abnormalities in peripheral B cell memory of patients with primary Sjogren’s syndrome. Arthritis Rheum. 2004, 50, 1897–1908. [Google Scholar] [CrossRef]
- Harrois, A.; Huet, O.; Duranteau, J. Alterations of mitochondrial function in sepsis and critical illness. Curr. Opin. Anesthesiol. 2009, 22, 143–149. [Google Scholar] [CrossRef]
- Barcante, J.M.P.; Barcante, T.A.; Peconick, A.P.; Pereira, L.J.; Lima, W.S. Parasitic infections and inflammatory diseases. In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 205–218. [Google Scholar]
- Serafin, W.E.; Austen, K.F. Mediators of immediate hypersensitivity reactions. N. Engl. J. Med. 1987, 317, 30–34. [Google Scholar] [CrossRef]
- Bischoff, S.C. Role of mast cells in allergic and non-allergic immune responses: Comparison of human and murine data. Nat. Rev. Immunol. 2007, 7, 93–104. [Google Scholar] [CrossRef]
- Arias, J.I.; Aller, M.A.; Arias, J. Cancer cell: Using inflammation to invade the host. Mol. Cancer 2007, 6, 29. [Google Scholar] [CrossRef]
- Eiró, N.; Vizoso, F.J. Inflammation and cancer. World J. Gastrointest. Surg. 2012, 27, 62–72. [Google Scholar]
- Azim, H.; Azim, H.A., Jr. Targeting RANKL in breast cancer: Bone metastasis and beyond. Exp. Rev. Anticancer Ther. 2013, 13, 195–201. [Google Scholar] [CrossRef]
- Moore, M.M.; Chua, W.; Charles, K.A.; Clarke, S.J. Inflammation and cancer: Causes and consequences. Clin. Pharmacol. Ther. 2010, 87, 504–508. [Google Scholar] [CrossRef]
- Lu, W.; Lu, L.; Feng, Y.; Chen, J.; Li, Y.; Kong, X.; Chen, S.; Li, X.; Chen, Q.; Zhang, P. Inflammation promotes oral squamous carcinoma immune evasion via induced programmed death ligand-1 surface expression. Oncol. Lett. 2013, 5, 1519–1526. [Google Scholar]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Akova, Y.A.; Gungar, S.G. Ocular involvement in Behcet’s disease. In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 391–416. [Google Scholar]
- Su, S.C.; Hua, K.F.; Lee, H.; Chao, L.K.; Tan, S.K; Yang, S.F.; Hsu, H.Y. LTA and LPS mediated activation of protein kinases in the regulation of inflammatory cytokines expression in macrophages. Clin. Chim. Acta 2006, 374, 106–115. [Google Scholar] [CrossRef]
- Issa, L.A. Biologic agents for inflammatory bowel disease: The current, the future and the controversy. In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 417–430. [Google Scholar]
- Chen, C.-J.; Chin, J.E.; Ueda, K. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting edge: Cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 2003, 170, 3939–3943. [Google Scholar]
- Gukovskaya, A.S.; Gukovsky, I. Autophagy and pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G993–G1003. [Google Scholar] [CrossRef]
- Drayton, D.L.; Liao, S.; Mounzer, R.H.; Ruddle, N.H. Lymphoid organ development: From ontology to neogenesis. Nat. Immunol. 2006, 7, 344–353. [Google Scholar]
- Harvey, B.P.; Quan, T.F.; Rudenga, B.J.; Roman, R.M.; Craft, J.; Mamula, M.J. Editing antigen presentation: Antigen transfer between B lymphocyte and macrophages mediated by Class A Scavenger receptors. J. Immunol. 2008, 181, 4043–4051. [Google Scholar]
- Sriskandan, S.; Cohen, J. Gram-positive sepsis. Mechanisms and differences from gram-negative sepsis. Infect. Dis. Clin. North Am. 1999, 12, 397–412. [Google Scholar] [CrossRef]
- Dranoff, G. Immune recognition and tumor protection. Curr. Opin. Immunol. 2002, 14, 161–164. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation and associated disease. Ann. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Heinz, C.; Fanihagh, F.; Steuhl, K.P. Squamous cell carcinoma of the conjunctiva in patients with atopic eczema. Cornea 2003, 22, 135–137. [Google Scholar] [CrossRef]
- Jabaut, J.; Ckless, K. Inflammation, immunity and redox signaling. In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 145–160. [Google Scholar]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson, W.R., Jr.; Muller, W.; Rudensky, A.Y. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar]
- Khatami, M.; Donnelly, J.J.; John, T.; Rockey, J.H. Vernal conjunctivitis. Model studies in guinea pigs immunized topically with fluoresceinyl ovalbumin. Arch. Ophthalmol. 1984, 102, 1683–1688. [Google Scholar] [CrossRef]
- Khatami, M.; Donnelly, J.J.; Haldar, J.P.; Rockey, J.H. Massive follicular lymphoid hyperplasia in experimental chronic recurrent allergic conjunctivitis. Arch. Ophthalmol. 1989, 107, 433–438. [Google Scholar] [CrossRef]
- Khatami, M. Developmental phases of inflammation-induced massive lymphoid hyperplasia and extensive changes in epithelium in an experimental model of allergy. Implications for a direct link between inflammation and carcinogenesis. Am. J. Ther. 2005, 12, 117–128. [Google Scholar] [CrossRef]
- Khatami, M. “Yin and Yang” in inflammation: Duality in innate immune cell function and tumorigenesis. Exp. Opin.Biol. Ther. 2008, 8, 1461–1472. [Google Scholar] [CrossRef]
- Khatami, M. Inflammation, aging and cancer: Tumoricidal vs tumorigenesis of immunity: A common denominator mapping chronic diseases. Cell Biochem. Biophys. 2009, 55, 55–79. [Google Scholar] [CrossRef]
- Khatami, M. Unresolved inflammation: “Immune tsunami” or erosion of integrity in immune-privileged and immune-responsive tissues and acute and chronic inflammatory diseases or cancer. Exp. Opin. Biol. Ther. 2011, 11, 1419–1432. [Google Scholar] [CrossRef]
- Innocenti, F.; Cox, N.J.; Dolan, M.E. The use of genomic information to optimize cancer chemotherapy. Semin. Oncol. 2011, 38, 186–195. [Google Scholar] [CrossRef]
- Liu, F.S. Mechanisms of chemotherapeutic drug resistance in cancer therapy—A quick review. Taiwan. J. Obstet. Gynecol. 2009, 48, 239–244. [Google Scholar] [CrossRef]
- Karwacz, K.; Arce, F.; Bricogne, C.; Kochan, G.; Escors, D. PD-L1 co-stimulation, ligand-induced TCR down-modulation and anti-tumor immunotherapy. Oncoimmunology 2012, 1, 86–88. [Google Scholar] [CrossRef]
- Steele, C.W.; Jamieson, N.B.; Evans, T.R.; McKaym, C.J.; Sansom, O.J.; Morton, J.P.; Carter, C.R. Cploiting inflammation for therapeutic gain in pancreatic cancer. Br. J. Cancer 2013, 108, 997–1003. [Google Scholar]
- Khatami, M. Induction of conjunctival-associated lymphoid hyperplasia by antigen and tumor promoting agents: Targeting mediators of inflammatory responses as biomarkers for early detection of tumor/cancer. In Special Conference Proceedings: The Biology and Genetics of Early Detection and Chemoprevention of Cancer, Am Assoc Cancer Research, Bal Harbour, FL, USA, 6–10 October 1999.
- Mierke, C.T. Physical break-down of the classical view on cancer cell invasion and metastasis. Eur. J. Cell Biol. 2013, 92, 89–104. [Google Scholar] [CrossRef]
- Schumann, J. The impact of macrophage membrane lipid composition on innate immune response mechanisms. In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 31–52. [Google Scholar]
- Khatami, M. (NCI) Standardizing Criteria on Cancer Biomarkers as Foundation of a Database: Creating a Common Language (Data Elements) for Cancer Biomarkers Tracking and Utilization for Professionals in Oncology Research; HHS Reference No. E-147–2005/0-Research Tool; Federal Register: Washington, DC, USA, 2005. [Google Scholar]
- Gurish, F.; Boyce, J.A. Mast cells: Ontogeny, homing, and recruitment of a unique innate effector cell. J. Allerg. Clin. Immunol. 2006, 117, 1285–1291. [Google Scholar] [CrossRef]
- Carow, B.; Rottenberg, M.E. “Suppressor of cytokine signalling”: Molecules in infection and inflammation. In Inflammation, Chronic Diseases and Cancer. Cell and Molecular Biology, Immunology and Clinical Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 279–306. [Google Scholar]
- Reichmann, N.T.; Gründling, A. Location, synthesis and function of glycolipids and polyglycerolphosphate lipoteichoic acid in Gram-positive bacteria of the phylum Firmicutes. FEMS Microbiol. Lett. 2011, 319, 97–105. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Tam, S.Y.; Tsai, M.; Yamaguchi, M.; Yano, K.; Butterfield, J.H.; Galli, S.J. Expression of functional TrkA receptor tyrosine kinase in the HMC-1 human mast cell line and in human mast cells. Blood 1997, 90, 1807–1820. [Google Scholar]
- Sansonetti, P.J. War and peace at mucosal surfaces. Nat. Rev. Immunol. 2004, 4, 953–964. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Wevand, C.M. T cell development and receptor diversity during aging. Curr. Opin. Immunol. 2005, 17, 468–475. [Google Scholar] [CrossRef]
- Khatami, M. Unresolved inflammation and cancer: Loss of natural immune surveillance as the correct “target” for therapy! Seeing the “Elephant” in the light of logic. Cell Biochem. Biophys. 2012, 62, 501–509. [Google Scholar] [CrossRef]
- Khatami, M. Standardizing cancer biomarkers criteria: Data elements as a foundation for a database. Inflammatory mediator/M-CSF as model marker. Cell Biochem. Biophysics 2007, 47, 187–198. [Google Scholar] [CrossRef]
- Gounaris, E.; Blatner, N.R.; Dennis, K.; Magnusson, F.; Gurish, M.F.; Storm, T.B.; Beckhove, P.; Gounari, F.; Khazaie, K. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res. 2009, 69, 5490–5497. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; Kalogeromitros, D. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef]
- Gounaris, E.; Erdman, S.E.; Restanio, C.; Gurish, M.F.; Friend, D.S.; Lee, D.M.; Zhang, G.; Glickman, J.N.; Shin, K.; Rao, V.P.; et al. Mast cells are an essential hematopoietic component for polyp development. Proc. Natl. Acad. Sci. USA 2007, 104, 19977–19982. [Google Scholar] [CrossRef]
- Kormelink, T.G.; Askenase, P.W.; Redegeld, F.A. Immunobiology of antigen-specific immunoglobulin free light chains in inflammatory diseases. Curr. Pharm. Design 2012, 18, 2278–2289. [Google Scholar] [CrossRef]
- Blank, U.; Rivera, J. The ins and outs of IgE-dependent mast-cell exocytosis. Trends Immunol. 2004, 25, 266–273. [Google Scholar] [CrossRef]
- Siraganian, R.P. Mast cell signal transduction from the high-affinity IgE receptor. Curr. Opin. Immunol. 2003, 15, 639–646. [Google Scholar] [CrossRef]
- Kraft, S.; Kinet, J.P. New developments in Fc epsilonRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef]
- Redegeld, F.A.; Nijkamp, F.P. Immunoglobulin free light chains and mast cells: Pivotal role in T-cell-mediated immune reactions? Trends Immunol. 2003, 24, 181–185. [Google Scholar] [CrossRef]
- Hamrah, P.; Hug, S.O.; Liu, Y.; Zhang, Q.; Dana, M.R. Corneal immunity is mediated by heterogenous population of antigen-presenting cells. J. Leukoc. Biol. 2003, 74, 172–178. [Google Scholar] [CrossRef]
- Stoecker, M.M.; Wang, E. Histiocytic/dendritic cell transformation of B-cell neoplasms: Pathologic evidence of lineage conversion in differentiated hematolymphoid malignancies. Arch. Pathol. Lab Med. 2013, 137, 865–870. [Google Scholar] [CrossRef]
- Rodriguez, L.L.; Schneider, I.C. Directed cell migration in multi-cue environments. Integr. Biol. Camb. 2013, 5, 1306–1323. [Google Scholar] [CrossRef]
- D’Amato, G.; Salzillo, A.; Piccolo, A.; D’Amato, M.; Liccardi, G. A review of anti-IgE monoclonal antibody (omalizumab) as add on therapy for severe allergic (IgE-mediated) asthma. Ther. Clin. Risk Manag. 2007, 3, 613–619. [Google Scholar]
- Bienenstock, J.; Tomioka, M.; Matsuda, H.; Stead, R.H.; Quinonez, G.; Simon, G.T.; Coughlin, M.D.; Denburg, J.A. The role of mast cells in inflammatory processes: Evidence for nerve mast cell interactions. Int. Arch. Allergy Appl. Immunol. 1987, 82, 238–243. [Google Scholar] [CrossRef]
- Tal, M.; Liberman, R. Local injection of nerve growth factor (NGF) triggers degranulation of mast cells in rat paw. Neurosci. Lett. 1997, 221, 129–132. [Google Scholar] [CrossRef]
- Hotamisisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- De Simone, R.; Alleva, E.; Tirassa, P.; Aloe, L. Nerve growth factor released into the bloodstream following intraspecific fighting induces mast cell degranulation in adult male mice. Brain Behav. Immun. 1990, 4, 74–81. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Comeau, M.R.; Jessup, H.K.; Yoon, B.P.; Breuer, A.; Chartier, S.; Paquette, N.; Ziegler, S.F.; Sarfati, M.; Delespesse, G. Thymic stromal lymphopoietin is released by human epithelial cell in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007, 19, 253–258. [Google Scholar]
- Kumar, V. Innate immune system in sepsis immunopathogenesis and its modulation as future therapeutic approach. In Inflammatory Diseases; Immunopathology, Clinical and Pharmacological Bases; Khatami, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 57–82. [Google Scholar]
- Matsuda, H.; Kawakita, K.; Kiso, Y.; Nakano, T.; Kitamura, Y. Substance P induces granulocyte infiltration through degranulation of mast cells. J. Immunol. 1989, 142, 927–931. [Google Scholar]
- Khatami, M. Cyclooxygenase inhibitor Ketorolac or mast cell stabilizers: Immunological challenges in cancer therapy. Letters to the Editor. Clin. Cancer Res. 2005, 11, 1350–1352. [Google Scholar]
- Ledford, H. Translational research: 4 ways to fix the clinical trial. Nature 2011, 477, 526–528. [Google Scholar] [CrossRef]
- Pathology of Incipient Neoplasia, 3rd ed.; Henson, D.E.; Alborez-Saavedra, J. (Eds.) Oxford: Oxford University Press: New York, NY, USA, 2001.
- Gargalionis, A.N.; Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G. Histone modifications as a pathogenic mechanism of colorectal tumorigenesis. Int. J. Biochem. Cell Biol. 2012, 44, 1276–1289. [Google Scholar] [CrossRef]
- Di Virgilio, F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol. Rev. 2013, 65, 872–905. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Hill, C.S. Beyond TGFβ: Roles of other TGFβ superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef]
- Ferrantini, M.; Capone, I.; Belardelli, F. Dendritic cells and cytokines in immune rejection of cancer. Cytokine Growth Rev. 2008, 19, 93–107. [Google Scholar] [CrossRef]
- Zack, T.R.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer pattern of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef]
- Zanetti, M. Cell-extrinsic effects of the tumor unfolded protein response on myeloid cells and T cells. Ann. NY Acad. Sci. 2013, 1284, 6–11. [Google Scholar] [CrossRef]
- Blatner, N.R.; Mulcahy, M.F.; Dennis, K.L.; Scholtens, D.; Bentrem, D.J.; Phillips, J.D.; Ham, S.; Sandall, B.P.; Khan, M.W.; Mahvi, D.M.; et al. Expression of RORγt marks a pathogenic regulatory T cell subset in human colon cancer. Sci. Transl. Med. 2012, 4, 164ra159. [Google Scholar]
- Khatami, M.; Donnelly, J.J.; Rockey, J.H. Induction and down-regulation of conjunctival type-1 hypersensitivity reactions in guinea pigs sensitized topically with fluoresceinyl ovalbumin. Ophthalmic Res. 1985, 17, 139–147. [Google Scholar] [CrossRef]
- Haldar, J.P.; Khatami, M.; Donnelly, J.J.; Rockey, J.H. Experimental allergic conjunctivitis: Production of different isotypes of antibody by conjunctival-associated lymphoid tissue in culture. Reg. Immunol. 1988, 1, 92–99. [Google Scholar]
- Helleboid, L.; Khatami, M.; Wei, Z.-G.; Rockey, J.H. Histamine and prostacyclin: Primary and secondary release in allergic conjunctivitis. Invest. Ophthalmol. Vis. Sci. 1991, 32, 2281–2289. [Google Scholar]
- Donnelly, J.J.; Taylor, J.R.; Young, E.; Khatami, M.; Lok, J.B.; Rockey, J.H. Experimental ocular onchocerciasis in cynomolgus monkeys. Invest. Ophthalmol. Vis. Sci. 1986, 27, 492–499. [Google Scholar]
- Topalkara, A.; Ben-Arie-Weintrob, Y.; Ferry, J.A.; Foster, C.S. Conjunctival marginal zone B-cell lymphoma (MALT lymphoma) with amyloid and relapse in the stomach. Ocul. Immunol. Inflamm. 2007, 15, 347–350. [Google Scholar]
- Choudry, H.A.; O’Malley, M.E.; Guo, Z.S.; Zeh, H.J.; Bartlett, D.L. Mucin as a therapeutic target in pseudomyxoma peritoneal. J. Surg. Oncol. 2012, 106, 911–917. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Ding, T.; Xu, J.; Wang, F.; Shi, M.; Zhang, Y.; Li, S.P.; Zheng, L. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Human Pathol. 2009, 40, 381–389. [Google Scholar]
- Werno, C.; Menrad, H.; Weigert, A.; Dehne, N.; Goerdt, S.; Schledzewski, K.; Kzhyshkowska, J.; Brune, B. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 2010, 31, 1863–1872. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187–204. [Google Scholar]
- Tataroğlu, C.; Kargi, A.; Ozkal, S.; Eşrefoğlu, N.; Akkoçlu, A. Association of macrophages, mast cells and eosinophil leukocytes with angiogenesis and tumor stage in non-small cell lung carcinomas (NSCLC). Lung Cancer 2004, 43, 47–54. [Google Scholar]
- Zhou, J.; Ding, T.; Pan, W.; Zhu, L.Y.; Li, A.; Zheng, L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer 2009, 125, 1640–1648. [Google Scholar] [CrossRef]
- Spear, P.; Barber, A.; Rynda-Apple, A.; Sentman, C.L. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J. Immunol. 2012, 188, 6389–6398. [Google Scholar] [CrossRef]
- Wang, R.; Ma, Z.; Wang, Y.; Cheng, Z.; Xu, H.; Li, W.; Wang, X. The interaction of coagulation factor XII and monocyte/macrophages mediating peritoneal metastasis of epithelial ovarian cancer. Gynecol. Oncol. 2010, 117, 460–466. [Google Scholar] [CrossRef]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.M.; Magnaudeix, A.; Yardin, C.; Terro, F. Autophagy dysfunction and its link to Alzheimer’s disease and Type II diabetes mellitus. CNS Neurol. Disord. Drug Targets 2013. PMID:24059314. [Google Scholar]
- Kondratskyi, A.; Yassine, M.; Kondratska, K.; Skryma, R.; Slomianny, C.; Prevarskaya, N. Calcium-permeable ion channels in control of autophagy and cancer. Front. Physiol. 2013, 4, 272. [Google Scholar]
- Tooze, S.A. Current views on the source of the autophagosome membrane. Essays Biochem. 2013, 55, 29–38. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khatami, M. Chronic Inflammation: Synergistic Interactions of Recruiting Macrophages (TAMs) and Eosinophils (Eos) with Host Mast Cells (MCs) and Tumorigenesis in CALTs. M-CSF, Suitable Biomarker for Cancer Diagnosis! Cancers 2014, 6, 297-322. https://doi.org/10.3390/cancers6010297
Khatami M. Chronic Inflammation: Synergistic Interactions of Recruiting Macrophages (TAMs) and Eosinophils (Eos) with Host Mast Cells (MCs) and Tumorigenesis in CALTs. M-CSF, Suitable Biomarker for Cancer Diagnosis! Cancers. 2014; 6(1):297-322. https://doi.org/10.3390/cancers6010297
Chicago/Turabian StyleKhatami, Mahin. 2014. "Chronic Inflammation: Synergistic Interactions of Recruiting Macrophages (TAMs) and Eosinophils (Eos) with Host Mast Cells (MCs) and Tumorigenesis in CALTs. M-CSF, Suitable Biomarker for Cancer Diagnosis!" Cancers 6, no. 1: 297-322. https://doi.org/10.3390/cancers6010297