Interplay of Stem Cell Characteristics, EMT, and Microtentacles in Circulating Breast Tumor Cells

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Breast Cancer Metastasis
1.2. Animal Models of Metastasis
1.3. Patient CTC Detection
2. The Cancer Stem Cell Hypothesis
2.1. Mammary Stem Cells
2.2. Breast Cancer Stem Cells
3. CSCs and Metastasis
4. Cytoskeletal Alterations as Breast Cancer Cells Enter Circulation
4.1. EMT and Circulating Tumor Cells
4.2. EMT-Directed Cytoskeletal Alterations May also Promote the Metastatic Potential of CTCs
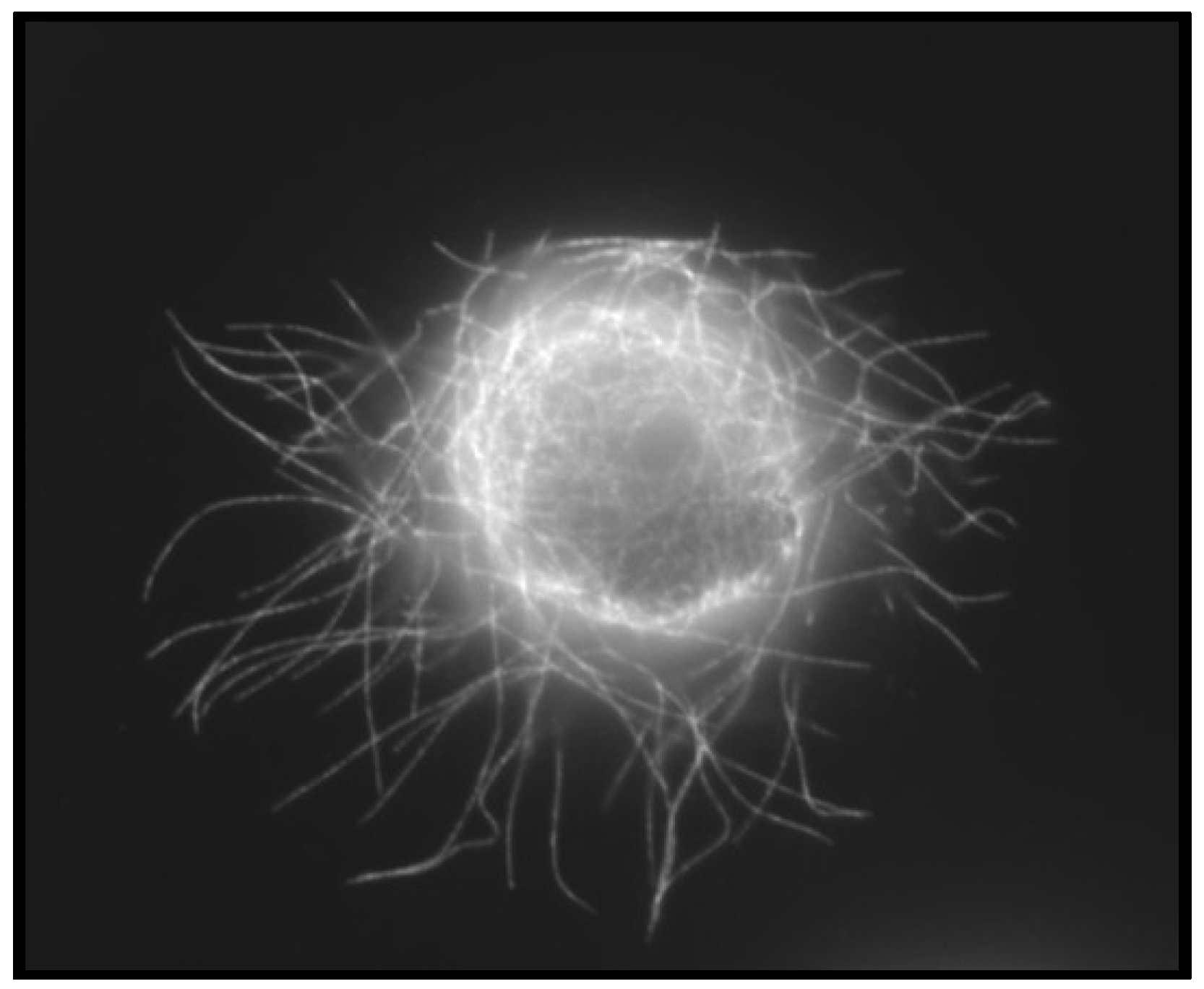
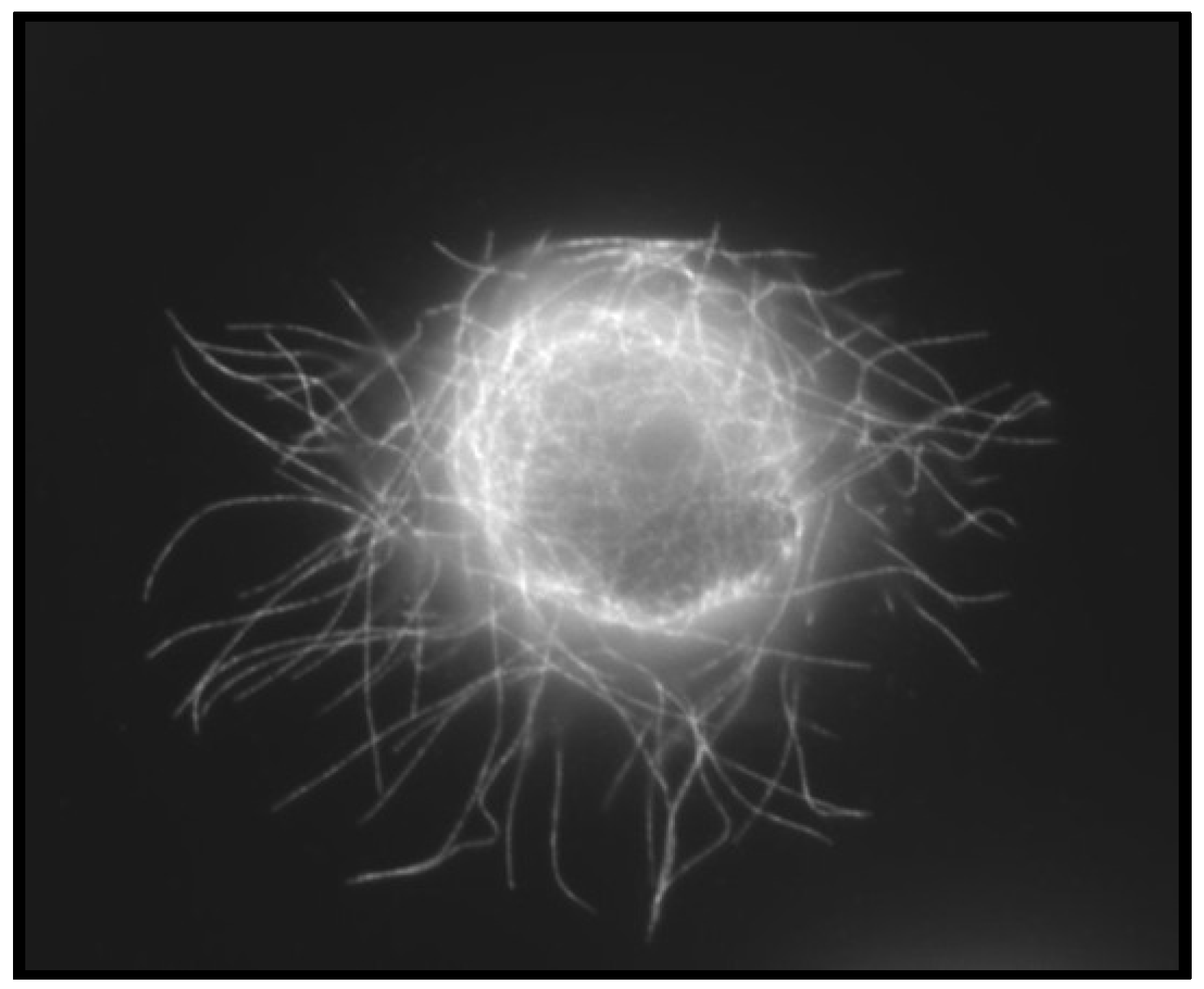
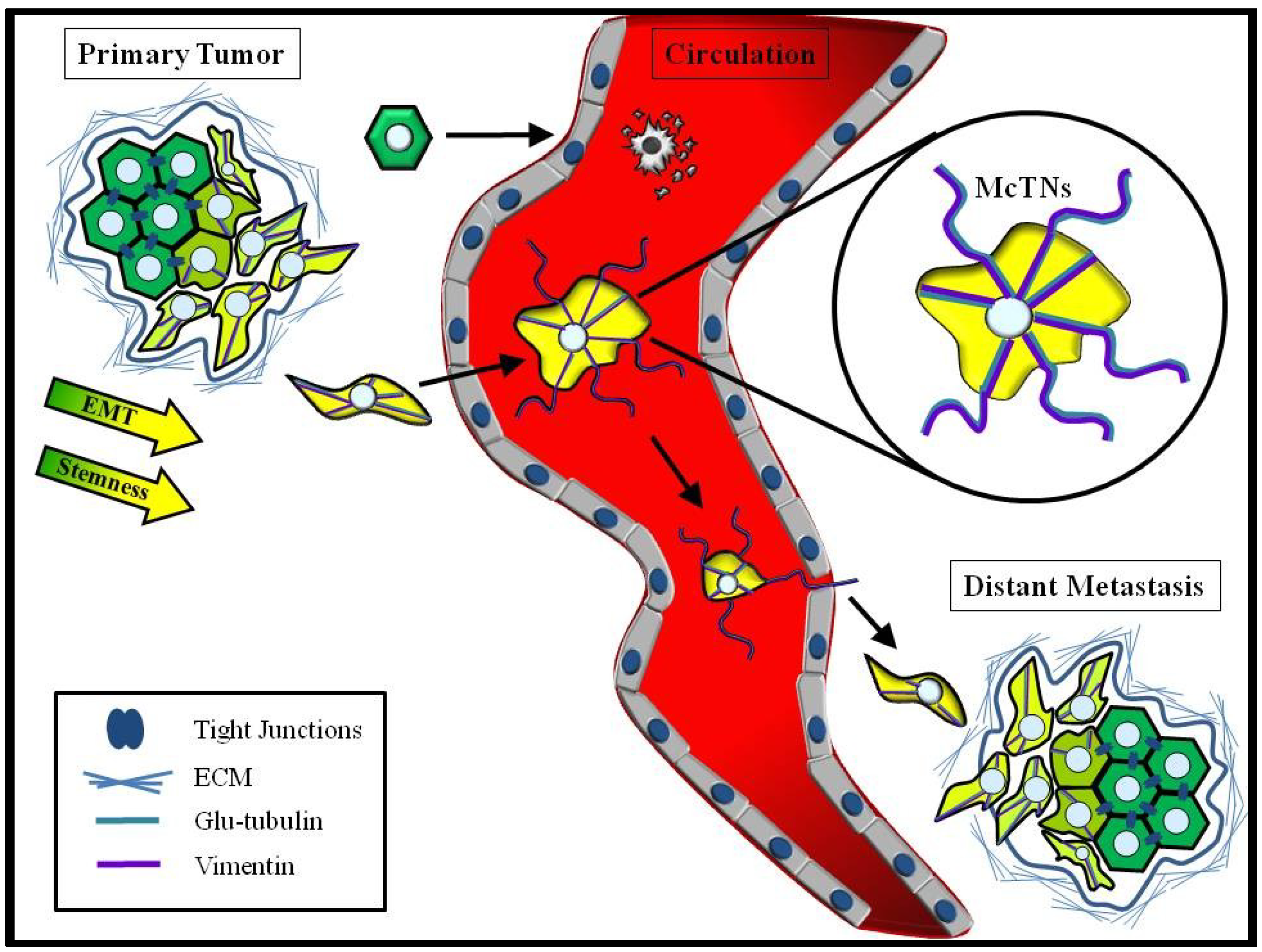
4.3. Convergence of EMT and CSCs
5. Emerging Evidence for EMT, CSC, and Related Cytoskeletal Alterations in CTCs from Breast Cancer Patients

6. Chemotherapeutics Targeting Primary Tumor Growth may have Unintended Consequences for CTCs
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- American Cancer Society, Inc, Breast Cancer Facts & Figures 2009–2010; Atlanta, GA, USA, 2009.
- Pagani, O.; Senkus, E.; Wood, W.; Colleoni, M.; Cufer, T.; Kyriakides, S.; Costa, A.; Winer, E.P.; Cardoso, F.; Force, E.-M.T. International guidelines for management of metastatic breast cancer: Can metastatic breast cancer be cured? J. Natl. Cancer Inst. 2010, 102, 456–463. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef]
- Weng, D.; Penzner, J.H.; Song, B.; Koido, S.; Calderwood, S.K.; Gong, J. Metastasis is an early event in mouse mammary carcinomas and is associated with cells bearing stem cell markers. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef]
- Schardt, J.A.; Meyer, M.; Hartmann, C.H.; Schubert, F.; Schmidt-Kittler, O.; Fuhrmann, C.; Polzer, B.; Petronio, M.; Eils, R.; Klein, C.A. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell 2005, 8, 227–239. [Google Scholar] [CrossRef]
- Vernon, A.E.; Bakewell, S.J.; Chodosh, L.A. Deciphering the molecular basis of breast cancer metastasis with mouse models. Rev. Endocr. Metab. Disord. 2007, 8, 199–213. [Google Scholar] [CrossRef]
- Jonkers, J.; Derksen, P.W. Modeling metastatic breast cancer in mice. J. Mammary Gland Biol. Neoplasia 2007, 12, 191–203. [Google Scholar] [CrossRef]
- Massoud, T.F.; Gambhir, S.S. Molecular imaging in living subjects: Seeing fundamental biological processes in a new light. Genes Dev. 2003, 17, 545–580. [Google Scholar] [CrossRef]
- Hoffman, R.M. Fluorescent proteins as visible in vivo sensors. Prog. Mol. Biol. Transl. Sci. 2013, 113, 389–402. [Google Scholar] [CrossRef]
- Glinskii, A.B.; Smith, B.A.; Jiang, P.; Li, X.M.; Yang, M.; Hoffman, R.M.; Glinsky, G.V. Viable circulating metastatic cells produced in orthotopic but not ectopic prostate cancer models. Cancer Res. 2003, 63, 4239–4243. [Google Scholar]
- Berezovskaya, O.; Schimmer, A.D.; Glinskii, A.B.; Pinilla, C.; Hoffman, R.M.; Reed, J.C.; Glinsky, G.V. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005, 65, 2378–2386. [Google Scholar] [CrossRef]
- Kolostova, K.; Pinterova, D.; Hoffman, R.M.; Bobek, V. Circulating human prostate cancer cells from an orthotopic mouse model rapidly captured by immunomagnetic beads and imaged by GFP expression. Anticancer Res. 2011, 31, 1535–1539. [Google Scholar]
- Eliane, J.P.; Repollet, M.; Luker, K.E.; Brown, M.; Rae, J.M.; Dontu, G.; Schott, A.F.; Wicha, M.; Doyle, G.V.; Hayes, D.F.; et al. Monitoring serial changes in circulating human breast cancer cells in murine xenograft models. Cancer Res. 2008, 68, 5529–5532. [Google Scholar] [CrossRef]
- Pantel, K.; Muller, V.; Auer, M.; Nusser, N.; Harbeck, N.; Braun, S. Detection and clinical implications of early systemic tumor cell dissemination in breast cancer. Clin. Cancer Res. 2003, 9, 6326–6334. [Google Scholar]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Riethdorf, S.; Fritsche, H.; Muller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Janicke, F.; et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: A validation study of the CellSearch system. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Budd, G.T.; Cristofanilli, M.; Ellis, M.J.; Stopeck, A.; Borden, E.; Miller, M.C.; Matera, J.; Repollet, M.; Doyle, G.V.; Terstappen, L.W.; et al. Circulating tumor cells versus imaging—Predicting overall survival in metastatic breast cancer. Clin. Cancer Res. 2006, 12, 6403–6409. [Google Scholar] [CrossRef]
- Budd, G.T.; Cristofanilli, M.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; Hayes, D.F. Monitoring circulating tumor cells (CTC) in non-measurable metastatic breast cancer (MBC). J. Clin. Oncol. 2005, 23. No. 16_suppl 503. [Google Scholar]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Monville, F.; Ginestier, C.; Dontu, G.; Birnbaum, D.; Wicha, M.S. Cancer stem cells in breast: Current opinion and future challenges. Pathobiology 2008, 75, 75–84. [Google Scholar] [CrossRef]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36, 59–72. [Google Scholar] [CrossRef]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef]
- Kordon, E.C.; Smith, G.H. An entire functional mammary gland may comprise the progeny from a single cell. Development 1998, 125, 1921–1930. [Google Scholar]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Sirard, C.; Lapidot, T.; Vormoor, J.; Cashman, J.D.; Doedens, M.; Murdoch, B.; Jamal, N.; Messner, H.; Addey, L.; Minden, M.; et al. Normal and leukemic SCID-repopulating cells (SRC) coexist in the bone marrow and peripheral blood from CML patients in chronic phase, whereas leukemic SRC are detected in blast crisis. Blood 1996, 87, 1539–1548. [Google Scholar]
- Dick, J.E. Normal and leukemic human stem cells assayed in SCID mice. Semin. Immunol. 1996, 8, 197–206. [Google Scholar] [CrossRef]
- Jones, R.J.; Barber, J.P.; Vala, M.S.; Collector, M.I.; Kaufmann, S.H.; Ludeman, S.M.; Colvin, O.M.; Hilton, J. Assessment of aldehyde dehydrogenase in viable cells. Blood 1995, 85, 2742–2746. [Google Scholar]
- Juopperi, T.A.; Schuler, W.; Yuan, X.; Collector, M.I.; Dang, C.V.; Sharkis, S.J. Isolation of bone marrow-derived stem cells using density-gradient separation. Exp. Hematol. 2007, 35, 335–341. [Google Scholar] [CrossRef]
- Jones, R.J.; Collector, M.I.; Barber, J.P.; Vala, M.S.; Fackler, M.J.; May, W.S.; Griffin, C.A.; Hawkins, A.L.; Zehnbauer, B.A.; Hilton, J.; et al. Characterization of mouse lymphohematopoietic stem cells lacking spleen colony-forming activity. Blood 1996, 88, 487–491. [Google Scholar]
- Kastan, M.B.; Schlaffer, E.; Russo, J.E.; Colvin, O.M.; Civin, C.I.; Hilton, J. Direct demonstration of elevated aldehyde dehydrogenase in human hematopoietic progenitor cells. Blood 1990, 75, 1947–1950. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef]
- Streuli, C.H.; Gilmore, A.P. Adhesion-mediated signaling in the regulation of mammary epithelial cell survival. J. Mammary Gland Biol. Neoplasia 1999, 4, 183–191. [Google Scholar] [CrossRef]
- Liao, M.J.; Zhang, C.C.; Zhou, B.; Zimonjic, D.B.; Mani, S.A.; Kaba, M.; Gifford, A.; Reinhardt, F.; Popescu, N.C.; Guo, W.; et al. Enrichment of a population of mammary gland cells that form mammospheres and have in vivo repopulating activity. Cancer Res. 2007, 67, 8131–8138. [Google Scholar] [CrossRef]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 2007, 317. [Google Scholar] [CrossRef]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef]
- Mine, T.; Matsueda, S.; Li, Y.; Tokumitsu, H.; Gao, H.; Danes, C.; Wong, K.K.; Wang, X.; Ferrone, S.; Ioannides, C.G. Breast cancer cells expressing stem cell markers CD44+ CD24 lo are eliminated by Numb-1 peptide-activated T cells. Cancer Immunol. Immunother. 2009, 58, 1185–1194. [Google Scholar] [CrossRef]
- Nishi, M.; Sakai, Y.; Akutsu, H.; Nagashima, Y.; Quinn, G.; Masui, S.; Kimura, H.; Perrem, K.; Umezawa, A.; Yamamoto, N.; et al. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene 2013. [Google Scholar] [CrossRef]
- Lawson, J.C.; Blatch, G.L.; Edkins, A.L. Cancer stem cells in breast cancer and metastasis. Breast Cancer Res. Treat. 2009, 118, 241–254. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef]
- Abraham, B.K.; Fritz, P.; McClellan, M.; Hauptvogel, P.; Athelogou, M.; Brauch, H. Prevalence of CD44+/CD24−/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin. Cancer Res. 2005, 11, 1154–1159. [Google Scholar]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef]
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11. [Google Scholar] [CrossRef]
- Idowu, M.O.; Kmieciak, M.; Dumur, C.; Burton, R.S.; Grimes, M.M.; Powers, C.N.; Manjili, M.H. CD44+/CD24−/low cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum. Pathol. 2012, 43, 364–373. [Google Scholar] [CrossRef]
- Berx, G.; Raspe, E.; Christofori, G.; Thiery, J.P.; Sleeman, J.P. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin. Exp. Metastasis 2007, 24, 587–597. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J. Cell Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef]
- Kumar, S.; Park, S.H.; Cieply, B.; Schupp, J.; Killiam, E.; Zhang, F.; Rimm, D.L.; Frisch, S.M. A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol. Cell. Biol. 2011, 31, 4036–4051. [Google Scholar] [CrossRef]
- Valsesia-Wittmann, S.; Magdeleine, M.; Dupasquier, S.; Garin, E.; Jallas, A.C.; Combaret, V.; Krause, A.; Leissner, P.; Puisieux, A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 2004, 6, 625–630. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Yamauchi, K.; Yang, M.; Jiang, P.; Yamamoto, N.; Xu, M.; Amoh, Y.; Tsuji, K.; Bouvet, M.; Tsuchiya, H.; Tomita, K.; et al. Real-time in vivo dual-color imaging of intracapillary cancer cell and nucleus deformation and migration. Cancer Res. 2005, 65, 4246–4252. [Google Scholar] [CrossRef]
- Ingber, D.E.; Tensegrity, I. Cell structure and hierarchical systems biology. J. Cell Sci. 2003, 116, 1157–1173. [Google Scholar] [CrossRef]
- Zhang, W.; Kai, K.; Choi, D.S.; Iwamoto, T.; Nguyen, Y.H.; Wong, H.; Landis, M.D.; Ueno, N.T.; Chang, J.; Qin, L. Microfluidics separation reveals the stem-cell-like deformability of tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2012, 109, 18707–18712. [Google Scholar] [CrossRef]
- Remmerbach, T.W.; Wottawah, F.; Dietrich, J.; Lincoln, B.; Wittekind, C.; Guck, J. Oral cancer diagnosis by mechanical phenotyping. Cancer Res. 2009, 69, 1728–1732. [Google Scholar] [CrossRef]
- Korb, T.; Schluter, K.; Enns, A.; Spiegel, H.U.; Senninger, N.; Nicolson, G.L.; Haier, J. Integrity of actin fibers and microtubules influences metastatic tumor cell adhesion. Exp. Cell Res. 2004, 299, 236–247. [Google Scholar] [CrossRef]
- Whipple, R.A.; Cheung, A.M.; Martin, S.S. Detyrosinated microtubule protrusions in suspended mammary epithelial cells promote reattachment. Exp. Cell Res. 2007, 313, 1326–1336. [Google Scholar] [CrossRef]
- Balzer, E.M.; Whipple, R.A.; Thompson, K.; Boggs, A.E.; Slovic, J.; Cho, E.H.; Matrone, M.A.; Yoneda, T.; Mueller, S.C.; Martin, S.S. c-Src differentially regulates the functions of microtentacles and invadopodia. Oncogene 2010, 29, 6402–6408. [Google Scholar] [CrossRef]
- Matrone, M.A.; Whipple, R.A.; Thompson, K.; Cho, E.H.; Vitolo, M.I.; Balzer, E.M.; Yoon, J.R.; Ioffe, O.B.; Tuttle, K.C.; Tan, M.; et al. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene 2010, 29, 3217–3227. [Google Scholar] [CrossRef]
- Whipple, R.A.; Balzer, E.M.; Cho, E.H.; Matrone, M.A.; Yoon, J.R.; Martin, S.S. Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Res. 2008, 68, 5678–5688. [Google Scholar] [CrossRef]
- Whipple, R.A.; Matrone, M.A.; Cho, E.H.; Balzer, E.M.; Vitolo, M.I.; Yoon, J.R.; Ioffe, O.B.; Tuttle, K.C.; Yang, J.; Martin, S.S. Epithelial-to-mesenchymal transition promotes tubulin detyrosination and microtentacles that enhance endothelial engagement. Cancer Res. 2010, 70, 8127–8137. [Google Scholar] [CrossRef]
- Janke, C.; Bulinski, J.C. Post-translational regulation of the microtubule cytoskeleton: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011, 12, 773–786. [Google Scholar] [CrossRef]
- Westermann, S.; Weber, K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4, 938–947. [Google Scholar] [CrossRef]
- Kreitzer, G.; Liao, G.; Gundersen, G.G. Detyrosination of tubulin regulates the interaction of intermediate filaments with microtubules in vivo via a kinesin-dependent mechanism. Mol. Biol. Cell 1999, 10, 1105–1118. [Google Scholar] [CrossRef]
- Kuroda, H.; Saito, K.; Kuroda, M.; Suzuki, Y. Differential expression of glu-tubulin in relation to mammary gland disease. Virchows Arch. 2010, 457, 477–482. [Google Scholar] [CrossRef]
- Mialhe, A.; Lafanechere, L.; Treilleux, I.; Peloux, N.; Dumontet, C.; Bremond, A.; Panh, M.H.; Payan, R.; Wehland, J.; Margolis, R.L.; et al. Tubulin detyrosination is a frequent occurrence in breast cancers of poor prognosis. Cancer Res. 2001, 61, 5024–5027. [Google Scholar]
- Willipinski-Stapelfeldt, B.; Riethdorf, S.; Assmann, V.; Woelfle, U.; Rau, T.; Sauter, G.; Heukeshoven, J.; Pantel, K. Changes in cytoskeletal protein composition indicative of an epithelial-mesenchymal transition in human micrometastatic and primary breast carcinoma cells. Clin. Cancer Res. 2005, 11, 8006–8014. [Google Scholar] [CrossRef]
- Yoon, J.R.; Whipple, R.A.; Balzer, E.M.; Cho, E.H.; Matrone, M.A.; Peckham, M.; Martin, S.S. Local anesthetics inhibit kinesin motility and microtentacle protrusions in human epithelial and breast tumor cells. Breast Cancer Res. Treat. 2011, 129, 691–701. [Google Scholar] [CrossRef]
- Yamashita, N.; Tokunaga, E.; Kitao, H.; Hisamatsu, Y.; Taketani, K.; Akiyoshi, S.; Okada, S.; Aishima, S.; Morita, M.; Maehara, Y. Vimentin as a poor prognostic factor for triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 739–746. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cellul. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Gilles, C.; Polette, M.; Zahm, J.M.; Tournier, J.M.; Volders, L.; Foidart, J.M.; Birembaut, P. Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci. 1999, 4615–4625. [Google Scholar]
- Taylor, M.A.; Parvani, J.G.; Schiemann, W.P. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J. Mammary Gland Biol. Neoplasia 2010, 15, 169–190. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Giuliano, M.; Giordano, A.; Jackson, S.; Hess, K.R.; de Giorgi, U.; Mego, M.; Handy, B.C.; Ueno, N.T.; Alvarez, R.H.; de Laurentiis, M.; et al. Circulating tumor cells as prognostic and predictive markers in metastatic breast cancer patients receiving first-line systemic treatment. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Giordano, A.; Gao, H.; Cohen, E.N.; Anfossi, S.; Khoury, J.; Hess, K.; Krishnamurthy, S.; Tin, S.; Cristofanilli, M.; Hortobagyi, G.N.; et al. Clinical relevance of cancer stem cells in bone marrow of early breast cancer patients. Ann. Oncol. 2013, 24, 2515–2521. [Google Scholar] [CrossRef]
- Wang, N.; Shi, L.; Li, H.; Hu, Y.; Du, W.; Liu, W.; Zheng, J.; Huang, S.; Qu, X. Detection of circulating tumor cells and tumor stem cells in patients with breast cancer by using flow cytometry: A valuable tool for diagnosis and prognosis evaluation. Tumour Biol. 2012, 33, 561–569. [Google Scholar] [CrossRef]
- Wang, J.; Cao, M.G.; You, C.Z.; Wang, C.L.; Liu, S.L.; Kai, C.; Dou, J. A preliminary investigation of the relationship between circulating tumor cells and cancer stem cells in patients with breast cancer. Cell. Mol. Biol. 2012, 58, 1641–1645. [Google Scholar]
- Theodoropoulos, P.A.; Polioudaki, H.; Agelaki, S.; Kallergi, G.; Saridaki, Z.; Mavroudis, D.; Georgoulias, V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010, 288, 99–106. [Google Scholar] [CrossRef]
- Reuben, J.M.; Lee, B.N.; Gao, H.; Cohen, E.N.; Mego, M.; Giordano, A.; Wang, X.; Lodhi, A.; Krishnamurthy, S.; Hortobagyi, G.N.; et al. Primary breast cancer patients with high risk clinicopathologic features have high percentages of bone marrow epithelial cells with ALDH activity and CD44+CD24lo cancer stem cell phenotype. Eur. J. Cancer 2011, 47, 1527–1536. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Fiska, A.; Koukourakis, M.I. The CD44+/CD24− phenotype relates to “triple-negative” state and unfavorable prognosis in breast cancer patients. Med. Oncol. 2011, 28, 745–752. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Kallergi, G.; Papadaki, M.A.; Politaki, E.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011, 13, R59. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Mego, M.; Gao, H.; Lee, B.N.; Cohen, E.N.; Tin, S.; Giordano, A.; Wu, Q.; Liu, P.; Nieto, Y.; Champlin, R.E.; et al. Prognostic value of EMT-circulating tumor cells in metastatic breast cancer patients undergoing high-dose chemotherapy with autologous hematopoietic stem cell transplantation. J. Cancer 2012, 3, 369–380. [Google Scholar] [CrossRef]
- Raimondi, C.; Gradilone, A.; Naso, G.; Vincenzi, B.; Petracca, A.; Nicolazzo, C.; Palazzo, A.; Saltarelli, R.; Spremberg, F.; Cortesi, E.; et al. Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res. Treat. 2011, 130, 449–455. [Google Scholar] [CrossRef]
- Giordano, A.; Gao, H.; Anfossi, S.; Cohen, E.; Mego, M.; Lee, B.N.; Tin, S.; de Laurentiis, M.; Parker, C.A.; Alvarez, R.H.; et al. Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol. Cancer Ther. 2012, 11, 2526–2534. [Google Scholar] [CrossRef]
- Steeg, P.S. Perspective: The right trials. Nature 2012, 485, S58–S59. [Google Scholar] [CrossRef]
- Balzer, E.M.; Whipple, R.A.; Cho, E.H.; Matrone, M.A.; Martin, S.S. Antimitotic chemotherapeutics promote adhesive responses in detached and circulating tumor cells. Breast Cancer Res. Treat. 2010, 121, 65–78. [Google Scholar] [CrossRef]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef]
- Mego, M.; Mani, S.A.; Lee, B.N.; Li, C.; Evans, K.W.; Cohen, E.N.; Gao, H.; Jackson, S.A.; Giordano, A.; Hortobagyi, G.N.; et al. Expression of epithelial-mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int. J. Cancer. 2012, 130, 808–816. [Google Scholar] [CrossRef]
- Hekimian, K.; Meisezahl, S.; Trompelt, K.; Rabenstein, C.; Pachmann, K. Epithelial cell dissemination and readhesion: analysis of factors contributing to metastasis formation in breast cancer. ISRN Oncol. 2012, 2012. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Charpentier, M.; Martin, S. Interplay of Stem Cell Characteristics, EMT, and Microtentacles in Circulating Breast Tumor Cells. Cancers 2013, 5, 1545-1565. https://doi.org/10.3390/cancers5041545
Charpentier M, Martin S. Interplay of Stem Cell Characteristics, EMT, and Microtentacles in Circulating Breast Tumor Cells. Cancers. 2013; 5(4):1545-1565. https://doi.org/10.3390/cancers5041545
Chicago/Turabian StyleCharpentier, Monica, and Stuart Martin. 2013. "Interplay of Stem Cell Characteristics, EMT, and Microtentacles in Circulating Breast Tumor Cells" Cancers 5, no. 4: 1545-1565. https://doi.org/10.3390/cancers5041545