Oxidative Stress in the Carcinogenicity of Chemical Carcinogens




Abstract
:1. Introduction
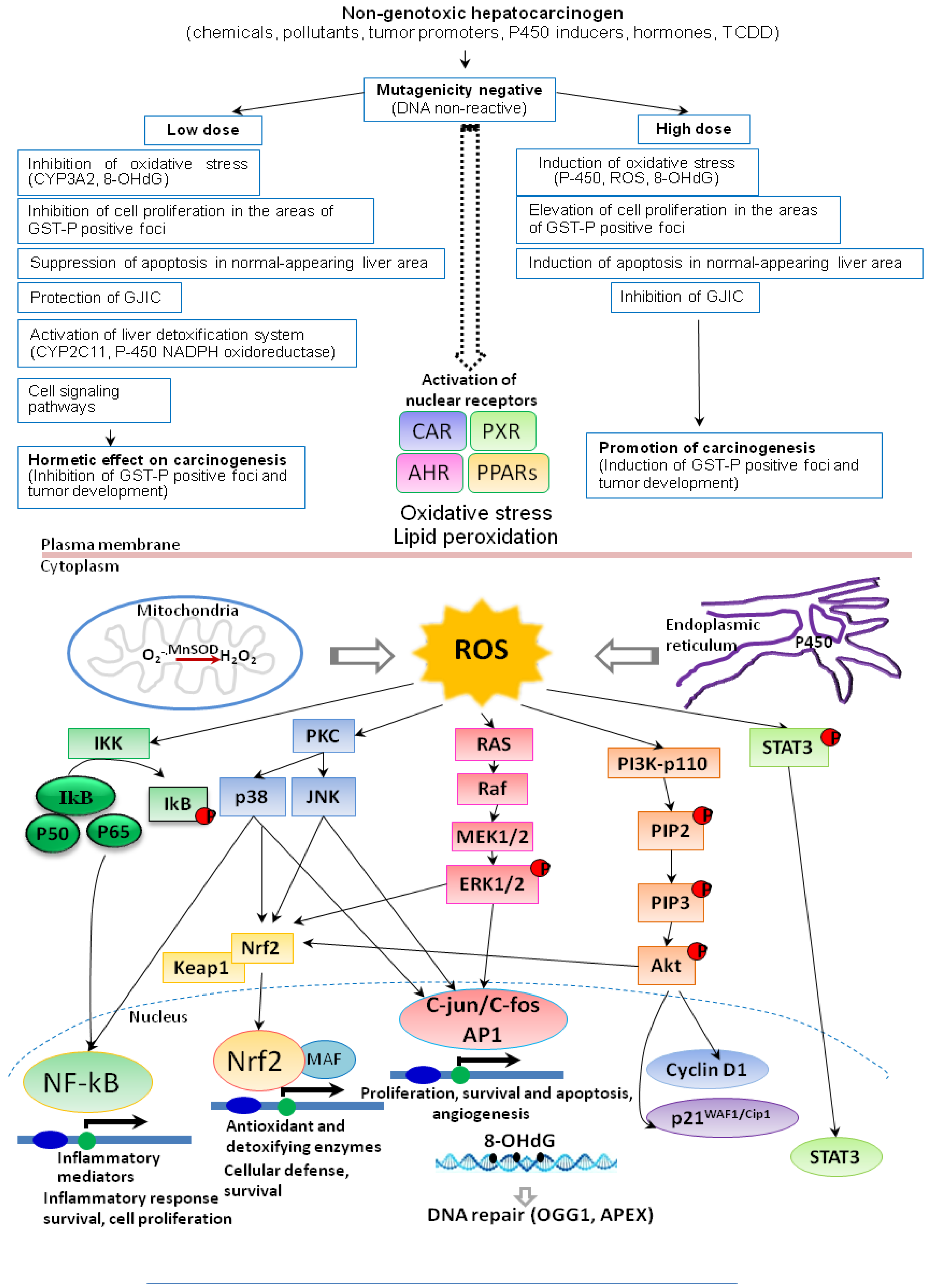
2. Induction of Oxidative Stress by Environmental Chemical Carcinogens and Toxicants
3. Hormesis in Carcinogenicity of Non-Genotoxic Carcinogens
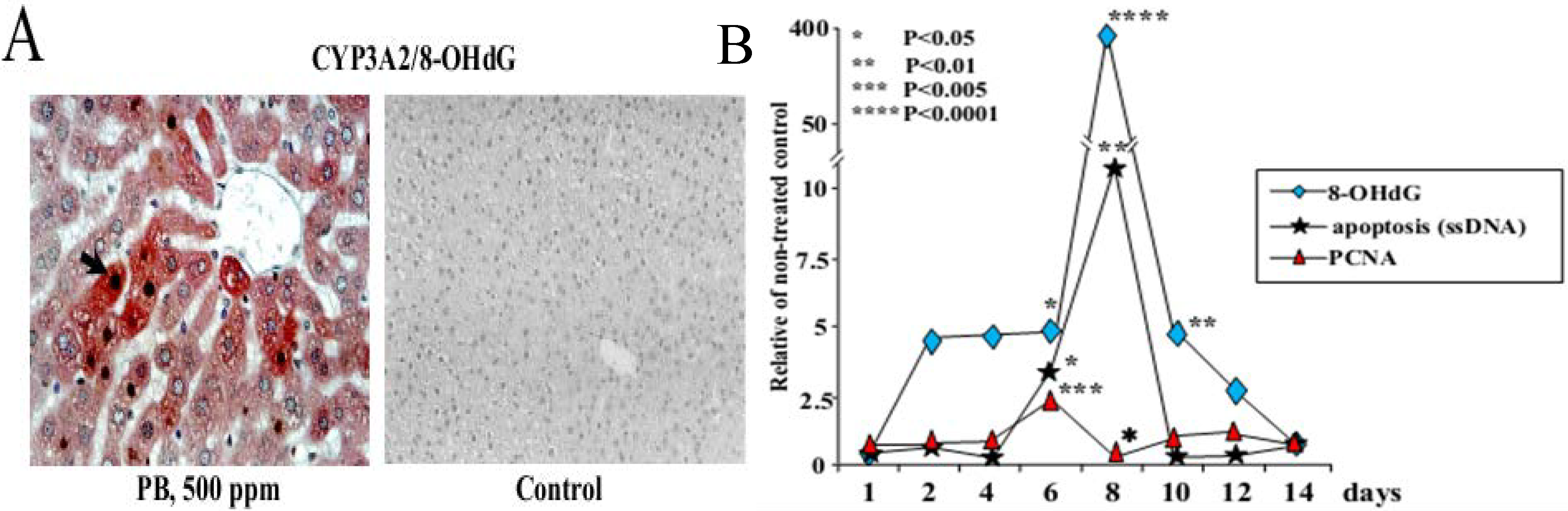
3.1. Carcinogenicity of Phenobarbital (PB)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Key in vivo findings |
|---|---|
| PB | Induction of P450 isoenzymes (e.g., CYP2B and CYP3A), 8-OHdG, cell cycle arrest and apoptosis in the rat liver (short-term study) Promotion of hepatocarcinogenesis in rats at high doses (60–500 ppm) and inhibition at low doses (1 and 2 ppm) (medium-term rat liver bioassay, 2-step carcinogenicity test) |
| α-BHC | Promotion of hepatocarcinogenesis in rats at high doses (0.5 to 500 ppm) and inhibition at low doses (0.01 and 0.1 ppm) (medium-term rat liver bioassay, 2-step carcinogenicity test) |
| DDT | Promotion of hepatocarcinogenesis in rats at high doses (20–500 ppm) and inhibition at low doses (0.005 and 0.01 ppm) (medium-term rat liver bioassay, 2-step carcinogenicity test) |
| ETBE | Induction of P450 (e.g., CYP2B, CYP3A, CYP2E1), 8-OHdG, cell cycle arrest and apoptosis in the rat liver (short-term study) Tumorigenicity in the rat liver (2-year carcinogenicity test) and promotion of hepatocarcinogenesis in rats (multi-organ carcinogenesis bioassay) |
| Organic arsenicals | |
| MMAV | Promotion of preneoplastic lesions development in the liver (medium-term rat liver bioassay) |
| DMAV | Induction of P450 isoenzymes (e.g., CYP2B1, CYP3A1 and CYP1A2), phase II metabolizing enzymes, 8-OHdG, cell cycle arrest and apoptosis in the rat liver (short-term study) Promotion of carcinogenesis in the rat urinary bladder, kidney, liver and thyroid gland (multi-organ carcinogenesis bioassay, medium-term rat liver bioassay) Carcinogenicity in the rat bladder (2-year carcinogenicity test) Carcinogenicity in the lung of Mmh/Ogg1-deficient mice (long-term carcinogenicity test) |
| TMAOV | Induction of P450 isoenzymes (e.g., CYP2B1 and CYP1A2), phase II metabolizing enzymes, 8-OHdG, cell cycle arrest and apoptosis in the rat liver (short-term study) Promotion of preneoplastic lesions in the rat liver (medium-term rat liver bioassay) Tumorigenicity in the rat liver (2-year carcinogenicity test) |
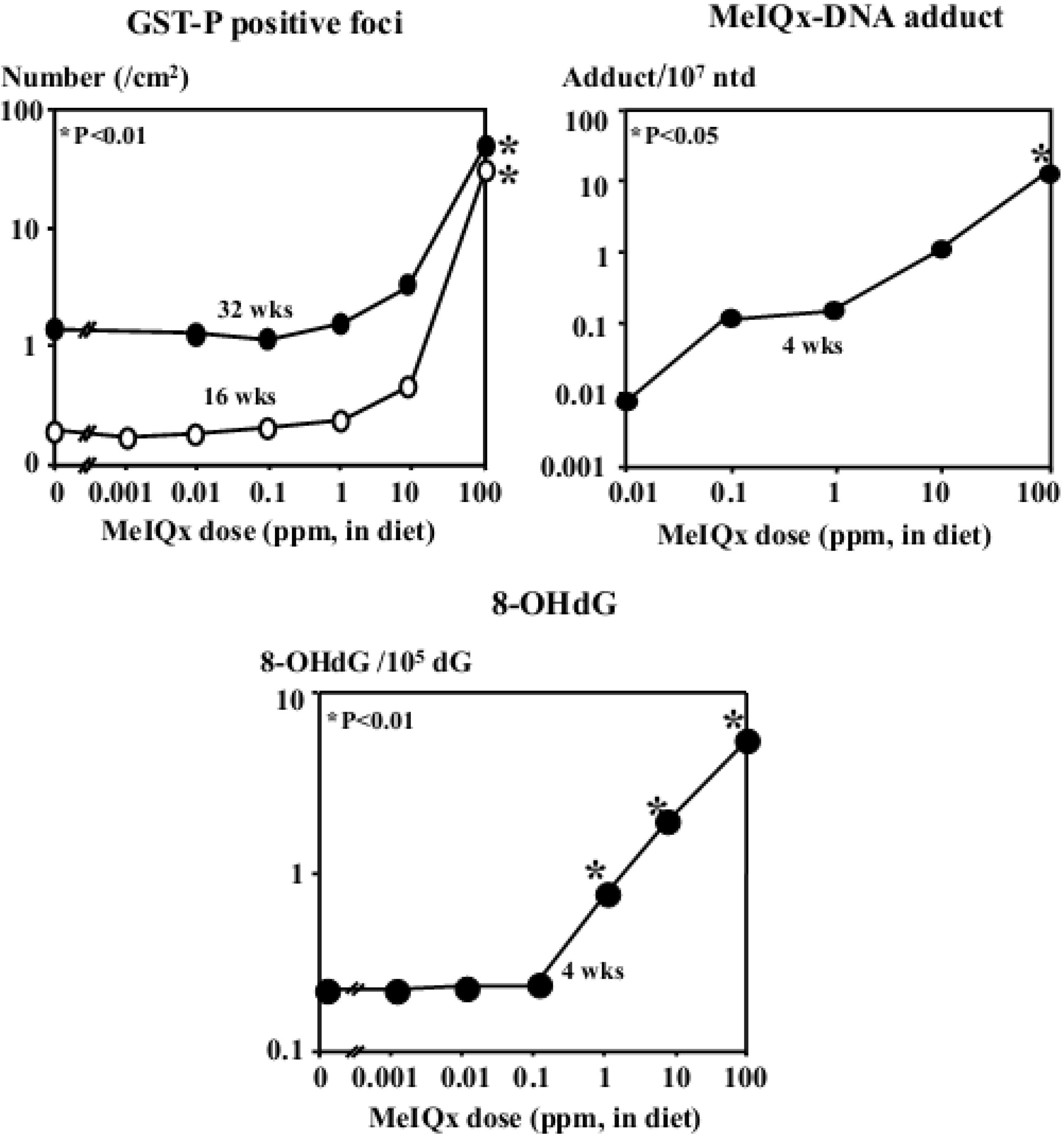
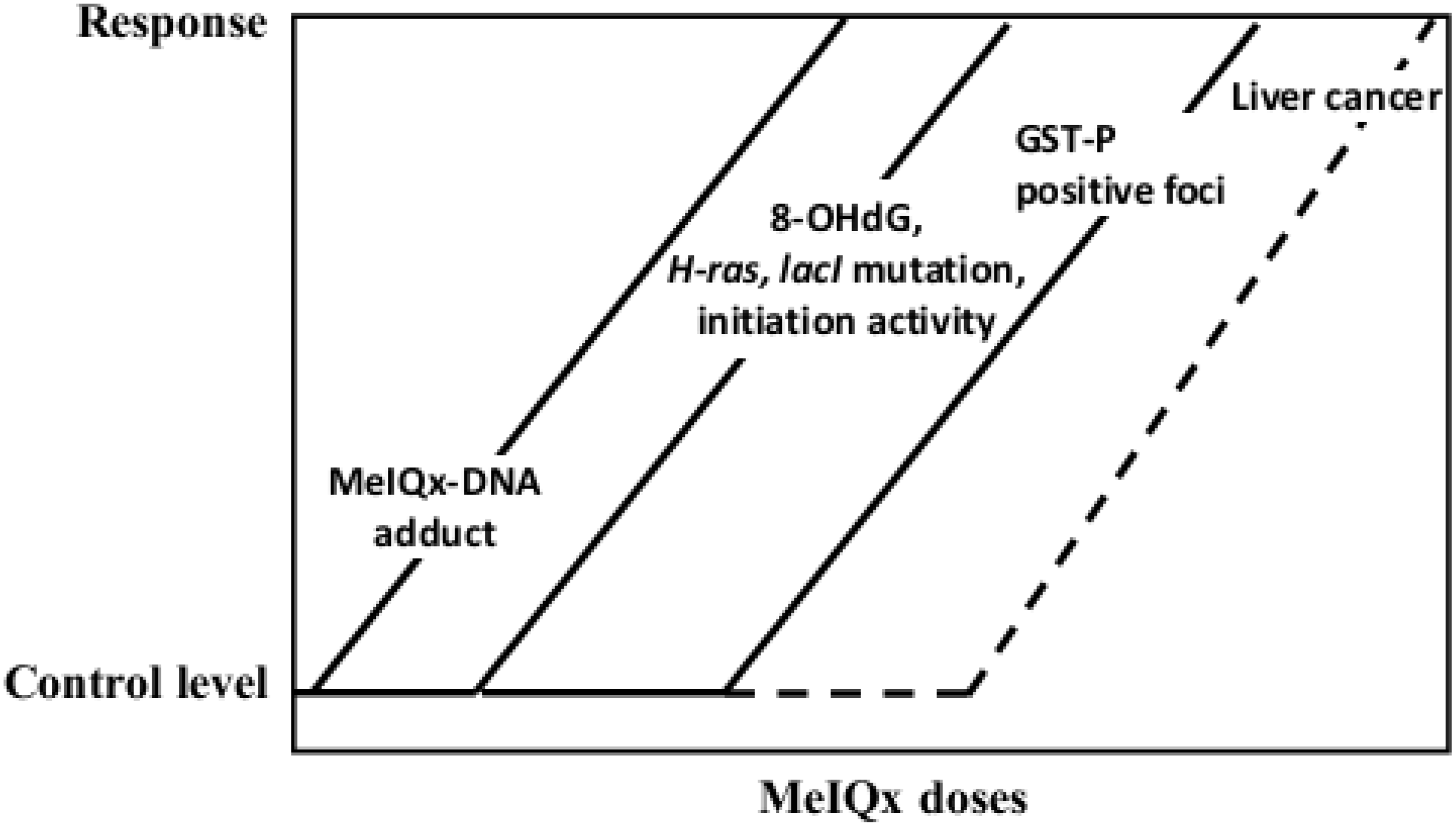
| MeIQx | Induction of GST-P positive foci and liver tumors (100 to 400 ppm), formation of MeIQx-DNA adducts, elevation of 8-OHdG, lacI and H-ras mutation levels (long-term carcinogenicity test) |
| KBrO3 | Increase of lacI mutations, 8-OHdG levels, and promotion of carcinogenesis in the rat kidney (2-step carcinogenicity test) |
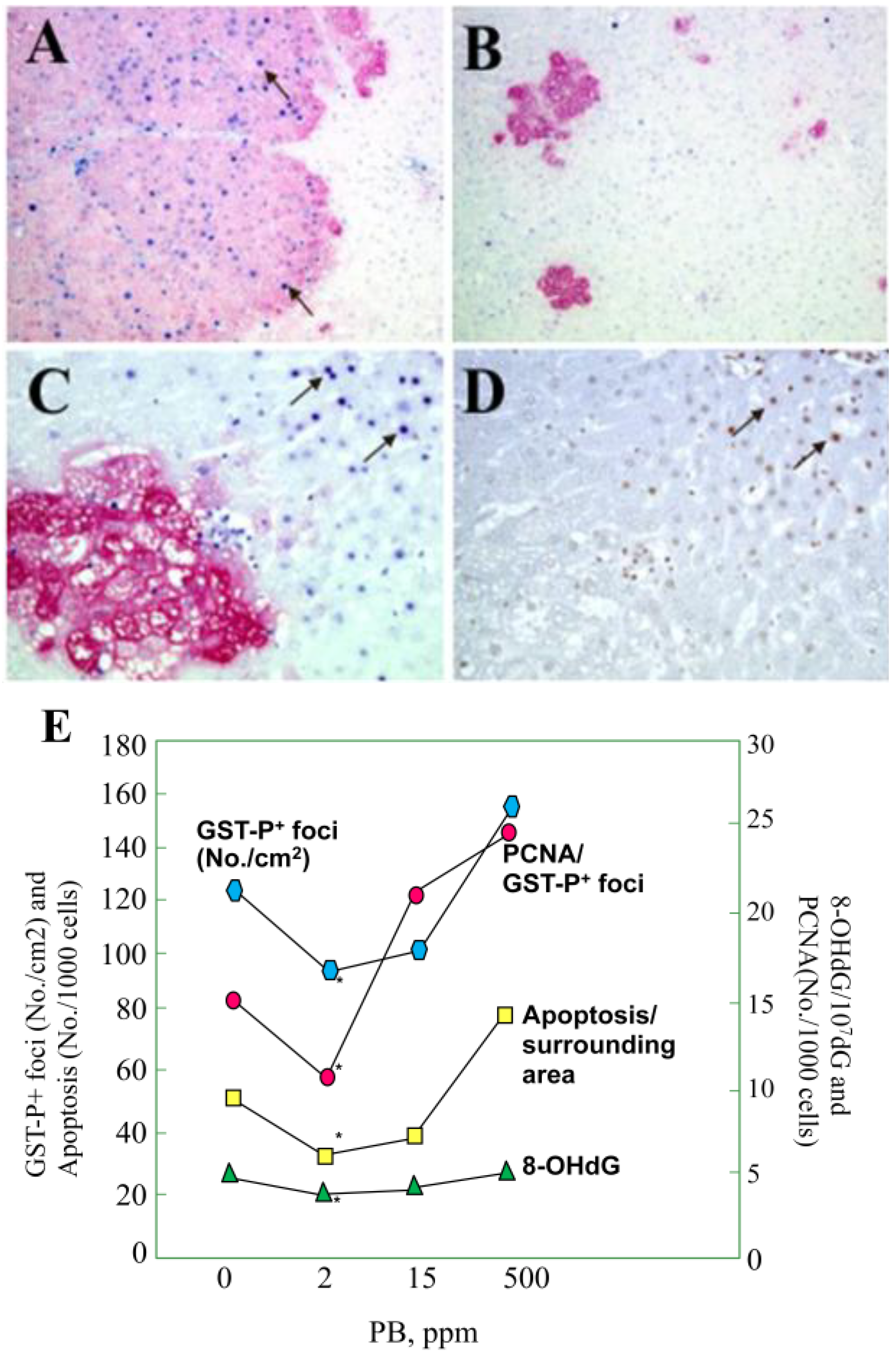
3.2. Carcinogenicity of α-Benzene Hexachloride (α-BHC)
3.3. Carcinogenicity of 1,1-bis(p-Chlorophenyl)-2,2,2-trichloroethane (DDT)
4. Carcinogenicity of Ethyl Tertiary-Butyl Ether (ETBE)
5. Carcinogenicity of Genotoxic Carcinogens and Involvement of Oxidative Stress
5.1. Arsenic Carcinogenicity
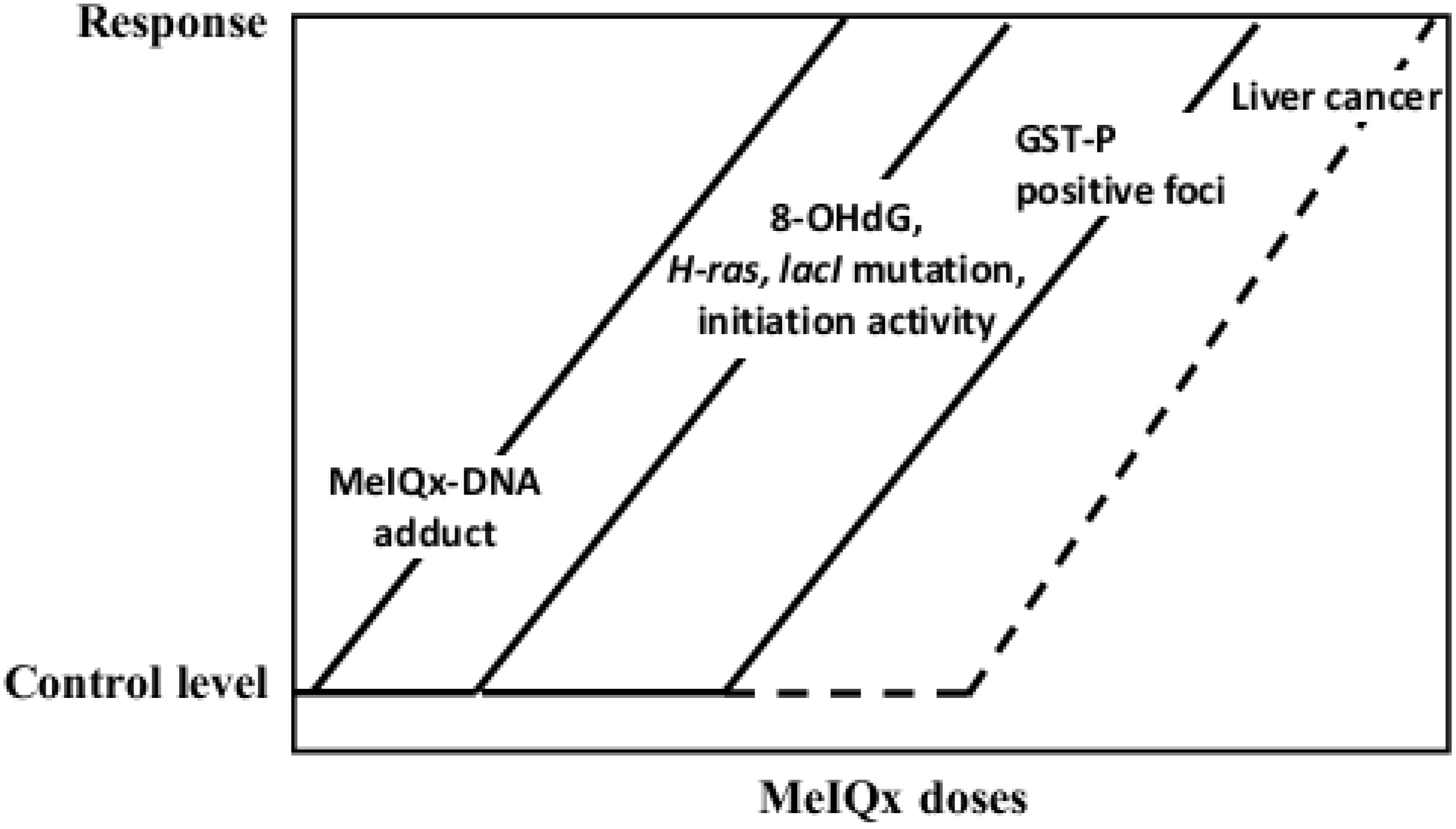
5.2. Carcinogenicity of 2-Amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx)
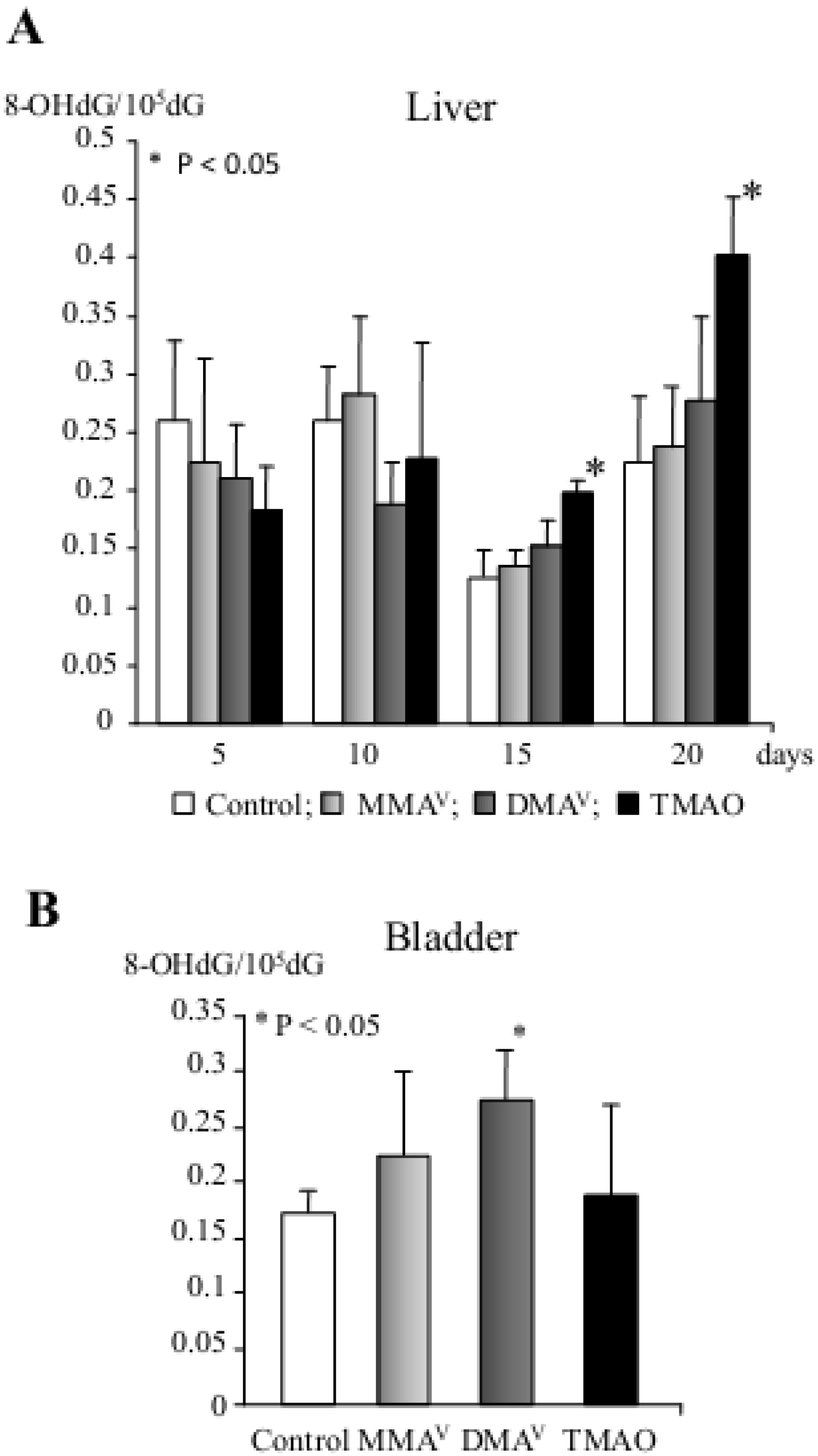

5.3. Carcinogenicity of Potassium Bromate (KBrO3)
6. Induction of ROS, Cell Proliferation, Apoptosis and DNA Repair
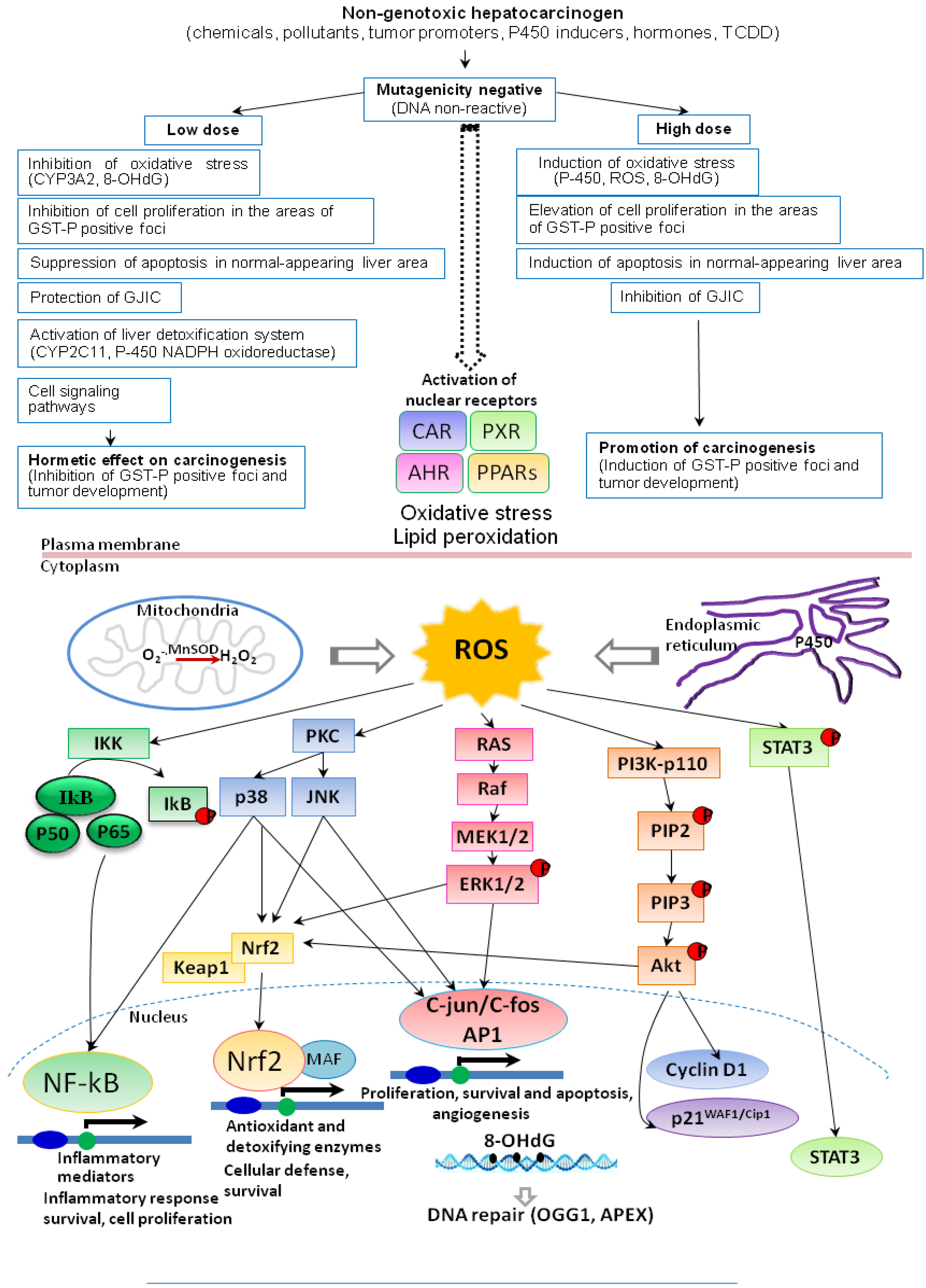
7. Conclusions
Acknowledgements
Conflicts of Interest
References
- Nowsheen, S.; Aziz, K.; Kryston, T.B.; Ferguson, N.F.; Georgakilas, A. The interplay between inflammation and oxidative stress in carcinogenesis. Curr. Mol. Med. 2012, 12, 672–680. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef]
- Loft, S.; Poulsen, H.E. Cancer risk and oxidative DNA damage in man. J. Mol. Med. 1996, 74, 297–312. [Google Scholar] [CrossRef]
- Wilson, K.T.; Fu, S.; Ramanujam, K.S.; Meltzer, S.J. Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett's esophagus and associated adenocarcinomas. Cancer Res. 1998, 58, 2929–2934. [Google Scholar]
- Bartsch, H.; Nair, J. Ultrasensitive and specific detection methods for exocylic DNA adducts: Markers for lipid peroxidation and oxidative stress. Toxicology 2000, 153, 105–114. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Nair, J.; Barbin, A.; Guichard, Y.; Bartsch, H. 1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytine in liver DNA from humans and untreated rodents detected by immunoaffinity/32P-postlabeling. Carcinogenesis 1995, 16, 613–617. [Google Scholar] [CrossRef]
- Hang, B.; Chenna, A.; Sagi, J.; Singer, B. Differential cleavage of oligonucleotides containing the benzene-derived adduct, 1,N6-benzetheno-dA, by the major human AP endonuclease HAP1 and Escherichia coli exonuclease III and endonuclease IV. Carcinogenesis 1998, 19, 1339–1343. [Google Scholar]
- Anisimov, V.N. Ageing and the mechanisms of carcinogenesis: Some practical implications. J. Exp. Clin. Cancer Res. 1998, 17, 263–268. [Google Scholar]
- Palut, D.; Kostka, G.; Strucinski, P. The role of nuclear receptors in cytochrome P-450 induction by xenochemicals. Rocz. Panstw. Zakl. Hig. 2002, 53, 321–332. [Google Scholar]
- Maglich, J.M.; Stoltz, C.M.; Goodwin, B.; Hawkins-Brown, D.; Moore, J.T.; Kliewer, S.A. Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002, 62, 638–646. [Google Scholar] [CrossRef]
- Bertilsson, G.; Heidrich, J.; Svensson, K.; Asman, M.; Jendeberg, L.; Sydow-Backman, M.; Ohlsson, R.; Postlind, H.; Blomquist, P.; Berkenstam, A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc. Natl. Acad. Sci. USA 1998, 95, 12208–12213. [Google Scholar] [CrossRef]
- Blumberg, B.; Evans, R.M. Orphan nuclear receptors-new ligands and new possibilities. Genes Dev. 1998, 12, 3149–3155. [Google Scholar] [CrossRef]
- Moore, L.B.; Parks, D.J.; Jones, S.A.; Bledsoe, R.K.; Consler, T.G.; Stimmel, J.B.; Goodwin, B.; Liddle, C.; Blanchard, S.G.; Willson, T.M.; et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 2000, 275, 15122–15127. [Google Scholar]
- Honkakoski, P.; Negishi, M. Regulatory DNA elements of phenobarbital-responsive cytochrome P450 CYP2B genes. J. Biochem. Mol. Toxicol. 1998, 12, 3–9. [Google Scholar] [CrossRef]
- Moore, L.B.; Maglich, J.M.; McKee, D.D.; Wisely, B.; Willson, T.M.; Kliewer, S.A.; Lambert, M.H.; Moore, J.T. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol. Endocrinol. 2002, 16, 977–986. [Google Scholar] [CrossRef]
- Sueyoshi, T.; Kawamoto, T.; Zelko, I.; Honkakoski, P.; Negishi, M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J. Biol. Chem. 1999, 274, 6043–6046. [Google Scholar]
- Dzhekova-Stojkova, S.; Bogdanska, J.; Stojkova, Z. Peroxisome proliferators: Their biological and toxicological effects. Clin. Chem. Lab. Med. 2001, 39, 468–474. [Google Scholar]
- O’Brien, M.L.; Spear, B.T.; Glauert, H.P. Role of oxidative stress in peroxisome proliferator-mediated carcinogenesis. Crit. Rev. Toxicol. 2005, 35, 61–88. [Google Scholar]
- Lim, Y.P.; Huang, J.D. Interplay of pregnane X receptor with other nuclear receptors on gene regulation. Drug Metab. Pharmacokinet. 2008, 23, 14–21. [Google Scholar] [CrossRef]
- Pascussi, J.M.; Gerbal-Chaloin, S.; Duret, C.; Daujat-Chavanieu, M.; Vilarem, M.J.; Maurel, P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: Crosstalk and consequences. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 1–32. [Google Scholar] [CrossRef]
- Moreau, A.; Vilarem, M.J.; Maurel, P.; Pascussi, J.M. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol. Pharm. 2008, 5, 35–41. [Google Scholar]
- Feig, D.I.; Reid, T.M.; Loeb, L.A. Reactive oxygen species in tumorigenesis. Cancer Res. 1994, 54, 1890s–1894s. [Google Scholar]
- Klaunig, J.E.; Xu, Y.; Isenberg, J.S.; Bachowski, S.; Kolaja, K.L.; Jiang, J.; Stevenson, D.E.; Walborg, E.F., Jr. The role of oxidative stress in chemical carcinogenesis. Environ. Health Perspect. 1998, 106, 289–295. [Google Scholar]
- Kim, Y.W.; West, X.Z.; Byzova, T.V. Inflammation and oxidative stress in angiogenesis and vascular disease. J. Mol. Med. 2013, 91, 323–328. [Google Scholar] [CrossRef]
- Shelton, P.; Jaiswal, A.K. The transcription factor NF-E2-related factor 2 (Nrf2): A protooncogene? FASEB J. 2013, 27, 414–423. [Google Scholar] [CrossRef]
- Nakae, D.; Kobayashi, Y.; Akai, H.; Andoh, N.; Satoh, H.; Ohashi, K.; Tsutsumi, M.; Konishi, Y. Involvement of 8-hydroxyguanine formation in the initiation of rat liver carcinogenesis by low dose levels of N-nitrosodiethylamine. Cancer Res. 1997, 57, 1281–1287. [Google Scholar]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-Hydroxy-2'-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef]
- Shen, H.M.; Ong, C.N.; Lee, B.L.; Shi, C.Y. Aflatoxin B1-induced 8-hydroxydeoxyguanosine formation in rat hepatic DNA. Carcinogenesis 1995, 16, 419–422. [Google Scholar] [CrossRef]
- Shen, H.M.; Ong, C.N.; Shi, C.Y. Involvement of reactive oxygen species in aflatoxin B1-induced cell injury in cultured rat hepatocytes. Toxicology 1995, 99, 115–123. [Google Scholar] [CrossRef]
- Kato, T.; Hasegawa, R.; Nakae, D.; Hirose, M.; Yaono, M.; Cui, L.; Kobayashi, Y.; Konishi, Y.; Ito, N.; Shirai, T. Dose-dependent induction of 8-hydroxyguanine and preneoplastic foci in rat liver by a food-derived carcinogen, 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline, at low dose levels. Jpn. J. Cancer Res. 1996, 87, 127–133. [Google Scholar] [CrossRef]
- Denda, A.; Endoh, T.; Nakae, D.; Konishi, Y. Effects of oxidative stress induced by redox-enzyme modulation on rat hepatocarcinogenesis. Toxicol. Lett. 1995, 82, 413–417. [Google Scholar]
- Kakehashi, A.; Hagiwara, A.; Imai, N.; Nagano, K.; Nishimaki, F.; Banton, M.; Wei, M.; Fukushima, S.; Wanibuchi, H. Mode of action of ethyl tertiary-butyl ether hepatotumorigenicity in the rat: Evidence for a role of oxidative stress via activation of CAR, PXR and PPARs signaling pathways. Toxicol. Appl. Pharmacol. 2013. [Google Scholar] [CrossRef]
- Kinoshita, A.; Wanibuchi, H.; Imaoka, S.; Ogawa, M.; Masuda, C.; Morimura, K.; Funae, Y.; Fukushima, S. Formation of 8-hydroxydeoxyguanosine and cell-cycle arrest in the rat liver via generation of oxidative stress by phenobarbital: Association with expression profiles of p21(WAF1/Cip1), cyclin D1 and Ogg1. Carcinogenesis 2002, 23, 341–349. [Google Scholar] [CrossRef]
- Floyd, R.A. The role of 8-hydroxyguanine in carcinogenesis. Carcinogenesis 1990, 11, 1447–1450. [Google Scholar] [CrossRef]
- Boiteux, S.; Radicella, J.P. Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress. Biochimie 1999, 81, 59–67. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013. [Google Scholar] [CrossRef]
- Kuraoka, I.; Endou, M.; Yamaguchi, Y.; Wada, T.; Handa, H.; Tanaka, K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J. Biol. Chem. 2003, 278, 7294–7299. [Google Scholar]
- Williams, G.M.; Whysner, J. Epigenetic carcinogens: Evaluation and risk assessment. Exp. Toxicol. Pathol. 1996, 48, 189–195. [Google Scholar] [CrossRef]
- Bombail, V.; Moggs, J.G.; Orphanides, G. Perturbation of epigenetic status by toxicants. Toxicol. Lett. 2004, 149, 51–58. [Google Scholar] [CrossRef]
- Kinoshita, A.; Wanibuchi, H.; Morimura, K.; Wei, M.; Shen, J.; Imaoka, S.; Funae, Y.; Fukushima, S. Phenobarbital at low dose exerts hormesis in rat hepatocarcinogenesis by reducing oxidative DNA damage, altering cell proliferation, apoptosis and gene expression. Carcinogenesis 2003, 24, 1389–1399. [Google Scholar]
- Masuda, C.; Wanibuchi, H.; Otori, K.; Wei, M.; Yamamoto, S.; Hiroi, T.; Imaoka, S.; Funae, Y.; Fukushima, S. Presence of a no-observed effect level for enhancing effects of development of the alpha-isomer of benzene hexachloride (alpha-BHC) on diethylnitrosamine-initiated hepatic foci in rats. Cancer Lett. 2001, 163, 179–185. [Google Scholar] [CrossRef]
- Puatanachokchai, R.; Kakuni, M.; Wanibuchi, H.; Kinoshita, A.; Kang, J.S.; Salim, E.I.; Morimura, K.; Tamano, S.; Merlino, G.T.; Fukushima, S. Lack of promoting effects of phenobarbital at low dose on diethylnitrosamine-induced hepatocarcinogenesis in TGF-alpha transgenic mice. Asian Pac. J. Cancer Prev. 2006, 7, 274–278. [Google Scholar]
- Kushida, M.; Sukata, T.; Uwagawa, S.; Ozaki, K.; Kinoshita, A.; Wanibuchi, H.; Morimura, K.; Okuno, Y.; Fukushima, S. Low dose DDT inhibition of hepatocarcinogenesis initiated by diethylnitrosamine in male rats: Possible mechanisms. Toxicol. Appl. Pharmacol. 2005, 208, 285–294. [Google Scholar]
- Peraino, C.; Fry, R.J.; Staffeldt, E. Reduction and enhancement by phenobarbital of hepatocarcinogenesis induced in the rat by 2-acetylaminofluorene. Cancer Res. 1971, 31, 1506–1512. [Google Scholar]
- Kaufmann, W.K.; Ririe, D.G.; Kaufman, D.G. Phenobarbital-dependent proliferation of putative initiated rat hepatocytes. Carcinogenesis 1988, 9, 779–782. [Google Scholar] [CrossRef]
- Butler, W.H.; Hempsall, V. Histochemical observations on nodules induced in the mouse liver by phenobarbitone. J. Pathol. 1978, 125, 155–161. [Google Scholar]
- Feldman, D.; Swarm, R.L.; Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 1981, 41, 2151–2162. [Google Scholar]
- Jirtle, R.L.; Meyer, S.A. Liver tumor promotion: Effect of phenobarbital on EGF and protein kinase C signal transduction and transforming growth factor-beta 1 expression. Dig. Dis. Sci. 1991, 36, 659–668. [Google Scholar] [CrossRef]
- Manjeshwar, S.; Rao, P.M.; Rajalakshmi, S.; Sarma, D.S. Inhibition of DNA synthesis by phenobarbital in primary cultures of hepatocytes from normal rat liver and from hepatic nodules. Carcinogenesis 1992, 13, 2287–2291. [Google Scholar] [CrossRef]
- Schulte-Hermann, R.; Timmermann-Trosiener, I.; Barthel, G.; Bursch, W. DNA synthesis, apoptosis, and phenotypic expression as determinants of growth of altered foci in rat liver during phenobarbital promotion. Cancer Res. 1990, 50, 5127–5135. [Google Scholar]
- Waxman, D.J.; Azaroff, L. Phenobarbital induction of cytochrome P-450 gene expression. Biochem. J. 1992, 281, 577–592. [Google Scholar]
- Ito, N.; Tamano, S.; Shirai, T. A medium-term rat liver bioassay for rapid in vivo detection of carcinogenic potential of chemicals. Cancer Sci. 2003, 94, 3–8. [Google Scholar] [CrossRef]
- Kitano, M.; Ichihara, T.; Matsuda, T.; Wanibuchi, H.; Tamano, S.; Hagiwara, A.; Imaoka, S.; Funae, Y.; Shirai, T.; Fukushima, S. Presence of a threshold for promoting effects of phenobarbital on diethylnitrosamine-induced hepatic foci in the rat. Carcinogenesis 1998, 19, 1475–1480. [Google Scholar] [CrossRef]
- Roos, D.; Winterbourn, C.C. Immunology. Lethal weapons. Science 2002, 296, 669–671. [Google Scholar]
- Whiteman, M.; Hong, H.S.; Jenner, A.; Halliwell, B. Loss of oxidized and chlorinated bases in DNA treated with reactive oxygen species: Implications for assessment of oxidative damage in vivo. Biochem. Biophys. Res. Commun. 2002, 296, 883–889. [Google Scholar] [CrossRef]
- Ito, N.; Hananouchi, M.; Sugihara, S.; Shirai, T.; Tsuda, H. Reversibility and irreversibility of liver tumors in mice induced by the alpha isomer of 1,2,3,4,5,6-hexachlorocyclohexane. Cancer Res. 1976, 36, 2227–2234. [Google Scholar]
- Ito, N.; Nagasaki, H.; Aoe, H.; Sugihara, S.; Miyata, Y. Development of hepatocellular carcinomas in rats treated with benzene hexachloride. J. Natl. Cancer Inst. 1975, 54, 801–805. [Google Scholar]
- Koransky, W.; Portig, J.; Vohland, H.W.; Klempau, I. Activation of microsome enzymes by hexachlorocyclohexane isomers. Its effect on scilliroside poisoning in rats. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1964, 247, 61–70. [Google Scholar] [CrossRef]
- Schlicht, I.; Koransky, W.; Magour, S.; Schulte-Hermann, R. Enlargement and DNA synthesis by the liver under in influence of substances alien to the body. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1968, 261, 26–41. [Google Scholar] [CrossRef]
- Puatanachokchai, R.; Morimura, K.; Wanibuchi, H.; Oka, M.; Kinoshita, A.; Mitsuru, F.; Yamaguchi, S.; Funae, Y.; Fukushima, S. Alpha-benzene hexachloride exerts hormesis in preneoplastic lesion formation of rat hepatocarcinogenesis with the possible role for hepatic detoxifying enzymes. Cancer Lett. 2006, 240, 102–113. [Google Scholar] [CrossRef]
- Wongpoomchai, R.; Morimura, K.; Wanibuchi, H.; Kakehashi, A.; Fukushima, S. Department of Pathology, Osaka City University Graduate School of Medicine: Osaka, Japan, Unpublished work. 2013.
- Gastel, J.A. Early indicators of response in biologically based risk assessment for nongenotoxic carcinogens. Regul. Toxicol. Pharmacol. 2001, 33, 393–398. [Google Scholar] [CrossRef]
- Honkakoski, P.; Zelko, I.; Sueyoshi, T.; Negishi, M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell Biol. 1998, 18, 5652–5658. [Google Scholar]
- Sukata, T.; Uwagawa, S.; Ozaki, K.; Ogawa, M.; Nishikawa, T.; Iwai, S.; Kinoshita, A.; Wanibuchi, H.; Imaoka, S.; Funae, Y.; et al. Detailed low-dose study of 1,1-bis(p-chlorophenyl)-2,2,2-trichloroethane carcinogenesis suggests the possibility of a hormetic effect. Int. J. Cancer 2002, 99, 112–118. [Google Scholar] [CrossRef]
- Chipman, J.K.; Mally, A.; Edwards, G.O. Disruption of gap junctions in toxicity and carcinogenicity. Toxicol. Sci. 2003, 71, 146–153. [Google Scholar] [CrossRef]
- Mally, A.; Chipman, J.K. Non-genotoxic carcinogens: Early effects on gap junctions, cell proliferation and apoptosis in the rat. Toxicology 2002, 180, 233–248. [Google Scholar] [CrossRef]
- Ahmed, F.E. Toxicology and human health effects following exposure to oxygenated or reformulated gasoline. Toxicol. Lett. 2001, 123, 89–113. [Google Scholar] [CrossRef]
- McGregor, D. Ethyl tertiary-butyl ether: A toxicological review. Crit. Rev. Toxicol. 2007, 37, 287–312. [Google Scholar]
- Saito, A.; Sasaki, T.; Kasai, T.; Katagiri, T.; Nishizawa, T.; Noguchi, T.; Aiso, S.; Nagano, K.; Fukushima, S. Hepatotumorigenicity of ethyl tertiary-butyl ether with 2-year inhalation exposure in F344 rats. Arch. Toxicol. 2013, 87, 905–914. [Google Scholar] [CrossRef]
- Hagiwara, A.; Doi, Y.; Imai, N.; Nakashima, H.; Ono, T.; Kawabe, M.; Furukawa, F.; Tamano, S.; Nagano, K.; Fukushima, S. Medium-term multi-organ carcinogenesis bioassay of ethyl tertiary-butyl ether in rats. Toxicology 2011, 289, 160–166. [Google Scholar] [CrossRef]
- Leonard, A.; Lauwerys, R.R. Carcinogenicity, teratogenicity and mutagenicity of arsenic. Mutat. Res. 1980, 75, 49–62. [Google Scholar] [CrossRef]
- Taylor, P.R.; Qiao, Y.L.; Schatzkin, A.; Yao, S.X.; Lubin, J.; Mao, B.L.; Rao, J.Y.; McAdams, M.; Xuan, X.Z.; Li, J.Y. Relation of arsenic exposure to lung cancer among tin miners in Yunnan Province, China. Br. J. Ind. Med. 1989, 46, 881–886. [Google Scholar]
- Kitchin, K.T.; Ahmad, S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol. Lett. 2003, 137, 3–13. [Google Scholar]
- Wanibuchi, H.; Yamamoto, S.; Chen, H.; Yoshida, K.; Endo, G.; Hori, T.; Fukushima, S. Promoting effects of dimethylarsinic acid on N-butyl-N-(4-hydroxybutyl)nitrosamine-induced urinary bladder carcinogenesis in rats. Carcinogenesis 1996, 17, 2435–2439. [Google Scholar] [CrossRef]
- Yamamoto, S.; Konishi, Y.; Matsuda, T.; Murai, T.; Shibata, M.A.; Matsui-Yuasa, I.; Otani, S.; Kuroda, K.; Endo, G.; Fukushima, S. Cancer induction by an organic arsenic compound, dimethylarsinic acid (cacodylic acid), in F344/DuCrj rats after pretreatment with five carcinogens. Cancer Res. 1995, 55, 1271–1276. [Google Scholar]
- Wei, M.; Wanibuchi, H.; Yamamoto, S.; Li, W.; Fukushima, S. Urinary bladder carcinogenicity of dimethylarsinic acid in male F344 rats. Carcinogenesis 1999, 20, 1873–1876. [Google Scholar] [CrossRef]
- Nishikawa, T.; Wanibuchi, H.; Ogawa, M.; Kinoshita, A.; Morimura, K.; Hiroi, T.; Funae, Y.; Kishida, H.; Nakae, D.; Fukushima, S. Promoting effects of monomethylarsonic acid, dimethylarsinic acid and trimethylarsine oxide on induction of rat liver preneoplastic glutathione S-transferase placental form positive foci: A possible reactive oxygen species mechanism. Int. J. Cancer 2002, 100, 136–139. [Google Scholar]
- Shen, J.; Wanibuchi, H.; Salim, E.I.; Wei, M.; Kinoshita, A.; Yoshida, K.; Endo, G.; Fukushima, S. Liver tumorigenicity of trimethylarsine oxide in male Fischer 344 rats—Association with oxidative DNA damage and enhanced cell proliferation. Carcinogenesis 2003, 24, 1827–1835. [Google Scholar] [CrossRef]
- Kitchin, K.T. Recent advances in arsenic carcinogenesis: Modes of action, animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Pharmacol. 2001, 172, 249–261. [Google Scholar] [CrossRef]
- Yamanaka, K.; Hoshino, M.; Okamoto, M.; Sawamura, R.; Hasegawa, A.; Okada, S. Induction of DNA damage by dimethylarsine, a metabolite of inorganic arsenics, is for the major part likely due to its peroxyl radical. Biochem. Biophys. Res. Commun. 1990, 168, 58–64. [Google Scholar] [CrossRef]
- Kinoshita, A.; Wanibuchi, H.; Wei, M.; Yunoki, T.; Fukushima, S. Elevation of 8-hydroxydeoxyguanosine and cell proliferation via generation of oxidative stress by organic arsenicals contributes to their carcinogenicity in the rat liver and bladder. Toxicol. Appl. Pharmacol. 2007, 221, 295–305. [Google Scholar] [CrossRef]
- Kinoshita, A.; Wanibuchi, H.; Morimura, K.; Wei, M.; Nakae, D.; Arai, T.; Minowa, O.; Noda, T.; Nishimura, S.; Fukushima, S. Carcinogenicity of dimethylarsinic acid in Ogg1-deficient mice. Cancer Sci. 2007, 98, 803–814. [Google Scholar] [CrossRef]
- Kato, T.; Ohgaki, H.; Hasegawa, H.; Sato, S.; Takayama, S.; Sugimura, T. Carcinogenicity in rats of a mutagenic compound, 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline. Carcinogenesis 1988, 9, 71–73. [Google Scholar]
- Fukushima, S.; Wanibuchi, H.; Morimura, K.; Wei, M.; Nakae, D.; Konishi, Y.; Tsuda, H.; Uehara, N.; Imaida, K.; Shirai, T.; et al. Lack of a dose-response relationship for carcinogenicity in the rat liver with low doses of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline or N-nitrosodiethylamine. Jpn. J. Cancer Res. 2002, 93, 1076–1082. [Google Scholar] [CrossRef]
- Yano, Y.; Yano, T.; Kinoshita, A.; Matoba, A.; Hasuma, T.; Wanibuchi, H.; Morimura, K.; Otani, S.; Fukushima, S. Sensitive quantitative assay for point mutations in the rat H-ras gene based on single nucleotide primer extension. Exp. Ther. Med. 2010, 1, 657–661. [Google Scholar]
- Hoshi, M.; Morimura, K.; Wanibuchi, H.; Wei, M.; Okochi, E.; Ushijima, T.; Takaoka, K.; Fukushima, S. No-observed effect levels for carcinogenicity and for in vivo mutagenicity of a genotoxic carcinogen. Toxicol. Sci. 2004, 81, 273–279. [Google Scholar] [CrossRef]
- Kurokawa, Y.; Maekawa, A.; Takahashi, M.; Hayashi, Y. Toxicity and carcinogenicity of potassium bromate—A new renal carcinogen. Environ. Health Perspect. 1990, 87, 309–335. [Google Scholar]
- Yamaguchi, T.; Wei, M.; Hagihara, N.; Omori, M.; Wanibuchi, H.; Fukushima, S. Lack of mutagenic and toxic effects of low dose potassium bromate on kidneys in the Big Blue rat. Mutat. Res. 2008, 652, 1–11. [Google Scholar] [CrossRef]
- Wei, M.; Hamoud, A.S.; Yamaguchi, T.; Kakehashi, A.; Morimura, K.; Doi, K.; Kushida, M.; Kitano, M.; Wanibuchi, H.; Fukushima, S. Potassium bromate enhances N-ethyl-N-hydroxyethylnitrosamine-induced kidney carcinogenesis only at high doses in Wistar rats: Indication of the existence of an enhancement threshold. Toxicol. Pathol. 2009, 37, 983–991. [Google Scholar]
- Biju, M.P.; Pyroja, S.; Rajeshkumar, N.V.; Paulose, C.S. Enhanced GABA(B) receptor in neoplastic rat liver: Induction of DNA synthesis by baclofen in hepatocyte cultures. J. Biochem. Mol. Biol. Biophys. 2002, 6, 209–214. [Google Scholar]
- Erlitzki, R.; Gong, Y.; Zhang, M.; Minuk, G. Identification of gamma-aminobutyric acid receptor subunit types in human and rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G733–G739. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kakehashi, A.; Wei, M.; Fukushima, S.; Wanibuchi, H. Oxidative Stress in the Carcinogenicity of Chemical Carcinogens. Cancers 2013, 5, 1332-1354. https://doi.org/10.3390/cancers5041332
Kakehashi A, Wei M, Fukushima S, Wanibuchi H. Oxidative Stress in the Carcinogenicity of Chemical Carcinogens. Cancers. 2013; 5(4):1332-1354. https://doi.org/10.3390/cancers5041332
Chicago/Turabian StyleKakehashi, Anna, Min Wei, Shoji Fukushima, and Hideki Wanibuchi. 2013. "Oxidative Stress in the Carcinogenicity of Chemical Carcinogens" Cancers 5, no. 4: 1332-1354. https://doi.org/10.3390/cancers5041332