Strategies in Gene Therapy for Glioblastoma

Abstract
:1. Introduction

{kind=link}
{kind=link}
| Country/Identifier | Model | Strategy/goals | Carrier | Phase |
|---|---|---|---|---|
| US/NCT00589875 | AdV-TK | Suicide gene | non-replicating virus | IIa |
| China/CT00870181 | AdV-TK | Suicide gene | non-replicating virus | II |
| US/NCT00634231 | AdV-TK (plus radiotherapy) | Suicide gene | non-replicating virus | I |
| US/NCT00751270 | AdV-TK (plus radiotherapy) | Suicide gene | non-replicating virus | Ib * |
| US/NCT00589875 | AdV-TK (plus radiotherapy) | Suicide gene | non-replicating virus | IIa * |
| US/NCT01811992 | (1) AdV-hCMV-TK and | (1) Suicide gene | non-replicating virus | I |
| (2) AdV-hCMV-Flt3L | (2) Immune stimulation | |||
| US/NCT01156584 | retroviral vector (Toca-511) carrying CDA | Suicide gene and viral oncolysis | replicating virus | I/II |
| US/NCT01174537 | New Castle Disease Virus | Viral oncolysis | replicating virus | I/II |
| US/NCT01301430 | H-1 parvovirus (ParvOryx-01) | Viral oncolysis | replicating virus | I/II |
| US/NCT01491893 | engineered chimeric poliovirus (PVS-RIPO) | Viral oncolysis and immune stimulation | replicating virus | I |
| US/NCT00390299 | Engineered measles virus (MV-CEA) | Viral oncolysis and immune activation | replicating virus | I |
| US/NCT01582516 | AdV-Delta-24-RGD | Viral oncolysis | replicating virus | I/II |
| delivered via CED | ||||
| US/NCT00805376 | AdV-Delta-24-RGD-4C | Viral oncolysis | replicating virus | I |
| UK/UK-0050 | HSV 1716 | Viral oncolysis | replicating virus | II |
| US/NCT01172964 | stem cells carrying CDA | Suicide gene | neural stem cells | Pilot |
2. Virus-Based Gene Therapy of GBM
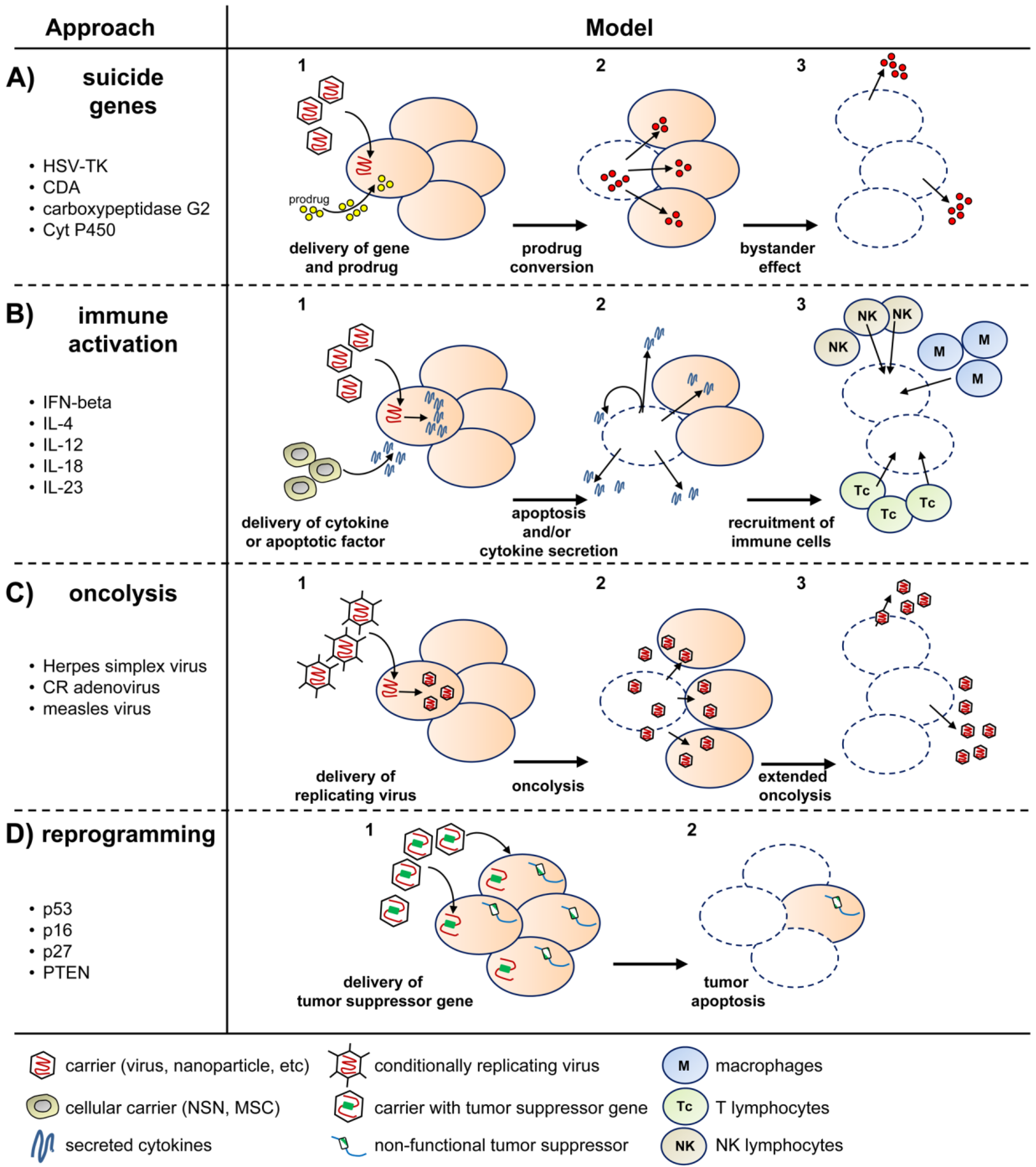
2.1. Viral Delivery of Suicide Genes
2.2. Viral Delivery of Tumor-Suppressor Genes
2.3. Viral Delivery of Immunomodulatory Genes
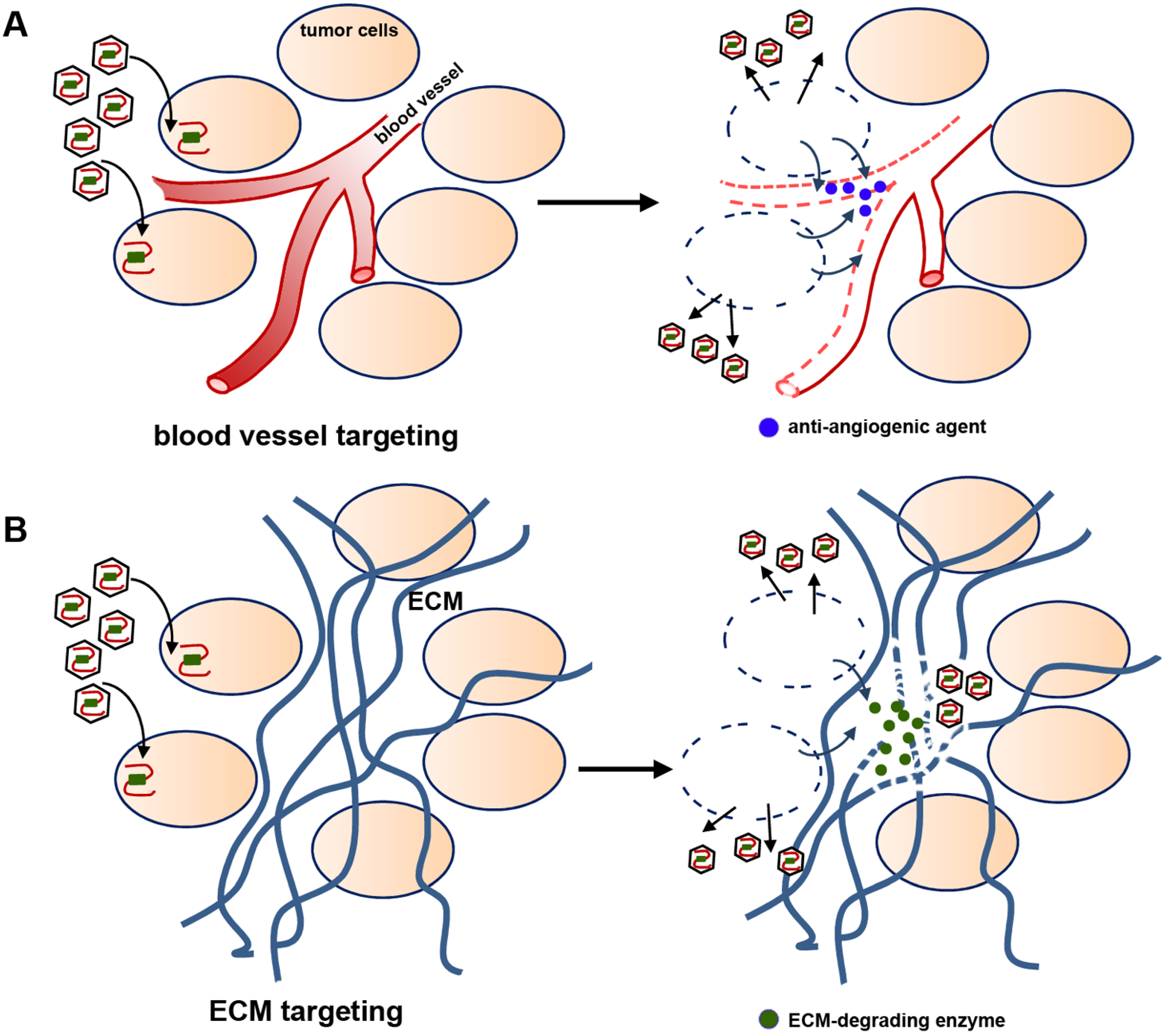
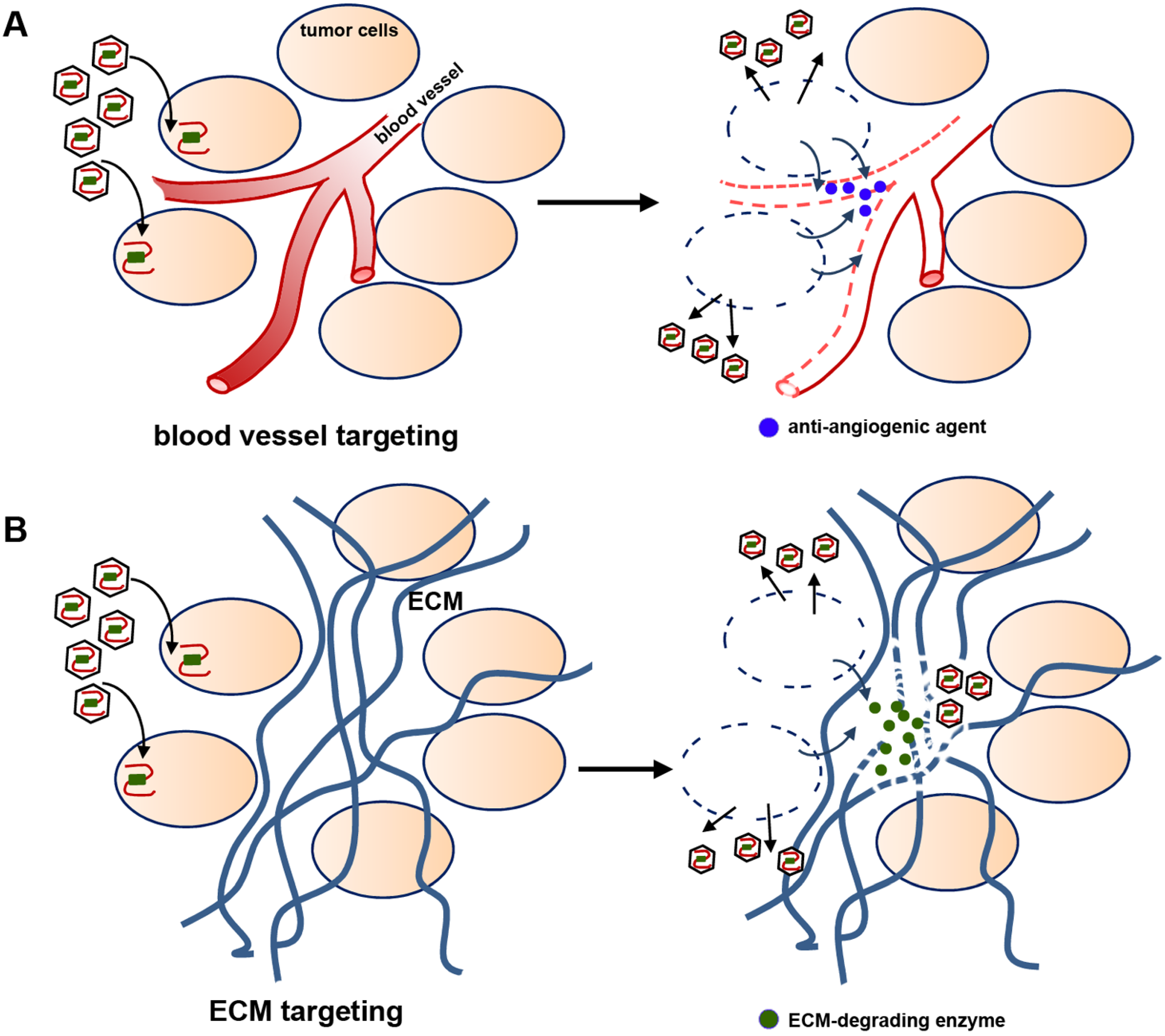
2.4. Viral Delivery of Genes That Modify the Tumor Stroma
2.5. Replication-Competent Oncolytic Viruses
2.6. Advantages and Challenges of Viral-Based Gene Therapy
3. Stem Cell-Based Gene Therapy of GBM
3.1. Neural Stem Cells
3.2. Mesenchymal Stem Cells
3.3. Embryonic Stem Cells
3.4. Advantages and Challenges of Cell-Based Gene Therapy
4. Nanotechnology-Based Gene Therapy of GBM
4.1. Liposomes
4.2. Polymers
4.3. Nanoparticles
4.4. Advantages and Challenges of Nanocarrier-Based Gene Therapy
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. Cbtrus statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2005–2009. Neuro-oncology 2012, 14, v1–v49. [Google Scholar] [CrossRef]
- Schwartzbaum, J.A.; Fisher, J.L.; Aldape, K.D.; Wrensch, M. Epidemiology and molecular pathology of glioma. Nat. Clin. Pract. Neurol. 2006, 2, 494–503. [Google Scholar]
- Louis, D.N. Molecular pathology of malignant gliomas. Ann. Rev. Pathol. Mech. Dis. 2006, 1, 97–117. [Google Scholar] [CrossRef]
- Nduom, E.K.; Hadjipanayis, C.G.; van Meir, E.G. Glioblastoma cancer stem-like cells: Implications for pathogenesis and treatment. Cancer J. 2012, 18, 100–106. [Google Scholar] [CrossRef]
- Keeler, C.E. Gene therapy. J. Hered. 1947, 38, 294–298. [Google Scholar]
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease? Science 1972, 175, 949–955. [Google Scholar]
- Sheridan, C. Gene therapy finds its niche. Nat. Biotechnol. 2011, 29, 121–128. [Google Scholar] [CrossRef]
- Natsume, A.; Yoshida, J. Gene therapy for high-grade glioma: Current approaches and future directions. Cell Adhes. Migr. 2008, 2, 186–191. [Google Scholar] [CrossRef]
- US National Institutes of Health. Index of clinical trials. Available online: http://clinicaltrials.gov (accessed on 3 October 2013).
- Journal of Gene Medicine. Gene Therapy Clinical Trials Worldwide. Available online: http://www.abedia.com/wiley/index.html (accessed on 3 October 2013).
- Zerboni, L.; Arvin, A. Investigation of varicella-zoster virus neurotropism and neurovirulence using scid mouse-human drg xenografts. J. Neurovirol. 2011, 17, 570–577. [Google Scholar] [CrossRef]
- Bertke, A.S.; Patel, A.; Krause, P.R. Herpes simplex virus latency-associated transcript sequence downstream of the promoter influences type-specific reactivation and viral neurotropism. J. Virol. 2007, 81, 6605–6613. [Google Scholar] [CrossRef]
- Lachmann, R. Herpes simplex virus-based vectors. Int. J. Exp. Pathol. 2004, 85, 177–190. [Google Scholar] [CrossRef]
- Muik, A.; Kneiske, I.; Werbizki, M.; Wilflingseder, D.; Giroglou, T.; Ebert, O.; Kraft, A.; Dietrich, U.; Zimmer, G.; Momma, S.; et al. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J. Virol. 2011, 85, 5679–5684. [Google Scholar] [CrossRef]
- Kim, S.H.; Xiao, S.; Shive, H.; Collins, P.L.; Samal, S.K. Replication, neurotropism, and pathogenicity of avian paramyxovirus serotypes 1–9 in chickens and ducks. PLoS One 2012, 7, e34927. [Google Scholar]
- Yumitori, K.; Handa, H.; Yamashita, J.; Suda, K.; Otsuka, S.; Shimizu, Y. Treatment of malignant glioma with mumps virus (author’s transl). No Shinkei Geka 1982, 10, 143–147. [Google Scholar]
- Short, M.P.; Choi, B.C.; Lee, J.K.; Malick, A.; Breakefield, X.O.; Martuza, R.L. Gene delivery to glioma cells in rat brain by grafting of a retrovirus packaging cell line. J. Neurosci. Res. 1990, 27, 427–439. [Google Scholar] [CrossRef]
- Culver, K.W.; Ram, Z.; Wallbridge, S.; Ishii, H.; Oldfield, E.H.; Blaese, R.M. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science 1992, 256, 1550–1552. [Google Scholar]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Dachs, G.U.; Tupper, J.; Tozer, G.M. From bench to bedside for gene-directed enzyme prodrug therapy of cancer. Anticancer Drugs 2005, 16, 349–359. [Google Scholar] [CrossRef]
- Duarte, S.; Carle, G.; Faneca, H.; de Lima, M.C.; Pierrefite-Carle, V. Suicide gene therapy in cancer: Where do we stand now? Cancer Lett. 2012, 324, 160–170. [Google Scholar] [CrossRef]
- Kaliberov, S.A.; Market, J.M.; Gillespie, G.Y.; Krendelchtchikova, V.; Della Manna, D.; Sellers, J.C.; Kaliberova, L.N.; Black, M.E.; Buchsbaum, D.J. Mutation of Escherichia coli cytosine deaminase significantly enhances molecular chemotherapy of human glioma. Gene Ther. 2007, 14, 1111–1119. [Google Scholar] [CrossRef]
- Moolten, F.L. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: Paradigm for a prospective cancer control strategy. Cancer Res. 1986, 46, 5276–5281. [Google Scholar]
- Valerie, K.; Hawkins, W.; Farnsworth, J.; Schmidt-Ullrich, R.; Lin, P.S.; Amir, C.; Feden, J. Substantially improved in vivo radiosensitization of rat glioma with mutant hsv-tk and acyclovir. Cancer Gene Ther. 2001, 8, 3–8. [Google Scholar]
- Ezzeddine, Z.D.; Martuza, R.L.; Platika, D.; Short, M.P.; Malick, A.; Choi, B.; Breakefield, X.O. Selective killing of glioma cells in culture and in vivo by retrovirus transfer of the herpes simplex virus thymidine kinase gene. New Biol. 1991, 3, 608–614. [Google Scholar]
- Maeda, M.; Moriuchi, S.; Sano, A.; Yoshimine, T. New drug delivery system for water-soluble drugs using silicone and its usefulness for local treatment: Application of gcv-silicone to gcv/hsv-tk gene therapy for brain tumor. J. Control. Release 2002, 84, 15–25. [Google Scholar] [CrossRef]
- Valerie, K.; Brust, D.; Farnsworth, J.; Amir, C.; Taher, M.M.; Hershey, C.; Feden, J. Improved radiosensitization of rat glioma cells with adenovirus-expressed mutant herpes simplex virus-thymidine kinase in combination with acyclovir. Cancer Gene Ther. 2000, 7, 879–884. [Google Scholar]
- Namba, H.; Tagawa, M.; Miyagawa, T.; Iwadate, Y.; Sakiyama, S. Treatment of rat experimental brain tumors by herpes simplex virus thymidine kinase gene-transduced allogeneic tumor cells and ganciclovir. Cancer Gene Ther. 2000, 7, 947–953. [Google Scholar]
- Mori, K.; Iwata, J.; Miyazaki, M.; Osada, H.; Tange, Y.; Yamamoto, T.; Aiko, Y.; Tamura, M.; Shiroishi, T. Bystander killing effect of tymidine kinase gene-transduced adult bone marrow stromal cells with ganciclovir on malignant glioma cells. Neurol. Med. Chir. (Tokyo) 2010, 50, 545–553. [Google Scholar] [CrossRef]
- Freeman, S.M.; Abboud, C.N.; Whartenby, K.A.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The “bystander effect”: Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993, 53, 5274–5283. [Google Scholar]
- Bi, W.L.; Parysek, L.M.; Warnick, R.; Stambrook, P.J. In vitro evidence that metabolic cooperation is responsible for the bystander effect observed with hsv tk retroviral gene therapy. Hum. Gene Ther. 1993, 4, 725–731. [Google Scholar] [CrossRef]
- Fick, J.; Barker, F.G., 2nd; Dazin, P.; Westphale, E.M.; Beyer, E.C.; Israel, M.A. The extent of heterocellular communication mediated by gap junctions is predictive of bystander tumor cytotoxicity in vitro. Proc. Natl. Acad. Sci. USA 1995, 92, 11071–11075. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, J.H.; Kolozsvary, A.; Brown, S.L.; Freytag, S.O. Preferential radiosensitization of 9l glioma cells transduced with hsv-tk gene by acyclovir. J. Neurooncol. 1997, 33, 189–194. [Google Scholar] [CrossRef]
- Wahlfors, T.; Hakkarainen, T.; Janne, J.; Alhonen, L.; Wahlfors, J. In vivo enhancement of herpes simplex virus thymidine kinase/ganciclovir cancer gene therapy with polyamine biosynthesis inhibition. Int. J. Cancer 2006, 118, 2907–2910. [Google Scholar] [CrossRef]
- Rainov, N.G.; Fels, C.; Droege, J.W.; Schafer, C.; Kramm, C.M.; Chou, T.C. Temozolomide enhances herpes simplex virus thymidine kinase/ganciclovir therapy of malignant glioma. Cancer Gene Ther. 2001, 8, 662–668. [Google Scholar] [CrossRef]
- Rainov, N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef]
- Goodman, J.C.; Trask, T.W.; Chen, S.H.; Woo, S.L.; Grossman, R.G.; Carey, K.D.; Hubbard, G.B.; Carrier, D.A.; Rajagopalan, S.; Aguilar-Cordova, E.; et al. Adenoviral-mediated thymidine kinase gene transfer into the primate brain followed by systemic ganciclovir: Pathologic, radiologic, and molecular studies. Hum. Gene Ther. 1996, 7, 1241–1250. [Google Scholar] [CrossRef]
- Dewey, R.A.; Morrissey, G.; Cowsill, C.M.; Stone, D.; Bolognani, F.; Dodd, N.J.; Southgate, T.D.; Klatzmann, D.; Lassmann, H.; Castro, M.G.; et al. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: Implications for clinical trials. Nat. Med. 1999, 5, 1256–1263. [Google Scholar] [CrossRef]
- Prados, M.D.; McDermott, M.; Chang, S.M.; Wilson, C.B.; Fick, J.; Culver, K.W.; van Gilder, J.; Keles, G.E.; Spence, A.; Berger, M. Treatment of progressive or recurrent glioblastoma multiforme in adults with herpes simplex virus thymidine kinase gene vector-producer cells followed by intravenous ganciclovir administration: A phase I/II multi-institutional trial. J. Neurooncol. 2003, 65, 269–278. [Google Scholar] [CrossRef]
- Germano, I.M.; Fable, J.; Gultekin, S.H.; Silvers, A. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: Preliminary results of a phase I trial in patients with recurrent malignant gliomas. J. Neurooncol. 2003, 65, 279–289. [Google Scholar] [CrossRef]
- Assi, H.; Candolfi, M.; Baker, G.; Mineharu, Y.; Lowenstein, P.R.; Castro, M.G. Gene therapy for brain tumors: Basic developments and clinical implementation. Neurosci. Lett. 2012, 527, 71–77. [Google Scholar] [CrossRef]
- Adachi, Y.; Tamiya, T.; Ichikawa, T.; Terada, K.; Ono, Y.; Matsumoto, K.; Furuta, T.; Hamada, H.; Ohmoto, T. Experimental gene therapy for brain tumors using adenovirus-mediated transfer of cytosine deaminase gene and uracil phosphoribosyltransferase gene with 5-fluorocytosine. Hum. Gene Ther. 2000, 11, 77–89. [Google Scholar] [CrossRef]
- Bourbeau, D.; Lavoie, G.; Nalbantoglu, J.; Massie, B. Suicide gene therapy with an adenovirus expressing the fusion gene cd::Uprt in human glioblastomas: Different sensitivities correlate with p53 status. J. Gene Med. 2004, 6, 1320–1332. [Google Scholar] [CrossRef]
- Ostertag, D.; Amundson, K.K.; Lopez Espinoza, F.; Martin, B.; Buckley, T.; Galvao da Silva, A.P.; Lin, A.H.; Valenta, D.T.; Perez, O.D.; Ibanez, C.E.; et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro-oncology 2012, 14, 145–159. [Google Scholar] [CrossRef]
- Ardiani, A.; Johnson, A.J.; Ruan, H.; Sanchez-Bonilla, M.; Serve, K.; Black, M.E. Enzymes to die for: Exploiting nucleotide metabolizing enzymes for cancer gene therapy. Curr. Gene Ther. 2012, 12, 77–91. [Google Scholar] [CrossRef]
- Tai, C.K.; Wang, W.; Lai, Y.H.; Logg, C.R.; Parker, W.B.; Li, Y.F.; Hong, J.S.; Sorscher, E.J.; Chen, T.C.; Kasahara, N. Enhanced efficiency of prodrug activation therapy by tumor-selective replicating retrovirus vectors armed with the escherichia coli purine nucleoside phosphorylase gene. Cancer Gene Ther. 2010, 17, 614–623. [Google Scholar] [CrossRef]
- Bharara, S.; Sorscher, E.J.; Gillespie, G.Y.; Lindsey, J.R.; Hong, J.S.; Curlee, K.V.; Allan, P.W.; Gadi, V.K.; Alexander, S.A.; Secrist, J.A., 3rd; et al. Antibiotic-mediated chemoprotection enhances adaptation of E. coli pnp for herpes simplex virus-based glioma therapy. Hum. Gene Ther. 2005, 16, 339–347. [Google Scholar] [CrossRef]
- Marais, R.; Spooner, R.A.; Stribbling, S.M.; Light, Y.; Martin, J.; Springer, C.J. A cell surface tethered enzyme improves efficiency in gene-directed enzyme prodrug therapy. Nat. Biotechnol. 1997, 15, 1373–1377. [Google Scholar] [CrossRef]
- Spooner, R.A.; Martin, J.; Friedlos, F.; Marais, R.; Springer, C.J. In suicide gene therapy, the site of subcellular localization of the activating enzyme is more important than the rate at which it activates prodrug. Cancer Gene Ther. 2000, 7, 1348–1356. [Google Scholar] [CrossRef]
- Cowen, R.L.; Williams, J.C.; Emery, S.; Blakey, D.; Darling, J.L.; Lowenstein, P.R.; Castro, M.G. Adenovirus vector-mediated delivery of the prodrug-converting enzyme carboxypeptidase g2 in a secreted or gpi-anchored form: High-level expression of this active conditional cytotoxic enzyme at the plasma membrane. Cancer Gene Ther. 2002, 9, 897–907. [Google Scholar]
- Wei, M.X.; Tamiya, T.; Chase, M.; Boviatsis, E.J.; Chang, T.K.; Kowall, N.W.; Hochberg, F.H.; Waxman, D.J.; Breakefield, X.O.; Chiocca, E.A. Experimental tumor therapy in mice using the cyclophosphamide-activating cytochrome p450 2b1 gene. Hum. Gene Ther. 1994, 5, 969–978. [Google Scholar] [CrossRef]
- Chase, M.; Chung, R.Y.; Chiocca, E.A. An oncolytic viral mutant that delivers the cyp2b1 transgene and augments cyclophosphamide chemotherapy. Nat. Biotechnol. 1998, 16, 444–448. [Google Scholar]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef]
- Mut, M.; Sherman, J.H.; Shaffrey, M.E.; Schiff, D. Cintredekin besudotox in treatment of malignant glioma. Expert Opin. Biol. Ther. 2008, 8, 805–812. [Google Scholar] [CrossRef]
- Candolfi, M.; Xiong, W.; Yagiz, K.; Liu, C.; Muhammad, A.K.; Puntel, M.; Foulad, D.; Zadmehr, A.; Ahlzadeh, G.E.; Kroeger, K.M.; et al. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proc. Natl. Acad. Sci. USA 2010, 107, 20021–20026. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Lang, F.F.; Yung, W.K.; Sawaya, R.; Tofilon, P.J. Adenovirus-mediated p53 gene therapy for human gliomas. Neurosurgery 1999, 45, 1093–1104. [Google Scholar] [CrossRef]
- Hong, Y.K.; Joe, Y.A.; Yang, Y.J.; Lee, K.S.; Son, B.C.; Jeun, S.S.; Chung, D.S.; Cho, K.K.; Park, C.K.; Kim, M.C.; et al. Potentials and limitations of adenovirus-p53 gene therapy for brain tumors. J. Korean Med. Sci. 2000, 15, 315–322. [Google Scholar]
- Lang, F.F.; Yung, W.K.; Raju, U.; Libunao, F.; Terry, N.H.; Tofilon, P.J. Enhancement of radiosensitivity of wild-type p53 human glioma cells by adenovirus-mediated delivery of the p53 gene. J. Neurosurg. 1998, 89, 125–132. [Google Scholar] [CrossRef]
- Li, H.; Alonso-Vanegas, M.; Colicos, M.A.; Jung, S.S.; Lochmuller, H.; Sadikot, A.F.; Snipes, G.J.; Seth, P.; Karpati, G.; Nalbantoglu, J. Intracerebral adenovirus-mediated p53 tumor suppressor gene therapy for experimental human glioma. Clin. Cancer Res. 1999, 5, 637–642. [Google Scholar]
- Biroccio, A.; Bufalo, D.D.; Ricca, A.; D’Angelo, C.; D’Orazi, G.; Sacchi, A.; Soddu, S.; Zupi, G. Increase of bcnu sensitivity by wt-p53 gene therapy in glioblastoma lines depends on the administration schedule. Gene Ther. 1999, 6, 1064–1072. [Google Scholar] [CrossRef]
- Kim, I.A.; Yang, Y.J.; Yoon, S.C.; Choi, I.B.; Kay, C.S.; Kwon, H.C.; Kim, C.M.; Joe, Y.A.; Kang, J.K.; Hong, Y.K. Potential of adenoviral p53 gene therapy and irradiation for the treatment of malignant gliomas. Int. J. Oncol. 2001, 19, 1041–1047. [Google Scholar]
- Chen, B.; Timiryasova, T.M.; Andres, M.L.; Kajioka, E.H.; Dutta-Roy, R.; Gridley, D.S.; Fodor, I. Evaluation of combined vaccinia virus-mediated antitumor gene therapy with p53, il-2, and il-12 in a glioma model. Cancer Gene Ther. 2000, 7, 1437–1447. [Google Scholar]
- Lang, F.F.; Bruner, J.M.; Fuller, G.N.; Aldape, K.; Prados, M.D.; Chang, S.; Berger, M.S.; McDermott, M.W.; Kunwar, S.M.; Junck, L.R.; et al. Phase i trial of adenovirus-mediated p53 gene therapy for recurrent glioma: Biological and clinical results. J. Clin. Oncol. 2003, 21, 2508–2518. [Google Scholar] [CrossRef]
- Chintala, S.K.; Fueyo, J.; Gomez-Manzano, C.; Venkaiah, B.; Bjerkvig, R.; Yung, W.K.; Sawaya, R.; Kyritsis, A.P.; Rao, J.S. Adenovirus-mediated p16/cdkn2 gene transfer suppresses glioma invasion in vitro. Oncogene 1997, 15, 2049–2057. [Google Scholar]
- Park, K.H.; Lee, J.; Yoo, C.G.; Kim, Y.W.; Han, S.K.; Shim, Y.S.; Kim, S.K.; Wang, K.C.; Cho, B.K.; Lee, C.T. Application of p27 gene therapy for human malignant glioma potentiated by using mutant p27. J. Neurosurg. 2004, 101, 505–510. [Google Scholar] [CrossRef]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef]
- Davies, M.A.; Lu, Y.; Sano, T.; Fang, X.; Tang, P.; LaPushin, R.; Koul, D.; Bookstein, R.; Stokoe, D.; Yung, W.K.; et al. Adenoviral transgene expression of mmac/pten in human glioma cells inhibits akt activation and induces anoikis. Cancer Res. 1998, 58, 5285–5290. [Google Scholar]
- Lu, W.; Zhou, X.; Hong, B.; Liu, J.; Yue, Z. Suppression of invasion in human u87 glioma cells by adenovirus-mediated co-transfer of timp-2 and pten gene. Cancer Lett. 2004, 214, 205–213. [Google Scholar] [CrossRef]
- Abe, T.; Terada, K.; Wakimoto, H.; Inoue, R.; Tyminski, E.; Bookstein, R.; Basilion, J.P.; Chiocca, E.A. Pten decreases in vivo vascularization of experimental gliomas in spite of proangiogenic stimuli. Cancer Res. 2003, 63, 2300–2305. [Google Scholar]
- Vervoorts, J.; Luscher, B. Post-translational regulation of the tumor suppressor p27(kip1). Cell Mol. Life Sci. 2008, 65, 3255–3264. [Google Scholar] [CrossRef]
- Schiappacassi, M.; Lovat, F.; Canzonieri, V.; Belletti, B.; Berton, S.; di Stefano, D.; Vecchione, A.; Colombatti, A.; Baldassarre, G. P27kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol. Cancer Ther. 2008, 7, 1164–1175. [Google Scholar] [CrossRef]
- Qin, X.Q.; Beckham, C.; Brown, J.L.; Lukashev, M.; Barsoum, J. Human and mouse ifn-beta gene therapy exhibits different anti-tumor mechanisms in mouse models. Mol. Ther. 2001, 4, 356–364. [Google Scholar] [CrossRef]
- Qin, X.Q.; Tao, N.; Dergay, A.; Moy, P.; Fawell, S.; Davis, A.; Wilson, J.M.; Barsoum, J. Interferon-beta gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 14411–14416. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Smith, K.M.; McKinney, B.; Palmer, C.A.; Rosenfeld, S.; Lillehei, K.; Hamilton, A.; DeMasters, B.K.; Judy, K.; Kirn, D. A phase I trial of Ad.Hifn-beta gene therapy for glioma. Mol. Ther. 2008, 16, 618–626. [Google Scholar] [CrossRef]
- Enderlin, M.; Kleinmann, E.V.; Struyf, S.; Buracchi, C.; Vecchi, A.; Kinscherf, R.; Kiessling, F.; Paschek, S.; Sozzani, S.; Rommelaere, J.; et al. TNF-alpha and the ifn-gamma-inducible protein 10 (ip-10/cxcl-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2009, 16, 149–160. [Google Scholar]
- Hellums, E.K.; Markert, J.M.; Parker, J.N.; He, B.; Perbal, B.; Roizman, B.; Whitley, R.J.; Langford, C.P.; Bharara, S.; Gillespie, G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-oncology 2005, 7, 213–224. [Google Scholar] [CrossRef]
- Chiu, T.L.; Wang, M.J.; Su, C.C. The treatment of glioblastoma multiforme through activation of microglia and trail induced by raav2-mediated il-12 in a syngeneic rat model. J. Biomed. Sci. 2012. [Google Scholar] [CrossRef]
- Markert, J.M.; Cody, J.J.; Parker, J.N.; Coleman, J.M.; Price, K.H.; Kern, E.R.; Quenelle, D.C.; Lakeman, A.D.; Schoeb, T.R.; Palmer, C.A.; et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J. Virol. 2012, 86, 5304–5313. [Google Scholar] [CrossRef]
- Tanaka, T.; Cao, Y.; Folkman, J.; Fine, H.A. Viral vector-targeted antiangiogenic gene therapy utilizing an angiostatin complementary DNA. Cancer Res. 1998, 58, 3362–3369. [Google Scholar]
- Ma, H.I.; Lin, S.Z.; Chiang, Y.H.; Li, J.; Chen, S.L.; Tsao, Y.P.; Xiao, X. Intratumoral gene therapy of malignant brain tumor in a rat model with angiostatin delivered by adeno-associated viral (aav) vector. Gene Ther. 2002, 9, 2–11. [Google Scholar] [CrossRef]
- Szentirmai, O.; Baker, C.H.; Bullain, S.S.; Lin, N.; Takahashi, M.; Folkman, J.; Mulligan, R.C.; Carter, B.S. Successful inhibition of intracranial human glioblastoma multiforme xenograft growth via systemic adenoviral delivery of soluble endostatin and soluble vascular endothelial growth factor receptor-2: Laboratory investigation. J. Neurosurg. 2008, 108, 979–988. [Google Scholar] [CrossRef]
- Hardcastle, J.; Kurozumi, K.; Dmitrieva, N.; Sayers, M.P.; Ahmad, S.; Waterman, P.; Weissleder, R.; Chiocca, E.A.; Kaur, B. Enhanced antitumor efficacy of vasculostatin (vstat120) expressing oncolytic hsv-1. Mol. Ther. 2010, 18, 285–294. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Haseley, A.; Bratasz, A.; Chiocca, E.A.; Zhang, J.; Powell, K.; Kaur, B. Antitumor efficacy of 34.5enve: A transcriptionally retargeted and “vstat120”-expressing oncolytic virus. Mol. Ther. 2012, 20, 287–297. [Google Scholar] [CrossRef]
- Haseley, A.; Alvarez-Breckenridge, C.; Chaudhury, A.R.; Kaur, B. Advances in oncolytic virus therapy for glioma. Recent Pat. CNS Drug Discov. 2009, 4, 1–13. [Google Scholar]
- Kuriyama, N.; Kuriyama, H.; Julin, C.M.; Lamborn, K.; Israel, M.A. Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum. Gene Ther. 2000, 11, 2219–2230. [Google Scholar] [CrossRef]
- Dmitrieva, N.; Yu, L.; Viapiano, M.; Cripe, T.P.; Chiocca, E.A.; Glorioso, J.C.; Kaur, B. Chondroitinase abc i-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin. Cancer Res. 2011, 17, 1362–1372. [Google Scholar] [CrossRef]
- Chiocca, E.A. Oncolytic viruses. Nat. Rev. Cancer 2002, 2, 938–950. [Google Scholar] [CrossRef]
- Selznick, L.A.; Shamji, M.F.; Fecci, P.; Gromeier, M.; Friedman, A.H.; Sampson, J. Molecular strategies for the treatment of malignant glioma—Genes, viruses, and vaccines. Neurosurg. Rev. 2008, 31, 141–155. [Google Scholar] [CrossRef]
- Allen, C.; Vongpunsawad, S.; Nakamura, T.; James, C.D.; Schroeder, M.; Cattaneo, R.; Giannini, C.; Krempski, J.; Peng, K.W.; Goble, J.M.; et al. Retargeted oncolytic measles strains entering via the egfrviii receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006, 66, 11840–11850. [Google Scholar] [CrossRef]
- Grandi, P.; Fernandez, J.; Szentirmai, O.; Carter, R.; Gianni, D.; Sena-Esteves, M.; Breakefield, X.O. Targeting hsv-1 virions for specific binding to epidermal growth factor receptor-viii-bearing tumor cells. Cancer Gene Ther. 2010, 17, 655–663. [Google Scholar] [CrossRef]
- Uchida, H.; Marzulli, M.; Nakano, K.; Goins, W.F.; Chan, J.; Hong, C.S.; Mazzacurati, L.; Yoo, J.Y.; Haseley, A.; Nakashima, H.; et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an egfr-retargeted oncolytic herpes simplex virus. Mol. Ther. 2013, 21, 561–569. [Google Scholar] [CrossRef]
- Tobias, A.; Ahmed, A.; Moon, K.S.; Lesniak, M.S. The art of gene therapy for glioma: A review of the challenging road to the bedside. J. Neurol. Neurosurg. Psychiatry 2013, 84, 213–222. [Google Scholar] [CrossRef]
- Terada, K.; Wakimoto, H.; Tyminski, E.; Chiocca, E.A.; Saeki, Y. Development of a rapid method to generate multiple oncolytic hsv vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 2006, 13, 705–714. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, g207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase ib trial of mutant herpes simplex virus g207 inoculated pre-and post-tumor resection for recurrent gbm. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef]
- Papanastassiou, V.; Rampling, R.; Fraser, M.; Petty, R.; Hadley, D.; Nicoll, J.; Harland, J.; Mabbs, R.; Brown, M. The potential for efficacy of the modified (icp 34.5(−)) herpes simplex virus hsv1716 following intratumoural injection into human malignant glioma: A proof of principle study. Gene Ther. 2002, 9, 398–406. [Google Scholar] [CrossRef]
- Harrow, S.; Papanastassiou, V.; Harland, J.; Mabbs, R.; Petty, R.; Fraser, M.; Hadley, D.; Patterson, J.; Brown, S.M.; Rampling, R. Hsv1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004, 11, 1648–1658. [Google Scholar] [CrossRef]
- Kanai, R.; Zaupa, C.; Sgubin, D.; Antoszczyk, S.J.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Effect of gamma34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J. Virol. 2012, 86, 4420–4431. [Google Scholar] [CrossRef]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic hsv-1 mutant expressing icp34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef]
- Hardcastle, J.; Kurozumi, K.; Chiocca, E.A.; Kaur, B. Oncolytic viruses driven by tumor-specific promoters. Curr. Cancer Drug Targets 2007, 7, 181–189. [Google Scholar] [CrossRef]
- Nandi, S.; Lesniak, M.S. Adenoviral virotherapy for malignant brain tumors. Expert Opin. Biol. Ther. 2009, 9, 737–747. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Geoerger, B.; Grill, J.; Opolon, P.; Morizet, J.; Aubert, G.; Terrier-Lacombe, M.J.; Bressac De-Paillerets, B.; Barrois, M.; Feunteun, J.; Kirn, D.H.; et al. Oncolytic activity of the e1b-55 kda-deleted adenovirus onyx-015 is independent of cellular p53 status in human malignant glioma xenografts. Cancer Res. 2002, 62, 764–772. [Google Scholar]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A phase i open-label, dose-escalation, multi-institutional trial of injection with an e1b-attenuated adenovirus, onyx-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef]
- Suzuki, K.; Fueyo, J.; Krasnykh, V.; Reynolds, P.N.; Curiel, D.T.; Alemany, R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 2001, 7, 120–126. [Google Scholar]
- Fueyo, J.; Alemany, R.; Gomez-Manzano, C.; Fuller, G.N.; Khan, A.; Conrad, C.A.; Liu, T.J.; Jiang, H.; Lemoine, M.G.; Suzuki, K.; et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J. Natl. Cancer Inst. 2003, 95, 652–660. [Google Scholar] [CrossRef]
- Auffinger, B.; Ahmed, A.U.; Lesniak, M.S. Oncolytic virotherapy for malignant glioma: Translating laboratory insights into clinical practice. Front. Oncol. 2013. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Zhu, Z.B.; Tyler, M.A.; Han, Y.; Rivera, A.A.; Khramtsov, A.; Curiel, D.T.; Lesniak, M.S. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum. Gene Ther. 2007, 18, 589–602. [Google Scholar] [CrossRef]
- Nandi, S.; Ulasov, I.V.; Tyler, M.A.; Sugihara, A.Q.; Molinero, L.; Han, Y.; Zhu, Z.B.; Lesniak, M.S. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008, 68, 5778–5784. [Google Scholar] [CrossRef]
- Zemp, F.J.; Corredor, J.C.; Lun, X.; Muruve, D.A.; Forsyth, P.A. Oncolytic viruses as experimental treatments for malignant gliomas: Using a scourge to treat a devil. Cytokine Growth Factor Rev. 2010, 21, 103–117. [Google Scholar] [CrossRef]
- Ochiai, H.; Campbell, S.A.; Archer, G.E.; Chewning, T.A.; Dragunsky, E.; Ivanov, A.; Gromeier, M.; Sampson, J.H. Targeted therapy for glioblastoma multiforme neoplastic meningitis with intrathecal delivery of an oncolytic recombinant poliovirus. Clin. Cancer Res. 2006, 12, 1349–1354. [Google Scholar] [CrossRef]
- Pasquinucci, G. Possible effect of measles on leukaemia. Lancet 1971, 1, 136. [Google Scholar] [CrossRef]
- Bluming, A.Z.; Ziegler, J.L. Regression of burkitt’s lymphoma in association with measles infection. Lancet 1971, 2, 105–106. [Google Scholar] [CrossRef]
- Allen, C.; Paraskevakou, G.; Iankov, I.; Giannini, C.; Schroeder, M.; Sarkaria, J.; Schroeder, M.; Puri, R.K.; Russell, S.J.; Galanis, E. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Mol. Ther. 2008, 16, 1556–1564. [Google Scholar] [CrossRef]
- Paraskevakou, G.; Allen, C.; Nakamura, T.; Zollman, P.; James, C.D.; Peng, K.W.; Schroeder, M.; Russell, S.J.; Galanis, E. Epidermal growth factor receptor (egfr)-retargeted measles virus strains effectively target egfr- or egfrviii expressing gliomas. Mol. Ther. 2007, 15, 677–686. [Google Scholar]
- Phuong, L.K.; Allen, C.; Peng, K.W.; Giannini, C.; Greiner, S.; TenEyck, C.J.; Mishra, P.K.; Macura, S.I.; Russell, S.J.; Galanis, E.C. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003, 63, 2462–2469. [Google Scholar]
- Myers, R.; Harvey, M.; Kaufmann, T.J.; Greiner, S.M.; Krempski, J.W.; Raffel, C.; Shelton, S.E.; Soeffker, D.; Zollman, P.; Federspiel, M.J.; et al. Toxicology study of repeat intracerebral administration of a measles virus derivative producing carcinoembryonic antigen in rhesus macaques in support of a phase I/II clinical trial for patients with recurrent gliomas. Hum. Gene Ther. 2008, 19, 690–698. [Google Scholar] [CrossRef]
- Allen, C.; Opyrchal, M.; Aderca, I.; Schroeder, M.A.; Sarkaria, J.N.; Domingo, E.; Federspiel, M.J.; Galanis, E. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Ther. 2013, 20, 444–449. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Aghi, M.; Fulci, G. Viral therapy for glioblastoma. Cancer J. 2003, 9, 167–179. [Google Scholar] [CrossRef]
- Allen, C.; Paraskevakou, G.; Liu, C.; Iankov, I.D.; Msaouel, P.; Zollman, P.; Myers, R.; Peng, K.W.; Russell, S.J.; Galanis, E. Oncolytic measles virus strains in the treatment of gliomas. Expert Opin. Biol. Ther. 2008, 8, 213–220. [Google Scholar] [CrossRef]
- Kurozumi, K.; Hardcastle, J.; Thakur, R.; Yang, M.; Christoforidis, G.; Fulci, G.; Hochberg, F.H.; Weissleder, R.; Carson, W.; Chiocca, E.A.; et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J. Natl. Cancer Inst. 2007, 99, 1768–1781. [Google Scholar] [CrossRef]
- Sena-Esteves, M.; Hampl, J.A.; Camp, S.M.; Breakefield, X.O. Generation of stable retrovirus packaging cell lines after transduction with herpes simplex virus hybrid amplicon vectors. J. Gene Med. 2002, 4, 229–239. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Read, T.A.; Vajkoczy, P.; Aebischer, P.; Pralong, W.; Platt, S.; Melvik, J.E.; Hagen, A.; Dornish, M. Cell therapy using encapsulated cells producing endostatin. Acta Neurochir. Suppl. 2003, 88, 137–141. [Google Scholar]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef]
- Aboody, K.S.; Najbauer, J.; Danks, M.K. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 2008, 15, 739–752. [Google Scholar] [CrossRef]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef]
- Binello, E.; Germano, I.M. Stem cells as therapeutic vehicles for the treatment of high-grade gliomas. Neuro-oncology 2012, 14, 256–265. [Google Scholar] [CrossRef]
- Benedetti, S.; Pirola, B.; Pollo, B.; Magrassi, L.; Bruzzone, M.G.; Rigamonti, D.; Galli, R.; Selleri, S.; di Meco, F.; de Fraja, C.; et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat. Med. 2000, 6, 447–450. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Alexiades, N.G.; Lesniak, M.S. The use of neural stem cells in cancer gene therapy: Predicting the path to the clinic. Curr. Opin. Mol. Ther. 2010, 12, 546–552. [Google Scholar]
- Herrlinger, U.; Woiciechowski, C.; Sena-Esteves, M.; Aboody, K.S.; Jacobs, A.H.; Rainov, N.G.; Snyder, E.Y.; Breakefield, X.O. Neural precursor cells for delivery of replication-conditional hsv-1 vectors to intracerebral gliomas. Mol. Ther. 2000, 1, 347–357. [Google Scholar] [CrossRef]
- Tyler, M.A.; Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Han, Y.; Marler, S.; Roth, J.; Lesniak, M.S. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2009, 16, 262–278. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Thaci, B.; Alexiades, N.G.; Han, Y.; Qian, S.; Liu, F.; Balyasnikova, I.V.; Ulasov, I.Y.; Aboody, K.S.; Lesniak, M.S. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol. Ther. 2011, 19, 1714–1726. [Google Scholar] [CrossRef]
- Barresi, V.; Belluardo, N.; Sipione, S.; Mudo, G.; Cattaneo, E.; Condorelli, D.F. Transplantation of prodrug-converting neural progenitor cells for brain tumor therapy. Cancer Gene Ther. 2003, 10, 396–402. [Google Scholar] [CrossRef]
- Rath, P.; Shi, H.; Maruniak, J.A.; Litofsky, N.S.; Maria, B.L.; Kirk, M.D. Stem cells as vectors to deliver hsv/tk gene therapy for malignant gliomas. Curr. Stem. Cell Res. Ther. 2009, 4, 44–49. [Google Scholar] [CrossRef]
- Ito, S.; Natsume, A.; Shimato, S.; Ohno, M.; Kato, T.; Chansakul, P.; Wakabayashi, T.; Kim, S.U. Human neural stem cells transduced with ifn-beta and cytosine deaminase genes intensify bystander effect in experimental glioma. Cancer Gene Ther. 2010, 17, 299–306. [Google Scholar]
- Ehtesham, M.; Kabos, P.; Kabosova, A.; Neuman, T.; Black, K.L.; Yu, J.S. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002, 62, 5657–5663. [Google Scholar]
- Yuan, X.; Hu, J.; Belladonna, M.L.; Black, K.L.; Yu, J.S. Interleukin-23-expressing bone marrow-derived neural stem-like cells exhibit antitumor activity against intracranial glioma. Cancer Res. 2006, 66, 2630–2638. [Google Scholar] [CrossRef]
- Shah, K.; Tung, C.H.; Yang, K.; Weissleder, R.; Breakefield, X.O. Inducible release of trail fusion proteins from a proapoptotic form for tumor therapy. Cancer Res. 2004, 64, 3236–3242. [Google Scholar] [CrossRef]
- Shah, K.; Bureau, E.; Kim, D.E.; Yang, K.; Tang, Y.; Weissleder, R.; Breakefield, X.O. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann. Neurol. 2005, 57, 34–41. [Google Scholar] [CrossRef]
- Hingtgen, S.; Ren, X.; Terwilliger, E.; Classon, M.; Weissleder, R.; Shah, K. Targeting multiple pathways in gliomas with stem cell and viral delivered s-trail and temozolomide. Mol. Cancer Ther. 2008, 7, 3575–3585. [Google Scholar] [CrossRef]
- Bagci-Onder, T.; Wakimoto, H.; Anderegg, M.; Cameron, C.; Shah, K. A dual pi3k/mtor inhibitor, pi-103, cooperates with stem cell-delivered trail in experimental glioma models. Cancer Res. 2011, 71, 154–163. [Google Scholar] [CrossRef]
- Balyasnikova, I.V.; Ferguson, S.D.; Han, Y.; Liu, F.; Lesniak, M.S. Therapeutic effect of neural stem cells expressing trail and bortezomib in mice with glioma xenografts. Cancer Lett. 2011, 310, 148–159. [Google Scholar] [CrossRef]
- Lorico, A.; Mercapide, J.; Solodushko, V.; Alexeyev, M.; Fodstad, O.; Rappa, G. Primary neural stem/progenitor cells expressing endostatin or cytochrome p450 for gene therapy of glioblastoma. Cancer Gene Ther. 2008, 15, 605–615. [Google Scholar] [CrossRef]
- Van Eekelen, M.; Sasportas, L.S.; Kasmieh, R.; Yip, S.; Figueiredo, J.L.; Louis, D.N.; Weissleder, R.; Shah, K. Human stem cells expressing novel tsp-1 variant have anti-angiogenic effect on brain tumors. Oncogene 2010, 29, 3185–3195. [Google Scholar] [CrossRef]
- Kim, S.K.; Cargioli, T.G.; Machluf, M.; Yang, W.; Sun, Y.; Al-Hashem, R.; Kim, S.U.; Black, P.M.; Carroll, R.S. Pex-producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clin. Cancer Res. 2005, 11, 5965–5970. [Google Scholar] [CrossRef]
- Oreffo, R.O.; Cooper, C.; Mason, C.; Clements, M. Mesenchymal stem cells: Lineage, plasticity, and skeletal therapeutic potential. Stem Cell Rev. 2005, 1, 169–178. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Bexell, D.; Scheding, S.; Bengzon, J. Toward brain tumor gene therapy using multipotent mesenchymal stromal cell vectors. Mol. Ther. 2010, 18, 1067–1075. [Google Scholar] [CrossRef]
- Nakamura, K.; Ito, Y.; Kawano, Y.; Kurozumi, K.; Kobune, M.; Tsuda, H.; Bizen, A.; Honmou, O.; Niitsu, Y.; Hamada, H. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004, 11, 1155–1164. [Google Scholar] [CrossRef]
- Chang, D.Y.; Yoo, S.W.; Hong, Y.; Kim, S.; Kim, S.J.; Yoon, S.H.; Cho, K.G.; Paek, S.H.; Lee, Y.D.; Kim, S.S.; et al. The growth of brain tumors can be suppressed by multiple transplantation of mesenchymal stem cells expressing cytosine deaminase. Int. J. Cancer 2010, 127, 1975–1983. [Google Scholar] [CrossRef]
- Choi, S.A.; Lee, J.Y.; Wang, K.C.; Phi, J.H.; Song, S.H.; Song, J.; Kim, S.K. Human adipose tissue-derived mesenchymal stem cells: Characteristics and therapeutic potential as cellular vehicles for prodrug gene therapy against brainstem gliomas. Eur. J. Cancer 2012, 48, 129–137. [Google Scholar]
- Gunnarsson, S.; Bexell, D.; Svensson, A.; Siesjo, P.; Darabi, A.; Bengzon, J. Intratumoral il-7 delivery by mesenchymal stromal cells potentiates ifngamma-transduced tumor cell immunotherapy of experimental glioma. J. Neuroimmunol. 2010, 218, 140–144. [Google Scholar] [CrossRef]
- Ryu, C.H.; Park, S.H.; Park, S.A.; Kim, S.M.; Lim, J.Y.; Jeong, C.H.; Yoon, W.S.; Oh, W.I.; Sung, Y.C.; Jeun, S.S. Gene therapy of intracranial glioma using interleukin 12-secreting human umbilical cord blood-derived mesenchymal stem cells. Hum. Gene Ther. 2011, 22, 733–743. [Google Scholar] [CrossRef]
- Xu, G.; Jiang, X.D.; Xu, Y.; Zhang, J.; Huang, F.H.; Chen, Z.Z.; Zhou, D.X.; Shang, J.H.; Zou, Y.X.; Cai, Y.Q.; et al. Adenoviral-mediated interleukin-18 expression in mesenchymal stem cells effectively suppresses the growth of glioma in rats. Cell Biol. Int. 2009, 33, 466–474. [Google Scholar] [CrossRef]
- Kim, S.M.; Lim, J.Y.; Park, S.I.; Jeong, C.H.; Oh, J.H.; Jeong, M.; Oh, W.; Park, S.H.; Sung, Y.C.; Jeun, S.S. Gene therapy using trail-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008, 68, 9614–9623. [Google Scholar] [CrossRef]
- Menon, L.G.; Kelly, K.; Yang, H.W.; Kim, S.K.; Black, P.M.; Carroll, R.S. Human bone marrow-derived mesenchymal stromal cells expressing s-trail as a cellular delivery vehicle for human glioma therapy. Stem Cells 2009, 27, 2320–2330. [Google Scholar] [CrossRef]
- Kim, S.M.; Oh, J.H.; Park, S.A.; Ryu, C.H.; Lim, J.Y.; Kim, D.S.; Chang, J.W.; Oh, W.; Jeun, S.S. Irradiation enhances the tumor tropism and therapeutic potential of tumor necrosis factor-related apoptosis-inducing ligand-secreting human umbilical cord blood-derived mesenchymal stem cells in glioma therapy. Stem Cells 2010, 28, 2217–2228. [Google Scholar] [CrossRef]
- Kim, S.M.; Woo, J.S.; Jeong, C.H.; Ryu, C.H.; Lim, J.Y.; Jeun, S.S. Effective combination therapy for malignant glioma with trail-secreting mesenchymal stem cells and lipoxygenase inhibitor mk886. Cancer Res. 2012, 72, 4807–4817. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Rolle, C.E.; Tyler, M.A.; Han, Y.; Sengupta, S.; Wainwright, D.A.; Balyasnikova, I.V.; Ulasov, I.V.; Lesniak, M.S. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus suppress the anti-adenoviral immune response in the cotton rat model. Mol. Ther. 2010, 18, 1846–1856. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Tyler, M.A.; Thaci, B.; Alexiades, N.G.; Han, Y.; Ulasov, I.V.; Lesniak, M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011, 8, 1559–1572. [Google Scholar] [CrossRef]
- Yong, R.L.; Shinojima, N.; Fueyo, J.; Gumin, J.; Vecil, G.G.; Marini, F.C.; Bogler, O.; Andreeff, M.; Lang, F.F. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus delta24-rgd to human gliomas. Cancer Res. 2009, 69, 8932–8940. [Google Scholar] [CrossRef]
- Keller, G. Embryonic stem cell differentiation: Emergence of a new era in biology and medicine. Genes Dev. 2005, 19, 1129–1155. [Google Scholar] [CrossRef]
- Germano, I.M.; Uzzaman, M.; Benveniste, R.J.; Zaurova, M.; Keller, G. Apoptosis in human glioblastoma cells produced using embryonic stem cell-derived astrocytes expressing tumor necrosis factor-related apoptosis-inducing ligand. J. Neurosurg. 2006, 105, 88–95. [Google Scholar] [CrossRef]
- Uzzaman, M.; Keller, G.; Germano, I.M. In vivo gene delivery by embryonic-stem-cell-derived astrocytes for malignant gliomas. Neuro-oncology 2009, 11, 102–108. [Google Scholar] [CrossRef]
- Germano, I.M.; Emdad, L.; Qadeer, Z.A.; Binello, E.; Uzzaman, M. Embryonic stem cell (esc)-mediated transgene delivery induces growth suppression, apoptosis and radiosensitization, and overcomes temozolomide resistance in malignant gliomas. Cancer Gene Ther. 2010, 17, 664–674. [Google Scholar] [CrossRef]
- Bak, X.Y.; Lam, D.H.; Yang, J.; Ye, K.; Wei, E.L.; Lim, S.K.; Wang, S. Human embryonic stem cell-derived mesenchymal stem cells as cellular delivery vehicles for prodrug gene therapy of glioblastoma. Hum. Gene Ther. 2011, 22, 1365–1377. [Google Scholar] [CrossRef]
- Zhao, Y.; Lam, D.H.; Yang, J.; Lin, J.; Tham, C.K.; Ng, W.H.; Wang, S. Targeted suicide gene therapy for glioma using human embryonic stem cell-derived neural stem cells genetically modified by baculoviral vectors. Gene Ther. 2012, 19, 189–200. [Google Scholar] [CrossRef]
- Nakashima, H.; Kaur, B.; Chiocca, E.A. Directing systemic oncolytic viral delivery to tumors via carrier cells. Cytokine Growth Factor Rev. 2010, 21, 119–126. [Google Scholar] [CrossRef]
- Tang, Y.; Shah, K.; Messerli, S.M.; Snyder, E.; Breakefield, X.; Weissleder, R. In vivo tracking of neural progenitor cell migration to glioblastomas. Hum. Gene Ther. 2003, 14, 1247–1254. [Google Scholar] [CrossRef]
- Reitz, M.; Demestre, M.; Sedlacik, J.; Meissner, H.; Fiehler, J.; Kim, S.U.; Westphal, M.; Schmidt, N.O. Intranasal delivery of neural stem/progenitor cells: A noninvasive passage to target intracerebral glioma. Stem Cells Transl. Med. 2012, 1, 866–873. [Google Scholar] [CrossRef]
- Assanah, M.; Lochhead, R.; Ogden, A.; Bruce, J.; Goldman, J.; Canoll, P. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J. Neurosci. 2006, 26, 6781–6790. [Google Scholar] [CrossRef]
- Yoshida, J.; Mizuno, M. Clinical gene therapy for brain tumors. Liposomal delivery of anticancer molecule to glioma. J. Neurooncol. 2003, 65, 261–267. [Google Scholar] [CrossRef]
- Niidome, T.; Huang, L. Gene therapy progress and prospects: Nonviral vectors. Gene Ther. 2002, 9, 1647–1652. [Google Scholar] [CrossRef]
- Barbu, E.; Molnar, E.; Tsibouklis, J.; Gorecki, D.C. The potential for nanoparticle-based drug delivery to the brain: Overcoming the blood-brain barrier. Expert Opin. Drug Deliv. 2009, 6, 553–565. [Google Scholar] [CrossRef]
- Reszka, R.C.; Jacobs, A.; Voges, J. Liposome-mediated suicide gene therapy in humans. Methods Enzymol. 2005, 391, 200–208. [Google Scholar] [CrossRef]
- Yoshida, J.; Mizuno, M.; Fujii, M.; Kajita, Y.; Nakahara, N.; Hatano, M.; Saito, R.; Nobayashi, M.; Wakabayashi, T. Human gene therapy for malignant gliomas (glioblastoma multiforme and anaplastic astrocytoma) by in vivo transduction with human interferon beta gene using cationic liposomes. Hum. Gene Ther. 2004, 15, 77–86. [Google Scholar] [CrossRef]
- Sun, X.; Pang, Z.; Ye, H.; Qiu, B.; Guo, L.; Li, J.; Ren, J.; Qian, Y.; Zhang, Q.; Chen, J.; et al. Co-delivery of pegfp-htrail and paclitaxel to brain glioma mediated by an angiopep-conjugated liposome. Biomaterials 2012, 33, 916–924. [Google Scholar]
- Ito, A.; Shinkai, M.; Bouhon, I.A.; Honda, H.; Kobayashi, T. Bystander-killing effect and cyclic induction of tnf-alpha gene under heat-inducible promoter gadd 153. J. Biosci. Bioeng. 2000, 90, 437–441. [Google Scholar]
- Ito, A.; Shinkai, M.; Honda, H.; Kobayashi, T. Heat-inducible tnf-alpha gene therapy combined with hyperthermia using magnetic nanoparticles as a novel tumor-targeted therapy. Cancer Gene Ther. 2001, 8, 649–654. [Google Scholar] [CrossRef]
- Yin, T.; Wang, P.; Li, J.; Zheng, R.; Zheng, B.; Cheng, D.; Li, R.; Lai, J.; Shuai, X. Ultrasound-sensitive sirna-loaded nanobubbles formed by hetero-assembly of polymeric micelles and liposomes and their therapeutic effect in gliomas. Biomaterials 2013, 34, 4532–4543. [Google Scholar] [CrossRef]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Poly(ethylenimine) and its role in gene delivery. J. Control. Release 1999, 60, 149–160. [Google Scholar] [CrossRef]
- Lu, X.; Ping, Y.; Xu, F.J.; Li, Z.H.; Wang, Q.Q.; Chen, J.H.; Yang, W.T.; Tang, G.P. Bifunctional conjugates comprising beta-cyclodextrin, polyethylenimine, and 5-fluoro-2'-deoxyuridine for drug delivery and gene transfer. Bioconjug. Chem. 2010, 21, 1855–1863. [Google Scholar] [CrossRef]
- Li, J.; Gu, B.; Meng, Q.; Yan, Z.; Gao, H.; Chen, X.; Yang, X.; Lu, W. The use of myristic acid as a ligand of polyethylenimine/DNA nanoparticles for targeted gene therapy of glioblastoma. Nanotechnology 2011, 22, 435101. [Google Scholar] [CrossRef]
- Zhan, C.; Meng, Q.; Li, Q.; Feng, L.; Zhu, J.; Lu, W. Cyclic rgd-polyethylene glycol-polyethylenimine for intracranial glioblastoma-targeted gene delivery. Chem. Asian J. 2012, 7, 91–96. [Google Scholar] [CrossRef]
- Dufes, C.; Uchegbu, I.F.; Schatzlein, A.G. Dendrimers in gene delivery. Adv. Drug Deliv. Rev. 2005, 57, 2177–2202. [Google Scholar] [CrossRef]
- Pettit, M.W.; Griffiths, P.; Ferruti, P.; Richardson, S.C. Poly(amidoamine) polymers: Soluble linear amphiphilic drug-delivery systems for genes, proteins and oligonucleotides. Ther. Deliv. 2011, 2, 907–917. [Google Scholar] [CrossRef]
- Han, L.; Zhang, A.; Wang, H.; Pu, P.; Jiang, X.; Kang, C.; Chang, J. Tat-bmps-pamam conjugates enhance therapeutic effect of small interference rna on u251 glioma cells in vitro and in vivo. Hum. Gene Ther. 2010, 21, 417–426. [Google Scholar] [CrossRef]
- Bai, C.Z.; Choi, S.; Nam, K.; An, S.; Park, J.S. Arginine modified pamam dendrimer for interferon beta gene delivery to malignant glioma. Int. J. Pharm. 2013, 445, 79–87. [Google Scholar] [CrossRef]
- Wang, H.; Su, W.; Wang, S.; Wang, X.; Liao, Z.; Kang, C.; Han, L.; Chang, J.; Wang, G.; Pu, P. Smart multifunctional core-shell nanospheres with drug and gene co-loaded for enhancing the therapeutic effect in a rat intracranial tumor model. Nanoscale 2012, 4, 6501–6508. [Google Scholar] [CrossRef]
- Kami, D.; Takeda, S.; Itakura, Y.; Gojo, S.; Watanabe, M.; Toyoda, M. Application of magnetic nanoparticles to gene delivery. Int. J. Mol. Sci. 2011, 12, 3705–3722. [Google Scholar] [CrossRef]
- Silva, A.C.; Oliveira, T.R.; Mamani, J.B.; Malheiros, S.M.; Malavolta, L.; Pavon, L.F.; Sibov, T.T.; Amaro, E., Jr.; Tannus, A.; Vidoto, E.L.; et al. Application of hyperthermia induced by superparamagnetic iron oxide nanoparticles in glioma treatment. Int. J. Nanomed. 2011, 6, 591–603. [Google Scholar]
- Wankhede, M.; Bouras, A.; Kaluzova, M.; Hadjipanayis, C.G. Magnetic nanoparticles: An emerging technology for malignant brain tumor imaging and therapy. Expert Rev. Clin. Pharmacol. 2012, 5, 173–186. [Google Scholar] [CrossRef]
- Yun, J.; Sonabend, A.M.; Ulasov, I.V.; Kim, D.H.; Rozhkova, E.A.; Novosad, V.; Dashnaw, S.; Brown, T.; Canoll, P.; Bruce, J.N.; et al. A novel adenoviral vector labeled with superparamagnetic iron oxide nanoparticles for real-time tracking of viral delivery. J. Clin. Neurosci. 2012, 19, 875–880. [Google Scholar] [CrossRef]
- Kievit, F.M.; Veiseh, O.; Fang, C.; Bhattarai, N.; Lee, D.; Ellenbogen, R.G.; Zhang, M. Chlorotoxin labeled magnetic nanovectors for targeted gene delivery to glioma. ACS Nano 2010, 4, 4587–4594. [Google Scholar] [CrossRef]
- Veiseh, O.; Kievit, F.M.; Fang, C.; Mu, N.; Jana, S.; Leung, M.C.; Mok, H.; Ellenbogen, R.G.; Park, J.O.; Zhang, M. Chlorotoxin bound magnetic nanovector tailored for cancer cell targeting, imaging, and sirna delivery. Biomaterials 2010, 31, 8032–8042. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kwiatkowska, A.; Nandhu, M.S.; Behera, P.; Chiocca, E.A.; Viapiano, M.S. Strategies in Gene Therapy for Glioblastoma. Cancers 2013, 5, 1271-1305. https://doi.org/10.3390/cancers5041271
Kwiatkowska A, Nandhu MS, Behera P, Chiocca EA, Viapiano MS. Strategies in Gene Therapy for Glioblastoma. Cancers. 2013; 5(4):1271-1305. https://doi.org/10.3390/cancers5041271
Chicago/Turabian StyleKwiatkowska, Aneta, Mohan S. Nandhu, Prajna Behera, E. Antonio Chiocca, and Mariano S. Viapiano. 2013. "Strategies in Gene Therapy for Glioblastoma" Cancers 5, no. 4: 1271-1305. https://doi.org/10.3390/cancers5041271
APA StyleKwiatkowska, A., Nandhu, M. S., Behera, P., Chiocca, E. A., & Viapiano, M. S. (2013). Strategies in Gene Therapy for Glioblastoma. Cancers, 5(4), 1271-1305. https://doi.org/10.3390/cancers5041271