Lentiviral Vectors for Cancer Immunotherapy and Clinical Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
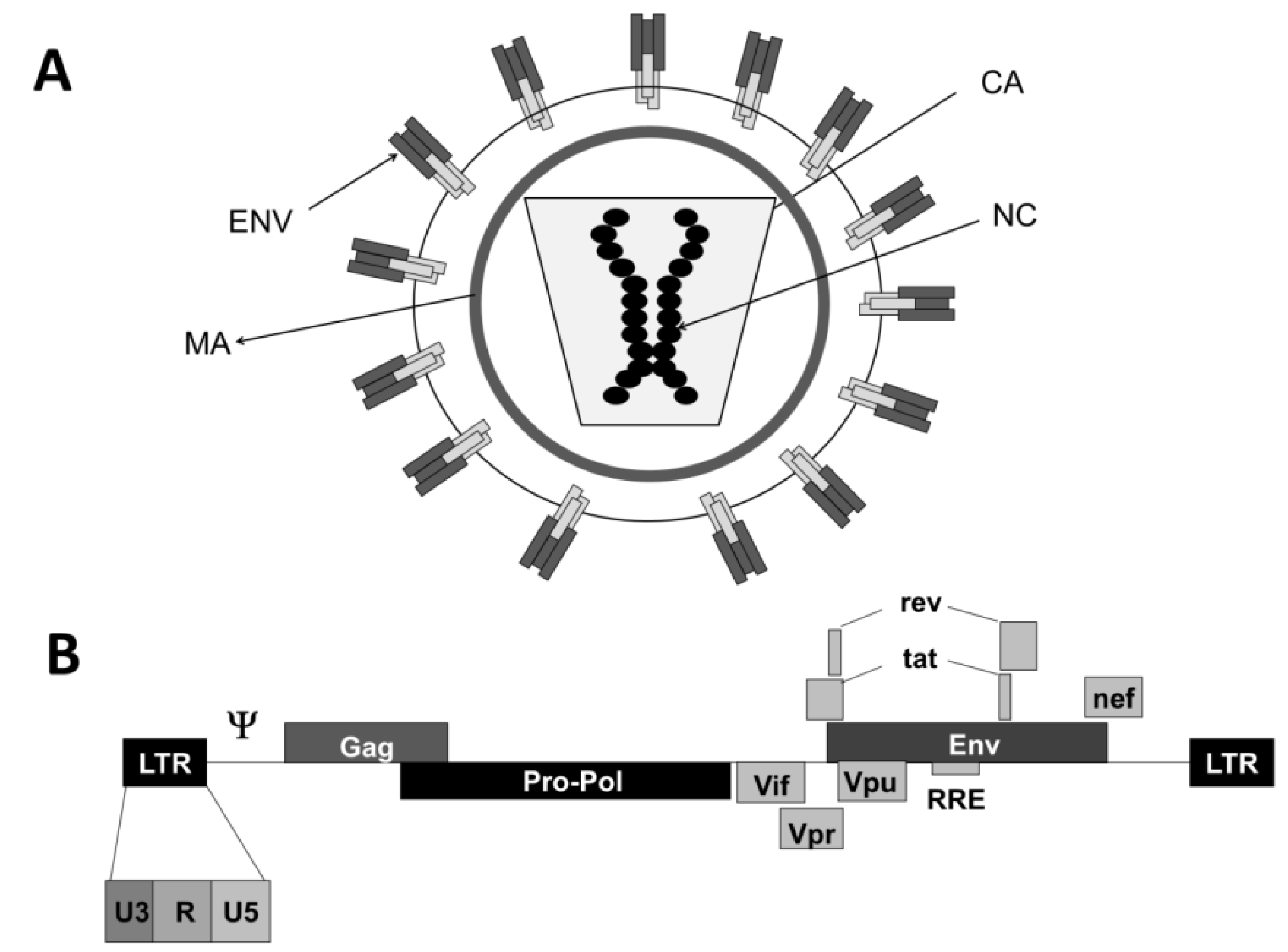
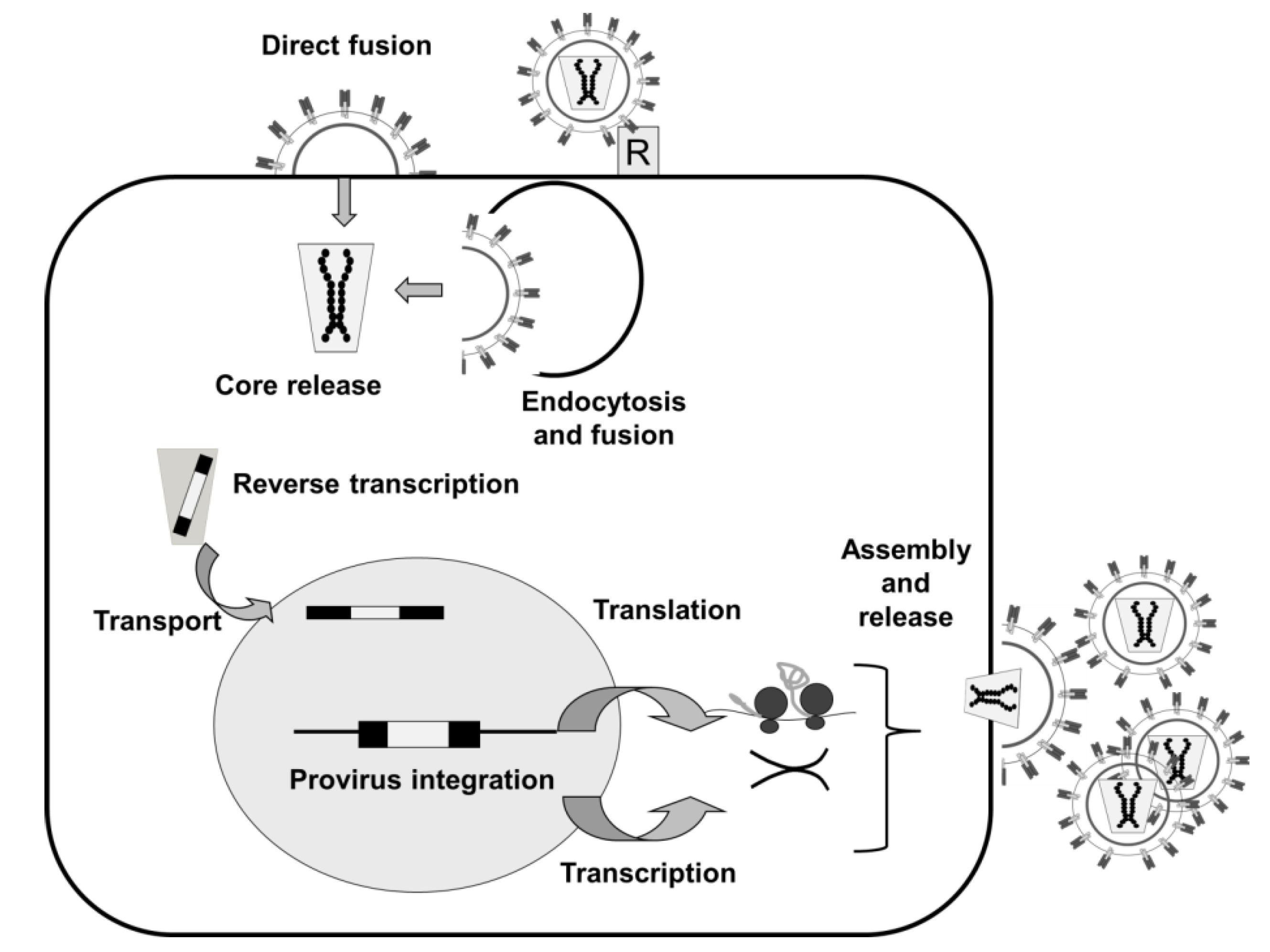
2. Lentiviruses and Lentiviral Vectors
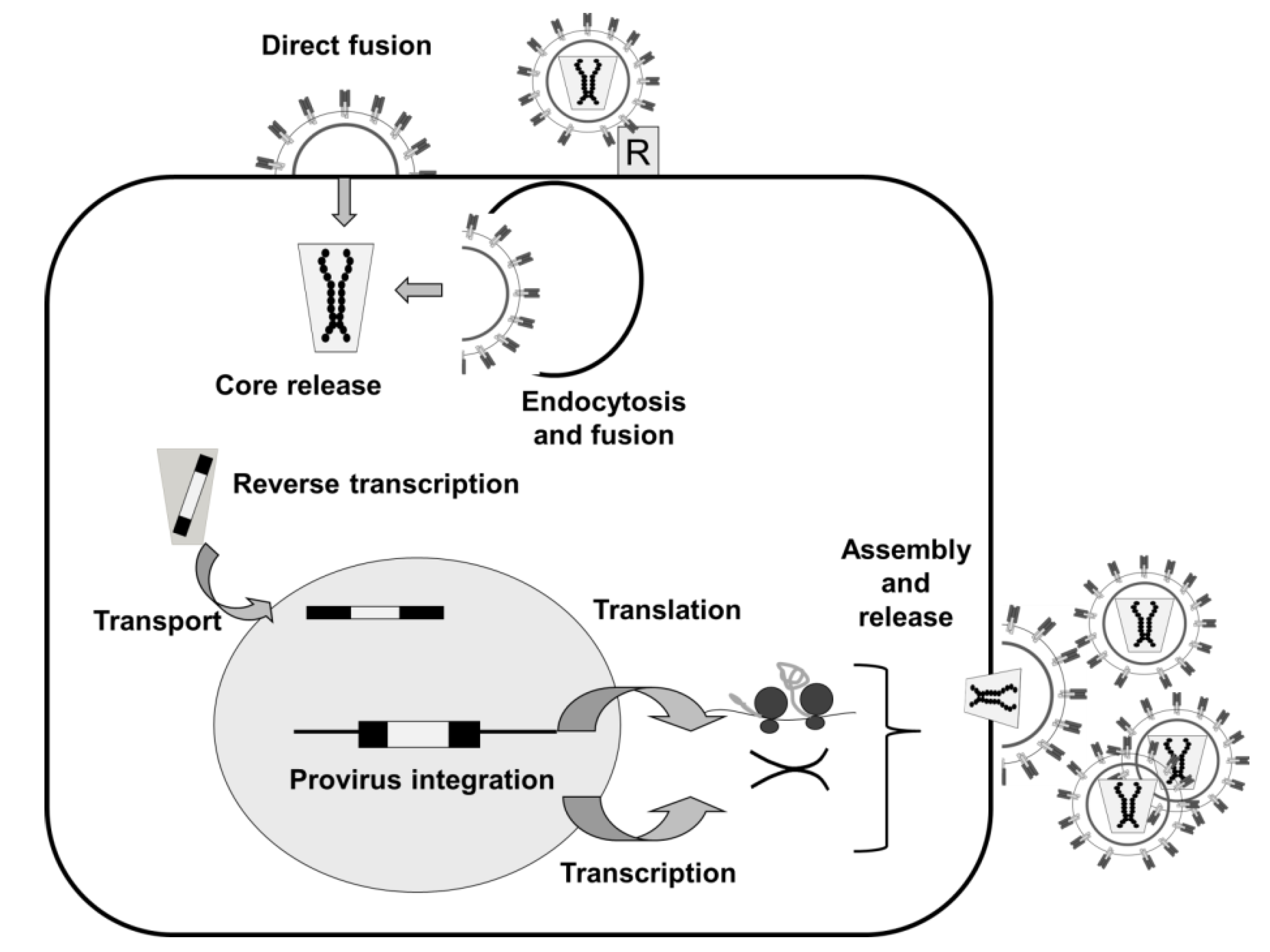
2.1. Lentiviruses

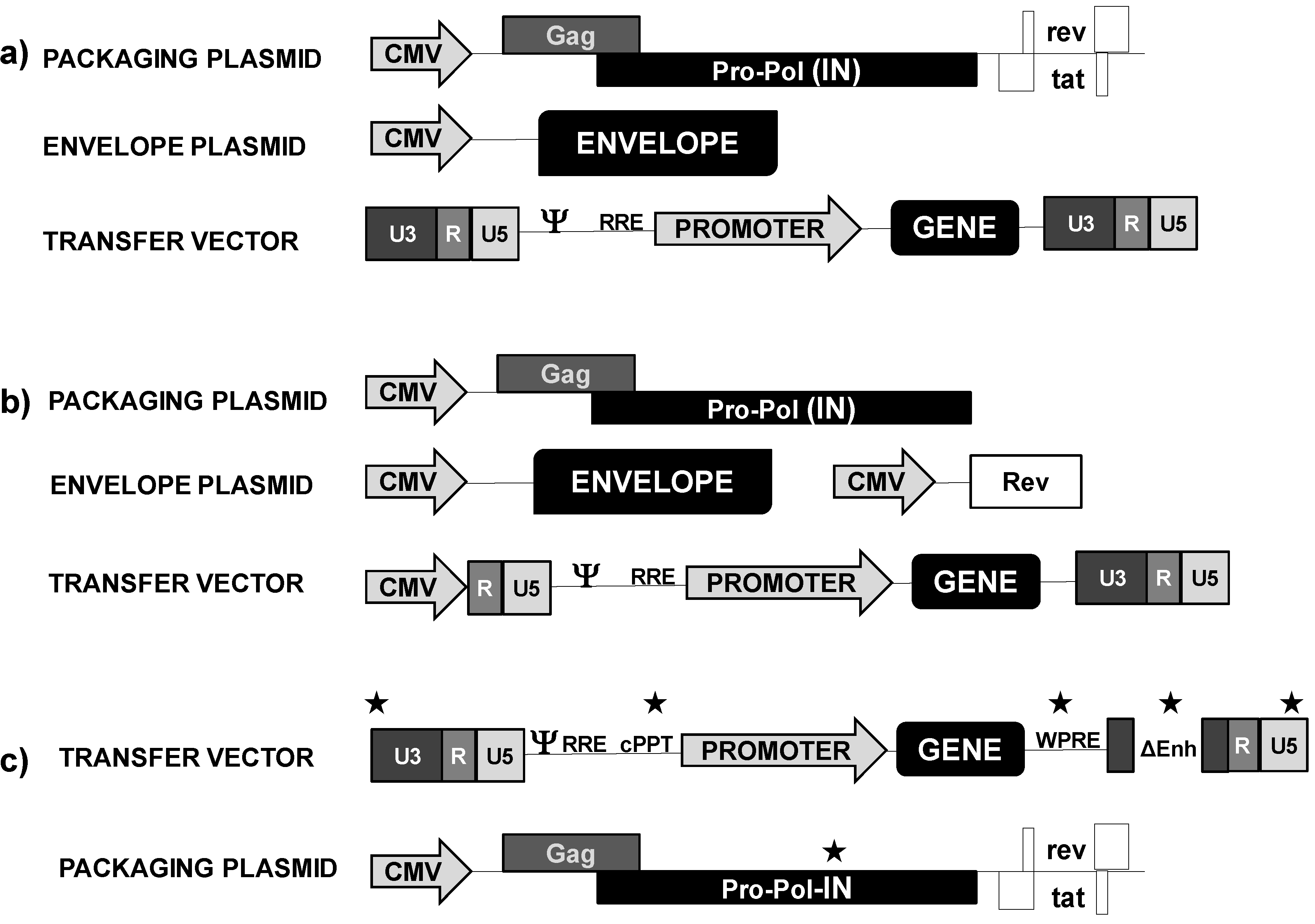
2.1.1. Lentivectors
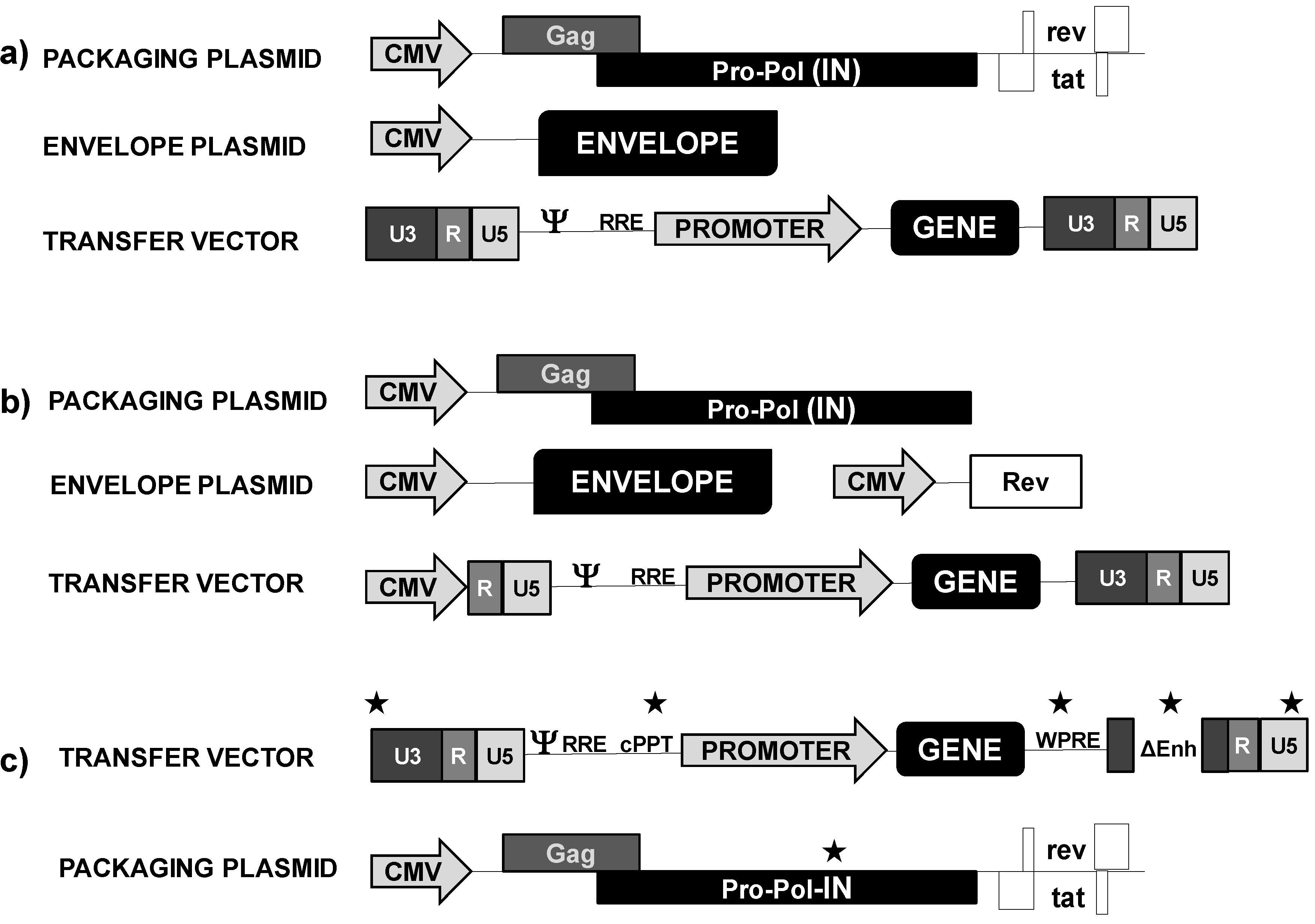
3. Lentivectors in Cancer Immunotherapy
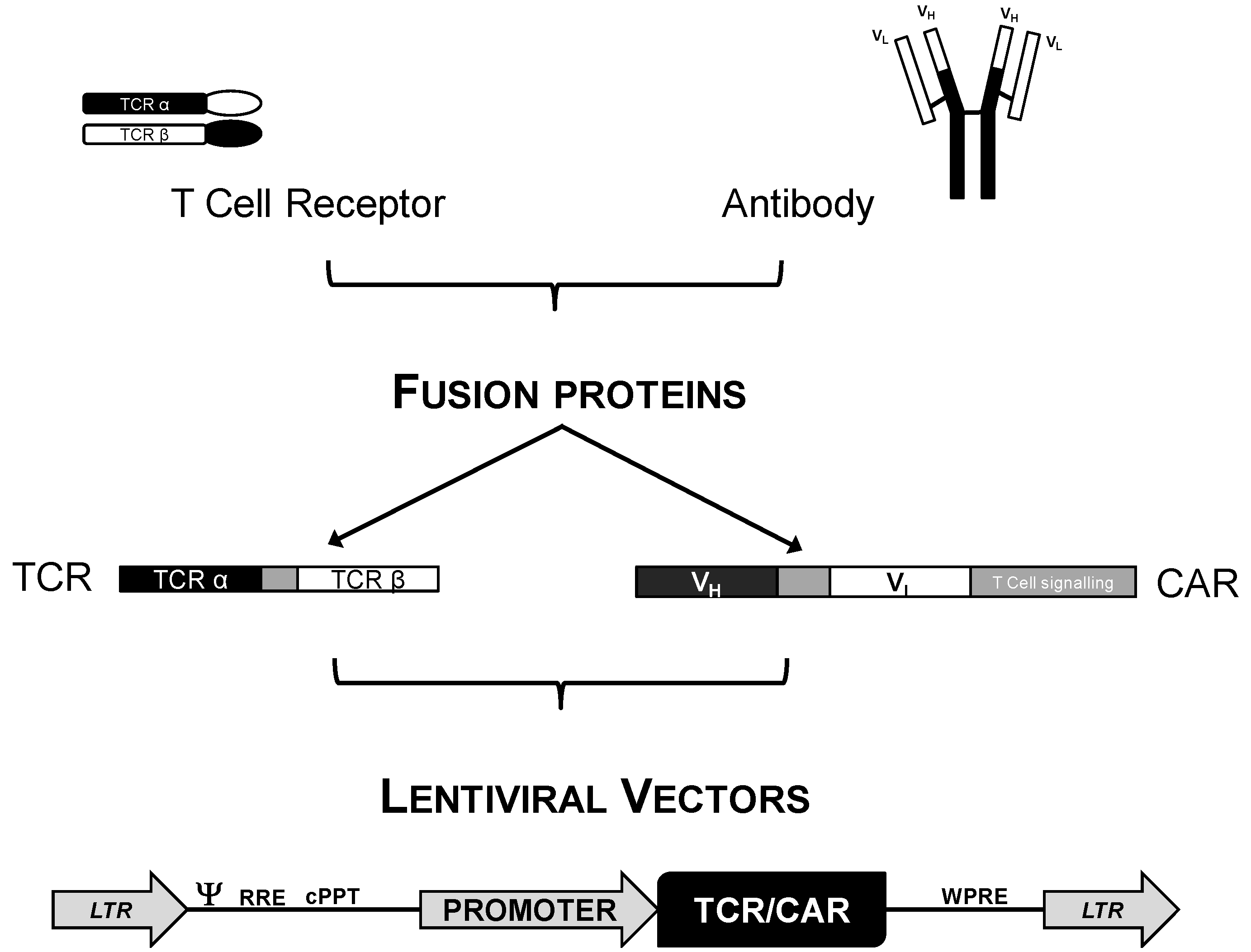
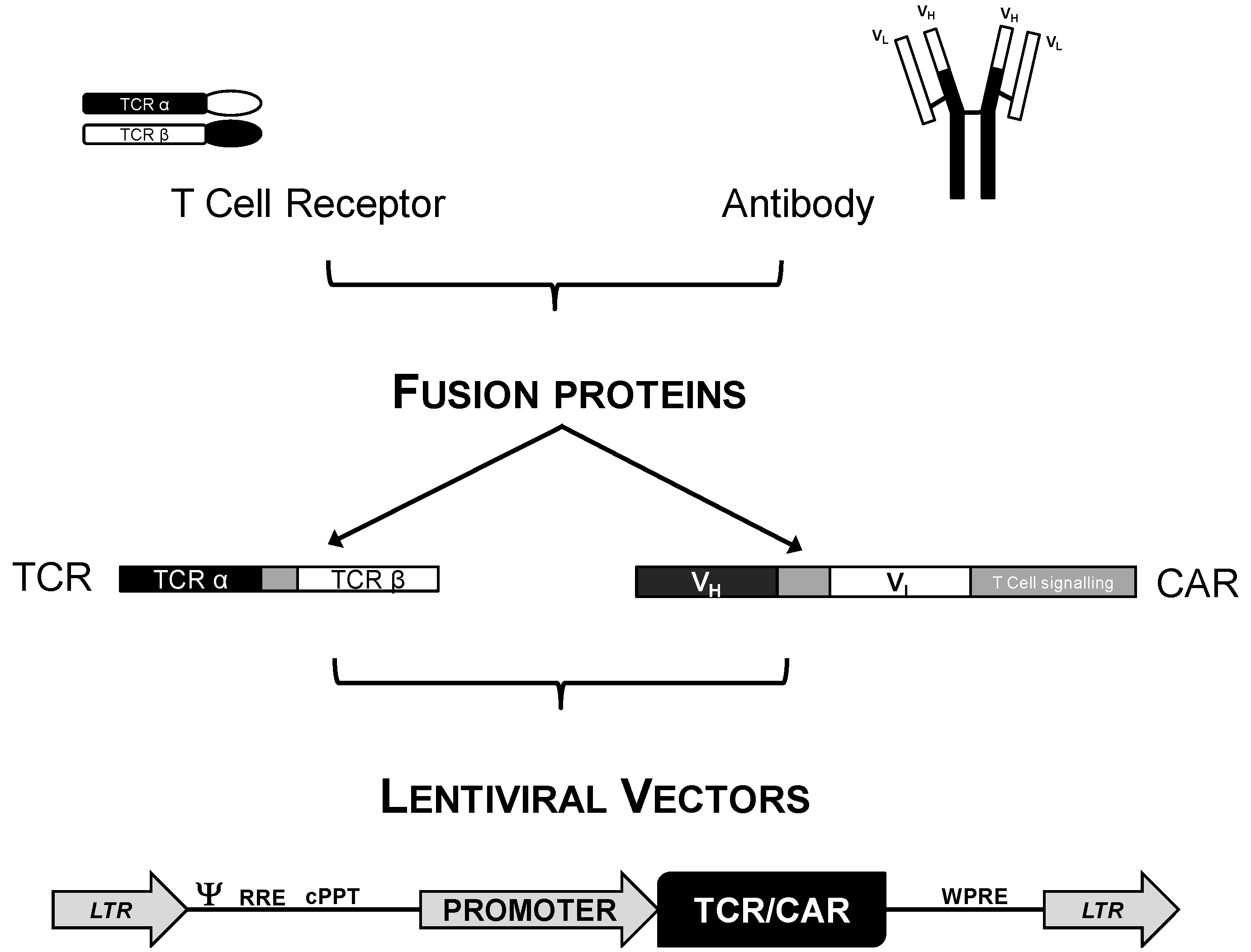
3.1. Genetic Modification of T Cells for Adoptive Cell Immunotherapy
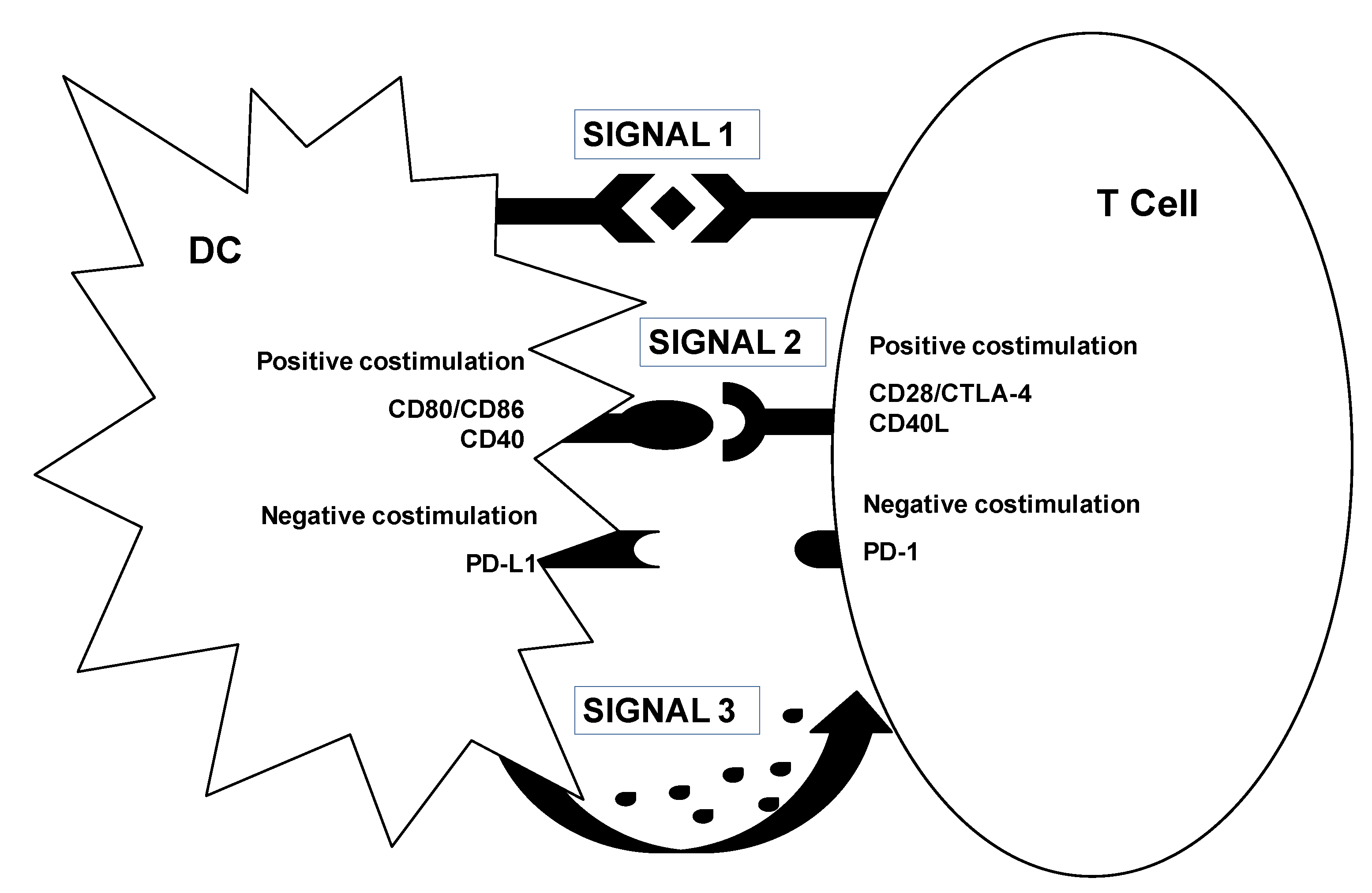
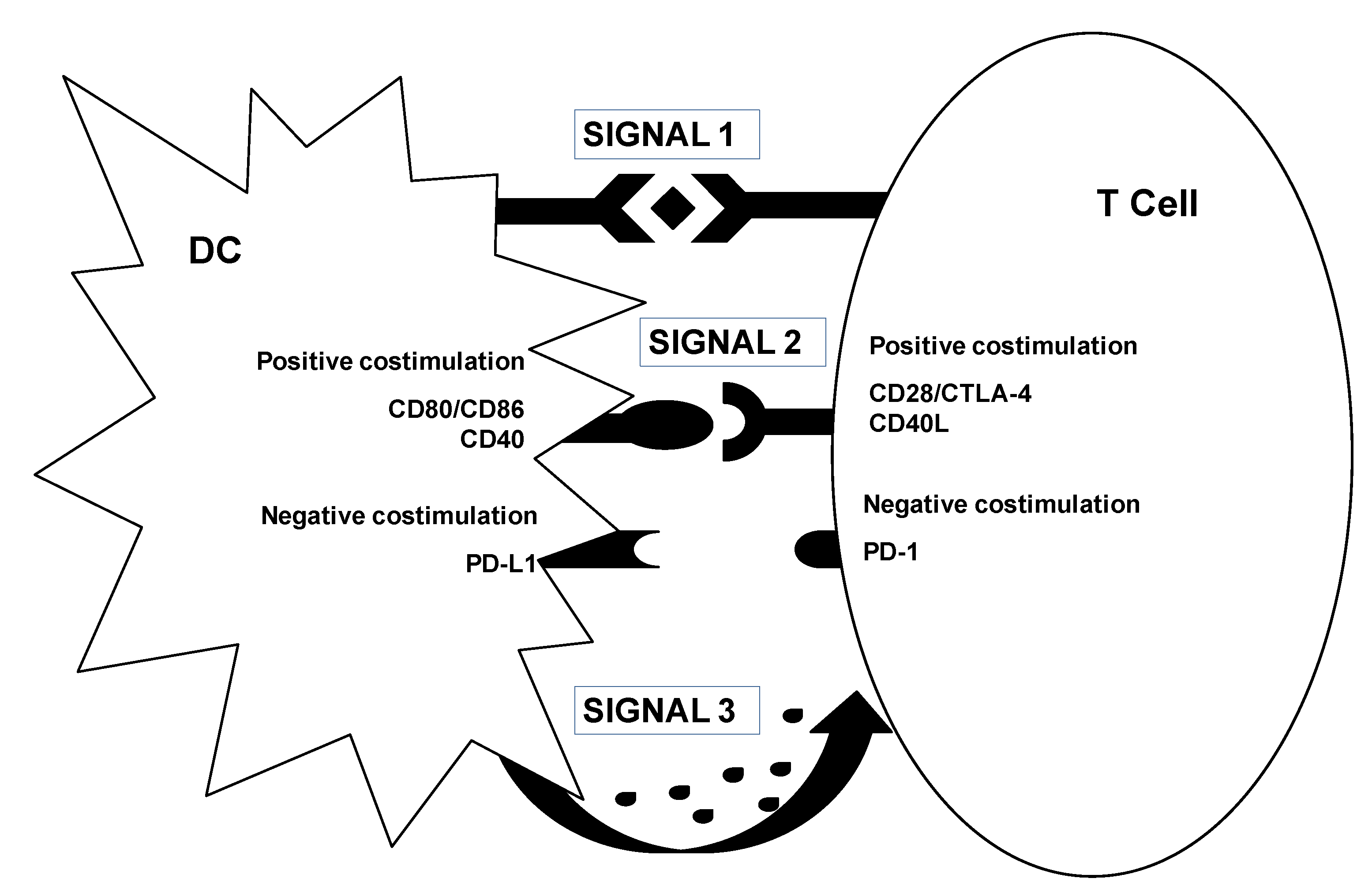
3.2. Genetic Modification of DCs for Cancer Immunotherapy
3.3. Myeloid-Derived Suppressor Cells. A New Target for Lentivector Immunotherapy?
4. Clinical Feasibility
5. Conclusions and Future Perspectives
Acknowledgments
Conflict of Interest
References
- Escors, D.; Breckpot, K. Lentiviral vectors in gene therapy: Their current status and future potential. Arch. Immunol. Ther. Exp. 2010, 58, 107–119. [Google Scholar] [CrossRef]
- Huang, X.; Guo, H.; Kang, J.; Choi, S.; Zhou, T.C.; Tammana, S.; Lees, C.J.; Li, Z.Z.; Milone, M.; Levine, B.L.; et al. Sleeping beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol. Ther. 2008, 16, 580–589. [Google Scholar] [CrossRef]
- Kitagawa, T.; Iwazawa, T.; Robbins, P.D.; Lotze, M.T.; Tahara, H. Advantages and limitations of particle-mediated transfection (gene gun) in cancer immuno-gene therapy using IL-10, IL-12 or B7-1 in murine tumor models. J. Gene Med. 2003, 5, 958–965. [Google Scholar] [CrossRef]
- Campos-Perez, J.; Rice, J.; Escors, D.; Collins, M.; Paterson, A.; Savelyeva, N.; Stevenson, F.K. DNA fusion vaccine designs to induce tumor-lytic CD8+ T-cell attack via the immunodominant cysteine-containing epitope of NY-ESO 1. Int. J. Cancer 2013. [Google Scholar] [CrossRef]
- Mitchell, M.S. Cancer vaccines, a critical review—Part II. Curr. Opin. Investig. Drugs 2002, 3, 150–158. [Google Scholar]
- Mitchell, M.S. Cancer vaccines, a critical review—Part I. Curr. Opin. Investig. Drugs 2002, 3, 140–149. [Google Scholar]
- Pachuk, C.J.; McCallus, D.E.; Weiner, D.B.; Satishchandran, C. DNA vaccines—Challenges in delivery. Curr. Opin. Mol. Ther. 2000, 2, 188–198. [Google Scholar]
- Kouraklis, G. Progress in cancer gene therapy. Acta Oncol. 1999, 38, 675–683. [Google Scholar]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar]
- Wang, H.; Kavanaugh, M.P.; North, R.A.; Kabat, D. Cell-surface receptor for ecotropic murine retroviruses is a basic amino-acid transporter. Nature 1991, 352, 729–731. [Google Scholar]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar]
- Wild, C.; Greenwell, T.; Matthews, T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res. Hum. Retroviruses 1993, 9, 1051–1053. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Haggerty, S.A.; Dempsey, M.P.; Sharova, N.; Adzhubel, A.; Spitz, L.; Lewis, P.F.; Goldfarb, D.; Emerman, M.; Stevenson, M. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature 1993, 365, 666–669. [Google Scholar]
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar]
- Palu, G.; Parolin, C.; Takeuchi, Y.; Pizzato, M. Progress with retroviral gene vectors. Rev. Med. Virol. 2000, 10, 185–202. [Google Scholar]
- Mann, R.; Mulligan, R.C.; Baltimore, D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 1983, 33, 153–159. [Google Scholar]
- He, Y.; Munn, D.; Falo, L.D., Jr. Recombinant lentivector as a genetic immunization vehicle for antitumor immunity. Expert Rev. Vaccines 2007, 6, 913–924. [Google Scholar]
- Wiznerowicz, M.; Trono, D. Harnessing HIV for therapy, basic research and biotechnology. Trends Biotechnol. 2005, 23, 42–47. [Google Scholar]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar]
- Yee, J.K.; Friedmann, T.; Burns, J.C. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 1994, 43, 99–112. [Google Scholar]
- Akkina, R.K.; Walton, R.M.; Chen, M.L.; Li, Q.X.; Planelles, V.; Chen, I.S. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J. Virol. 1996, 70, 2581–2585. [Google Scholar]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar]
- Bokhoven, M.; Stephen, S.L.; Knight, S.; Gevers, E.F.; Robinson, I.C.; Takeuchi, Y.; Collins, M.K. Insertional gene activation by lentiviral and gammaretroviral vectors. J. Virol. 2009, 83, 283–294. [Google Scholar]
- He, Y.; Zhang, J.; Donahue, C.; Falo, L.D., Jr. Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity 2006, 24, 643–656. [Google Scholar]
- He, Y.; Falo, L.D. Induction of T cell immunity by cutaneous genetic immunization with recombinant lentivector. Immunol. Res. 2006, 36, 101–117. [Google Scholar]
- Barouch, D.H.; Nabel, G.J. Adenovirus vector-based vaccines for human immunodeficiency virus type 1. Hum. Gene Ther. 2005, 16, 149–156. [Google Scholar] [CrossRef]
- Arce, F.; Breckpot, K.; Stephenson, H.; Karwacz, K.; Ehrenstein, M.R.; Collins, M.; Escors, D. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011, 63, 84–95. [Google Scholar] [CrossRef]
- Dufait, I.; Liechtenstein, T.; Lanna, A.; Bricogne, C.; Laranga, R.; Padella, A.; Breckpot, K.; Escors, D. Retroviral and lentiviral vectors for the induction of immunological tolerance. Scientifica. 2012. [Google Scholar] [CrossRef]
- Arce, F.; Breckpot, K.; Collins, M.; Escors, D. Targeting lentiviral vectors for cancer immunotherapy. Curr. Cancer Ther. Rev. 2011, 7, 248–260. [Google Scholar] [CrossRef]
- Esslinger, C.; Chapatte, L.; Finke, D.; Miconnet, I.; Guillaume, P.; Levy, F.; MacDonald, H.R. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J. Clin. Invest. 2003, 111, 1673–1681. [Google Scholar]
- Breckpot, K.; Emeagi, P.; Dullaers, M.; Michiels, A.; Heirman, C.; Thielemans, K. Activation of immature monocyte-derived dendritic cells after transduction with high doses of lentiviral vectors. Hum. Gene Ther. 2007, 18, 536–546. [Google Scholar]
- Rossetti, M.; Gregori, S.; Hauben, E.; Brown, B.D.; Sergi, L.S.; Naldini, L.; Roncarolo, M.G. HIV-1-derived lentiviral vectors directly activate plasmacytoid dendritic cells, which in turn induce the maturation of myeloid dendritic cells. Hum. Gene Ther. 2011, 22, 177–188. [Google Scholar]
- Breckpot, K.; Escors, D.; Arce, F.; Lopes, L.; Karwacz, K.; van Lint, S.; Keyaerts, M.; Collins, M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636. [Google Scholar] [CrossRef]
- Pichlmair, A.; Diebold, S.S.; Gschmeissner, S.; Takeuchi, Y.; Ikeda, Y.; Collins, M.K.; Reis e Sousa, C. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via Toll-like receptor 9. J. Virol. 2007, 81, 539–547. [Google Scholar]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef]
- Hematti, P.; Hong, B.K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004, 2, e423. [Google Scholar] [CrossRef] [Green Version]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is stronly modulated by vector design and integration site selesction in a mouse model of HSC gene therapy. J. Clin. Invest. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Cesana, D.; Sgualdino, J.; Rudilosso, L.; Merella, S.; Naldini, L.; Montini, E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J. Clin. Invest. 2012, 122, 1667–1676. [Google Scholar] [CrossRef]
- Knight, S.; Bokhoven, M.; Collins, M.; Takeuchi, Y. Effect of the internal promoter on insertional gene activation by lentiviral vectors with an intact HIV long terminal repeat. J. Virol. 2010, 84, 4856–4859. [Google Scholar] [CrossRef]
- Ginn, S.L.; Liao, S.H.; Dane, A.P.; Hu, M.; Hyman, J.; Finnie, J.W.; Zheng, M.; Cavazzana-Calvo, M.; Alexander, S.I.; Thrasher, A.J.; et al. Lymphomagenesis in SCID-X1 mice following lentivirus-mediated phenotype correction independent of insertional mutagenesis and gammac overexpression. Mol. Ther. 2010, 18, 965–976. [Google Scholar] [CrossRef]
- Maruggi, G.; Porcellini, S.; Facchini, G.; Perna, S.K.; Cattoglio, C.; Sartori, D.; Ambrosi, A.; Schambach, A.; Baum, C.; Bonini, C.; et al. Transcriptional enhancers induce insertional gene deregulation independently from the vector type and design. Mol. Ther. 2009, 17, 851–856. [Google Scholar] [CrossRef]
- Kustikova, O.S.; Schiedlmeier, B.; Brugman, M.H.; Stahlhut, M.; Bartels, S.; Li, Z.; Baum, C. Cell-intrinsic and vector-related properties cooperate to determine the incidence and consequences of insertional mutagenesis. Mol. Ther. 2009, 17, 1537–1547. [Google Scholar] [CrossRef]
- Arumugam, P.I.; Higashimoto, T.; Urbinati, F.; Modlich, U.; Nestheide, S.; Xia, P.; Fox, C.; Corsinotti, A.; Baum, C.; Malik, P. Genotoxic potential of lineage-specific lentivirus vectors carrying the beta-globin locus control region. Mol. Ther. 2009, 17, 1929–1937. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Morizono, K.; Xie, Y.; Ringpis, G.E.; Johnson, M.; Nassanian, H.; Lee, B.; Wu, L.; Chen, I.S. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 2005, 11, 346–352. [Google Scholar] [CrossRef]
- Morizono, K.; Pariente, N.; Xie, Y.; Chen, I.S. Redirecting lentiviral vectors by insertion of integrin-tageting peptides into envelope proteins. J. Gene Med. 2009, 11, 549–558. [Google Scholar] [CrossRef]
- Yang, L.; Bailey, L.; Baltimore, D.; Wang, P. Targeting lentiviral vectors to specific cell types in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 11479–11484. [Google Scholar]
- Escors, D.; Breckpot, K.; Arce, F.; Kochan, G.; Stephenson, H. Lentiviral vectors and gene therapy, 1st ed.; Springer: Basel, Switzerland, 2012. [Google Scholar]
- He, Y.; Falo, L.D.J. Lentivirus as a potent and mechanistically distinct vector for genetic immunization. Curr. Opin. Mol. Ther. 2007, 9, 439–446. [Google Scholar]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat. Med. 2013. [Google Scholar] [CrossRef]
- Breckpot, K.; Escors, D. Dendritic cells for active anti-cancer immunotherapy: Targeting activation pathways through genetic modification. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 328–343. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, Y.; Mi, M.; Guevara-Patino, J.; Munn, D.H.; Fu, N.; He, Y. Lentivector immunization stimulates potent CD8 T cell responses against melanoma self-antigen tyrosinase-related protein 1 and generates antitumor immunity in mice. J. Immunol. 2009, 182, 5960–5969. [Google Scholar] [CrossRef]
- Frecha, C.; Costa, C.; Negre, D.; Gauthier, E.; Russell, S.J.; Cosset, F.L.; Verhoeyen, E. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood 2008, 112, 4843–4852. [Google Scholar] [CrossRef]
- Perro, M.; Tsang, J.; Xue, S.A.; Escors, D.; Cesco-Gaspere, M.; Pospori, C.; Gao, L.; Hart, D.; Collins, M.; Stauss, H.; et al. Generation of multi-functional antigen-specific human T-cells by lentiviral TCR gene transfer. Gene Ther. 2010, 17, 721–732. [Google Scholar] [CrossRef]
- Bobisse, S.; Zanovello, P.; Rosato, A. T-cell receptor gene transfer by lentiviral vectors in adoptive cell therapy. Expert Opin. Biol. Ther. 2007, 7, 893–906. [Google Scholar] [CrossRef]
- Coccoris, M.; Straetemans, T.; Govers, C.; Lamers, C.; Sleijfer, S.; Debets, R. T cell receptor (TCR) gene therapy to treat melanoma: Lessons from clinical and preclinical studies. Expert Opin. Biol. Ther. 2010, 10, 547–562. [Google Scholar] [CrossRef]
- Thomas, S.; Stauss, H.J.; Morris, E.C. Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology 2010, 129, 10–17. [Google Scholar]
- Johnson, L.A.; Heemskerk, B.; Powell, D.J.J.; Cohen, C.J.; Morgan, R.A.; Dudley, M.E.; Robbins, P.F.; Rosenberg, S.A. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J. Immunol. 2006, 177, 6548–6559. [Google Scholar]
- Park, T.S.; Rosenberg, S.A.; Morgan, R.A. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011, 29, 550–557. [Google Scholar] [CrossRef]
- Escors, D.; Lopes, L.; Lin, R.; Hiscott, J.; Akira, S.; Davis, R.J.; Collins, M.K. Targeting dendritic cell signalling to regulate the response to immunisation. Blood 2008, 111, 3050–3061. [Google Scholar]
- Gross, G.; Waks, T.; Eshar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar]
- Kochenderfer, J.N.; Yu, Z.; Frasheri, D.; Restifo, N.P.; Rosenberg, S.A. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 2010, 116, 3875–3886. [Google Scholar]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Lamers, C.H.; Seijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.L.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgenic rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar]
- Lamers, C.H.; Willemsen, R.; van Elzakker, P.; van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar]
- Bobisse, S.; Rondina, M.; Merlo, A.; Tisato, V.; Mandruzzato, S.; Amendola, M.; Naldini, L.; Willemsen, R.A.; Debets, R.; Zanovello, P.; et al. Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res. 2009, 69, 9385–9394. [Google Scholar]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Dufait, I.; Lanna, A.; Breckpot, K.; Escors, D. Modulating co-stimulation during antigen presentation to enhance cancer immunotherapy. Immunol. Endocr. Metab. Agents Med. Chem. 2012, 12, 224–235. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Dufait, I.; Bricogne, C.; Lanna, A.; Pen, J.; Breckpot, K.; Escors, D. PD-L1/PD-1 co-stimulation, a brake for T cell activation and a T cell differentiation signal. J. Clin. Cell. Immunol. 2012. [Google Scholar] [CrossRef]
- Rowe, H.M.; Lopes, L.; Brown, N.; Efklidou, S.; Smallie, T.; Karrar, S.; Kaye, P.M.; Collins, M.K. Expression of vFLIP in a lentiviral vaccine vector activates NF-{kappa}B, matures dendritic cells, and increases CD8+ T-cell responses. J. Virol. 2009, 83, 1555–1562. [Google Scholar] [CrossRef]
- Karwacz, K.; Bricogne, C.; Macdonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8(+) T cells. EMBO Mol. Med. 2011, 3, 581–592. [Google Scholar] [CrossRef]
- Karwacz, K.; Mukherjee, S.; Apolonia, L.; Blundell, M.P.; Bouma, G.; Escors, D.; Collins, M.K.; Thrasher, A.J. Nonintegrating lentivector vaccines stimulate prolonged T-cell and antibody responses and are effective in tumor therapy. J. Virol. 2009, 83, 3094–3103. [Google Scholar] [CrossRef]
- Collins, M.K.; Cerundolo, V. Gene therapy meets vaccine development. Trends Biotechnol. 2004, 22, 623–626. [Google Scholar] [CrossRef]
- Esslinger, C.; Romero, P.; MacDonald, H.R. Efficient transduction of dendritic cells and induction of a T-cell response by third-generation lentivectors. Hum. Gene Ther. 2002, 13, 1091–1100. [Google Scholar] [CrossRef]
- Gruber, A.; Kan-Mitchell, J.; Kuhen, K.L.; Mukai, T.; Wong-Staal, F. Dendritic cells transduced by multiply deleted HIV-1 vectors exhibit normal phenotypes and functions and elicit an HIV-specific cytotoxic T-lymphocyte response in vitro. Blood 2000, 96, 1327–1333. [Google Scholar]
- He, Y.; Zhang, J.; Mi, Z.; Robbins, P.; Falo, L.D., Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J. Immunol. 2005, 174, 3808–3817. [Google Scholar]
- Breckpot, K.; Dullaers, M.; Bonehill, A.; van Meirvenne, S.; Heirman, C.; de Greef, C.; van der Bruggen, P.; Thielemans, K. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J. Gene Med. 2003, 5, 654–667. [Google Scholar] [CrossRef]
- Dyall, J.; Latouche, J.B.; Schnell, S.; Sadelain, M. Lentivirus-transduced human monocyte-derived dendritic cells efficiently stimulate antigen-specific cytotoxic T lymphocytes. Blood 2001, 97, 114–121. [Google Scholar] [CrossRef]
- Karwacz, K.; Arce, F.; Bricogne, C.; Kochan, G.; Escors, D. PD-L1 co-stimulation, ligand-induced TCR down-modulation and anti-tumor immunotherapy. Oncoimmunology 2012, 1, 86–88. [Google Scholar] [CrossRef]
- Zarei, S.; Leuba, F.; Arrighi, J.F.; Hauser, C.; Piguet, V. Transduction of dendritic cells by antigen-encoding lentiviral vectors permits antigen processing and MHC class I-dependent presentation. J. Allergy Clin. Immunol. 2002, 109, 988–994. [Google Scholar] [CrossRef]
- Akazawa, T.; Shingai, M.; Sasai, M.; Ebihara, T.; Inoue, N.; Matsumoto, M.; Seya, T. Tumor immunotherapy using bone marrow-derived dendritic cells overexpressing Toll-like receptor adaptors. FEBS Lett. 2007, 581, 3334–3340. [Google Scholar] [CrossRef]
- Bagneris, C.; Ageichik, A.V.; Cronin, N.; Wallace, B.; Collins, M.; Boshoff, C.; Waksman, G.; Barrett, T. Crystal structure of a vFlip-IKKgamma complex: Insights into viral activation of the IKK signalosome. Mol. Cell 2008, 30, 620–631. [Google Scholar] [CrossRef]
- Shimizu, A.; Baratchian, M.; Takeuchi, Y.; Escors, D.; Macdonald, D.; Barrett, T.; Bagneris, C.; Collins, M.; Noursadeghi, M. Kaposi’s sarcoma-associated herpesvirus vFLIP and human T cell lymphotropic virus type 1 Tax oncogenic proteins activate IkappaB kinase subunit gamma by different mechanisms independent of the physiological cytokine-induced pathways. J. Virol. 2011, 85, 7444–7448. [Google Scholar] [CrossRef]
- Efklidou, S.; Bailey, R.; Field, N.; Noursadeghi, M.; Collins, M.K. vFLIP from KSHV inhibits anoikis of primary endothelial cells. J. Cell Sci. 2008, 121, 450–457. [Google Scholar] [CrossRef]
- Field, N.; Low, W.; Daniels, M.; Howell, S.; Daviet, L.; Boshoff, C.; Collins, M. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 2003, 116, 3721–3728. [Google Scholar] [CrossRef]
- VandenDriessche, T.; Thorrez, L.; Naldini, L.; Follenzi, A.; Moons, L.; Berneman, Z.; Collen, D.; Chuah, M.K. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood 2002, 100, 813–822. [Google Scholar] [CrossRef]
- Goold, H.D.; Escors, D.; Conlan, T.J.; Chakraverty, R.; Bennett, C.L. Conventional dendritic cells are required for the activation of helper-dependent CD8 T cell responses to a model antigen after cutaneous vaccination with lentiviral vectors. J. Immunol. 2011, 186, 4565–4572. [Google Scholar] [CrossRef]
- Dullaers, M.; van Meirvenne, S.; Heirman, C.; Straetman, L.; Bonehill, A.; Aerts, J.L.; Thielemans, K.; Breckpot, K. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther. 2006, 13, 630–640. [Google Scholar] [CrossRef]
- Chapatte, L.; Colombetti, S.; Cerottini, J.C.; Levy, F. Efficient induction of tumor antigen-specific CD8+ memory T cells by recombinant lentivectors. Cancer Res. 2006, 66, 1155–1160. [Google Scholar] [CrossRef]
- Arce, F.; Rowe, H.; Lopes, L.; Escors, D.; Chain, B.; Collins, M.K. Sustained antigen presentation after lentiviral immunization. Human Gene Ther. 2008, 19, 1141–1142. [Google Scholar]
- Gruschwitz, M.S.; Vieth, G. Up-regulation of class II major histocompatibility complex and intercellular adhesion molecule 1 expression on scleroderma fibroblasts and endothelial cells by interferon-gamma and tumor necrosis factor alpha in the early disease stage. Arthritis Rheum. 1997, 40, 540–550. [Google Scholar] [CrossRef]
- Annoni, A.; Brown, B.D.; Cantore, A.; Sergi, L.S.; Naldini, L.; Roncarolo, M.G. In vivo delivery of a microRNA-regulated transgene induces antigen-specific regulatory T cells and promotes immunologic tolerance. Blood 2009, 114, 5152–5161. [Google Scholar] [CrossRef]
- Brown, B.D.; Venneri, M.A.; Zingale, A.; Sergi Sergi, L.; Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 2006, 12, 585–591. [Google Scholar] [CrossRef]
- Brown, B.D.; Sitia, G.; Annoni, A.; Hauben, E.; Sergi Sergi, L.; Zingale, A.; Roncarolo, M.G.; Guidotti, L.G.; Naldini, L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood 2006, 109, 2797–2805. [Google Scholar]
- Bronte, V.; Serafini, P.; Apolloni, E.; Zanovello, P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J. Immunother. 2001, 24, 431–446. [Google Scholar] [CrossRef]
- Bronte, V.; Apolloni, E.; Cabrelle, A.; Ronca, R.; Serafini, P.; Zamboni, P.; Restifo, N.P.; Zanovello, P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood 2000, 96, 3838. [Google Scholar]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Thrasher, A.J.; Gaspar, H.B.; Baum, C.; Modlich, U.; Schambach, A.; Candotti, F.; Otsu, M.; Sorrentino, B.; Scobie, L.; Cameron, E.; et al. Gene therapy: X-SCID transgene leukaemogenicity. Nature 2006, 443, E5–E6. [Google Scholar]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi Sergi, L.; Benedicenti, F.; Ambrosi, A.; di Serio, C.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar]
- Moiani, A.; Paleari, Y.; Sartori, D.; Mezzadra, R.; Miccio, A.; Cattoglio, C.; Cocchiarella, F.; Lidonnici, M.R.; Ferrari, G.; Mavilio, F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Invest. 2012, 122, 1653–1666. [Google Scholar] [CrossRef] [Green Version]
- Yanez-Munoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.E.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef]
- Negri, D.R.; Michelini, Z.; Baroncelli, S.; Spada, M.; Vendetti, S.; Buffa, V.; Bona, R.; Leone, P.; Klotman, M.E.; Cara, A. Successful immunization with a single injetion of non-integrating lentiviral vector. Mol. Ther. 2007, 15, 1716–1723. [Google Scholar] [CrossRef]
- Coutant, F.; Sanchez David, R.Y.; Felix, T.; Boulay, A.; Caleechurn, L.; Souque, P.; Thouvenot, C.; Bourgouin, C.; Beignon, A.S.; Charneau, P. A nonintegrative lentiviral vector-based vaccine provides long-term sterile protection against malaria. PLoS One 2012, 7, e48644. [Google Scholar] [CrossRef]
- Wang, G.P.; Levine, B.L.; Binder, G.K.; Berry, C.C.; Malani, N.; McGarrity, G.; Tebas, P.; June, C.H.; Bushman, F.D. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol. Ther. 2009, 17, 844–850. [Google Scholar] [CrossRef]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R.; et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Binder-Scholl, G.; Mukherjee, R.; Brady, T.; Rebello, T.; Humeau, L.; Kalos, M.; Papasavvas, E.; Montaner, L.J.; et al. Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 2013, 121, 1524–1533. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Rowe, H.M.; Lopes, L.; Ikeda, Y.; Bailey, R.; Barde, I.; Zenke, M.; Chain, B.M.; Collins, M.K. Immunization with a lentiviral vector stimulates both CD4 and CD8 T cell responses to an ovalbumin transgene. Mol. Ther. 2006, 13, 310–319. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liechtenstein, T.; Perez-Janices, N.; Escors, D. Lentiviral Vectors for Cancer Immunotherapy and Clinical Applications. Cancers 2013, 5, 815-837. https://doi.org/10.3390/cancers5030815
Liechtenstein T, Perez-Janices N, Escors D. Lentiviral Vectors for Cancer Immunotherapy and Clinical Applications. Cancers. 2013; 5(3):815-837. https://doi.org/10.3390/cancers5030815
Chicago/Turabian StyleLiechtenstein, Therese, Noemi Perez-Janices, and David Escors. 2013. "Lentiviral Vectors for Cancer Immunotherapy and Clinical Applications" Cancers 5, no. 3: 815-837. https://doi.org/10.3390/cancers5030815
APA StyleLiechtenstein, T., Perez-Janices, N., & Escors, D. (2013). Lentiviral Vectors for Cancer Immunotherapy and Clinical Applications. Cancers, 5(3), 815-837. https://doi.org/10.3390/cancers5030815