Dynamic Length Changes of Telomeres and Their Nuclear Organization in Chronic Myeloid Leukemia

Abstract
:1. Telomere Overview
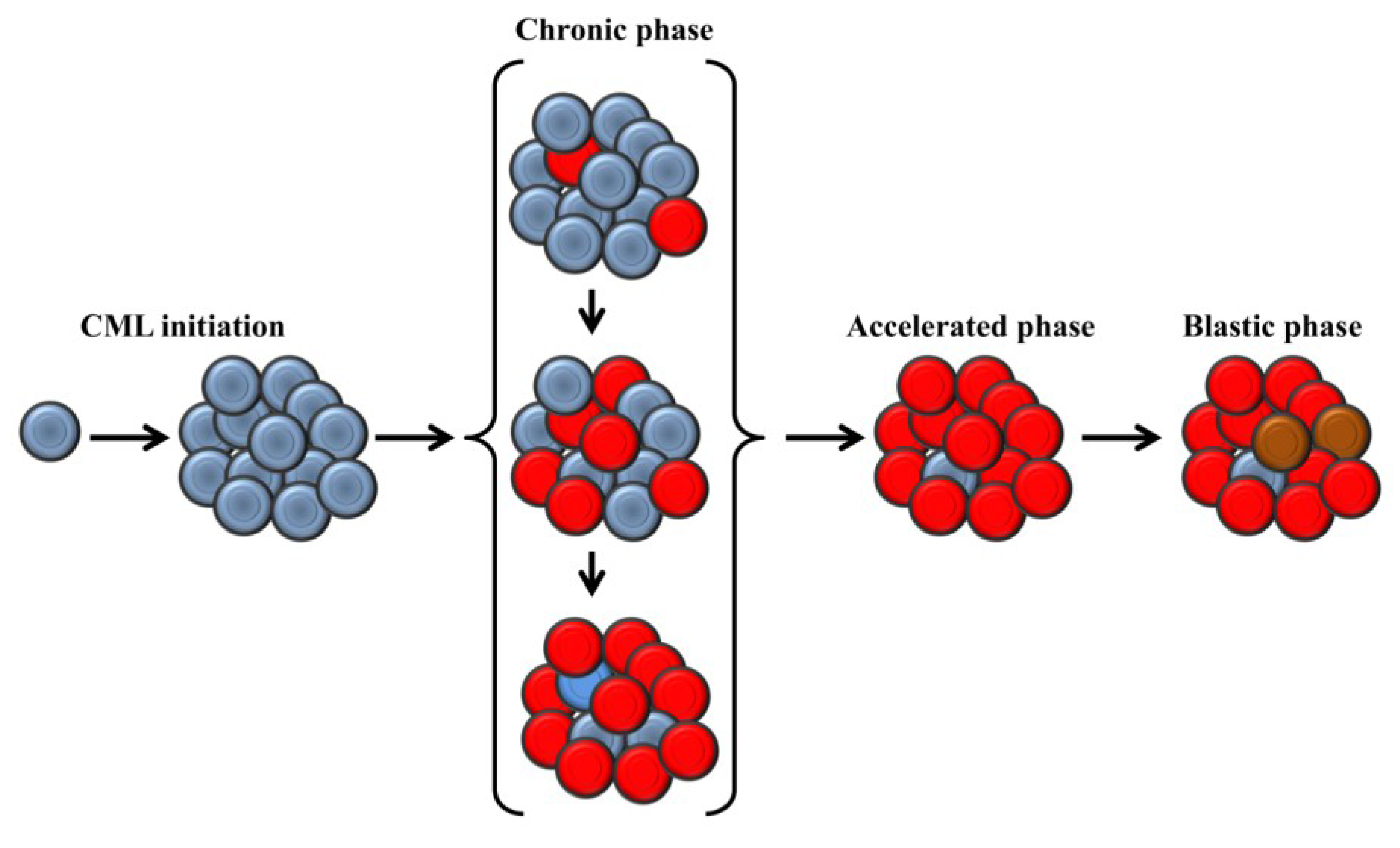
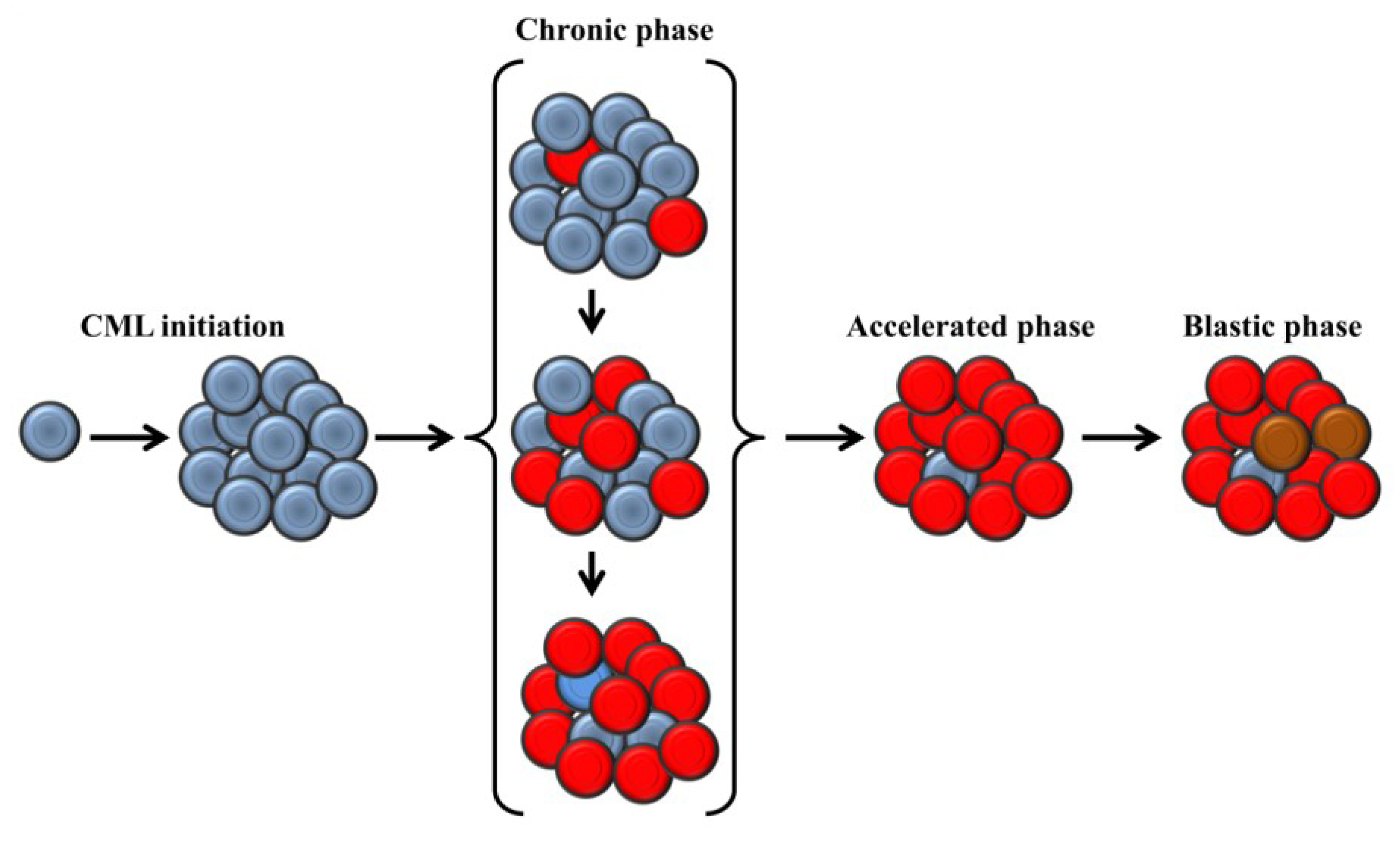
2. Clinical Presentation and Molecular Biology of Chronic Myeloid Leukemia
3. Telomere Length in Chronic Myeloid Leukemia
3.1. Average Length of Telomeres

{kind=link}
| Techniques | Materials | Advantages | Disadvantages | |
|---|---|---|---|---|
| Measurements of average telomeres | TRF | DNA | Comparison between studies | Large amount of DNA Labor intense |
| Flow FISH | Interphase cells | Can measure different cell subsets | Require cytological preparation | |
| Measurements of individual telomeres | Q-FISH | Metaphases | Can identify all individual telomeres | Requires metaphases |
| 3D telomere FISH | Interphase cells | Assess the number, intensity and the position of individual telomeres in interphase nuclei | Cannot identify individual telomeres |
3.2. Length of Individual Telomeres
4. Mechanisms of Telomere Maintenance
4.1. Telomerase
4.2. Alternative Lengthening of Telomere
5. Telomeric Nuclear Organization
6. Conclusions
Acknowledgements
Conflicts of Interest
References
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef]
- Samassekou, O.; Gadji, M.; Drouin, R.; Yan, J. Sizing the ends: Normal length of human telomeres. Ann. Anat. 2010, 192, 284–291. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Vaziri, H.; Schachter, F.; Uchida, I.; Wei, L.; Zhu, X.; Effros, R.; Cohen, D.; Harley, C.B. Loss of telomeric DNA during aging of normal and trisomy 21 human lymphocytes. Am. J. Hum. Genet. 1993, 52, 661–667. [Google Scholar]
- Vaziri, H.; Dragowska, W.; Allsopp, R.C.; Thomas, T.E.; Harley, C.B.; Lansdorp, P.M. Evidence for a mitotic clock in human hematopoietic stem cells: Loss of telomeric DNA with age. Proc. Natl. Acad. Sci. USA 1994, 91, 9857–9860. [Google Scholar]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. The telomere terminal transferase of tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987, 51, 887–898. [Google Scholar] [CrossRef]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Abdallah, P.; Luciano, P.; Runge, K.W.; Lisby, M.; Geli, V.; Gilson, E.; Teixeira, M.T. A two-step model for senescence triggered by a single critically short telomere. Nat. Cell Biol. 2009, 11, 988–993. [Google Scholar] [CrossRef]
- Di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Ouellette, M.M.; Liao, M.; Herbert, B.S.; Johnson, M.; Holt, S.E. Subsenescent telomere lengths in fibroblasts immortalized by limiting amounts of telomerase. J. Biol. Chem. 2000, 275, 10072–10076. [Google Scholar]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar]
- Wu, G.; Lee, W.H.; Chen, P.L. NBS1 and TRF1 colocalize at promyelocytic leukemia bodies during late S/G2 phases in immortalized telomerase-negative cells. Implication of NBS1 in alternative lengthening of telomeres. J. Biol. Chem. 2000, 275, 30618–30622. [Google Scholar] [CrossRef]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Muntoni, A.; Neumann, A.A.; Hills, M.; Reddel, R.R. Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum. Mol. Genet. 2009, 18, 1017–1027. [Google Scholar] [CrossRef]
- Wang, R.C.; Smogorzewska, A.; de Lange, T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 2004, 119, 355–368. [Google Scholar] [CrossRef]
- Henson, J.D.; Reddel, R.R. Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Bailey, S.M.; Brenneman, M.A.; Goodwin, E.H. Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 2004, 32, 3743–3751. [Google Scholar] [CrossRef]
- Londono-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef]
- Svenson, U.; Roos, G. Telomere length as a biological marker in malignancy. Biochim. Biophys. Acta 2009, 1792, 317–323. [Google Scholar] [CrossRef]
- Avigad, S.; Naumov, I.; Ohali, A.; Jeison, M.; Berco, G.H.; Mardoukh, J.; Stark, B.; Ash, S.; Cohen, I.J.; Meller, I.; et al. Short telomeres: A novel potential predictor of relapse in ewing sarcoma. Clin. Cancer Res. 2007, 13, 5777–5783. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Baumgartner, K.B.; Bisoffi, M.; Baumgartner, R.N.; Griffith, J.K. Telomere DNA content predicts breast cancer-free survival interval. Clin. Cancer Res. 2007, 13, 7037–7043. [Google Scholar] [CrossRef]
- Fordyce, C.A.; Heaphy, C.M.; Joste, N.E.; Smith, A.Y.; Hunt, W.C.; Griffith, J.K. Association between cancer-free survival and telomere DNA content in prostate tumors. J. Urol. 2005, 173, 610–614. [Google Scholar] [CrossRef]
- Mai, S. Initiation of telomere-mediated chromosomal rearrangements in cancer. J. Cell. Biochem. 2010, 109, 1095–1102. [Google Scholar]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.; Moshir, S.; Mougey, V.; et al. C-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 9613–9618. [Google Scholar] [CrossRef]
- Knecht, H.; Bruderlein, S.; Mai, S.; Moller, P.; Sawan, B. 3D structural and functional characterization of the transition from hodgkin to reed-sternberg cells. Ann. Anat. 2010, 192, 302–308. [Google Scholar] [CrossRef]
- Gadji, M.; Fortin, D.; Tsanaclis, A.M.; Garini, Y.; Katzir, N.; Wienburg, Y.; Yan, J.; Klewes, L.; Klonisch, T.; Drouin, R.; et al. Three-dimensional nuclear telomere architecture is associated with differential time to progression and overall survival in glioblastoma patients. Neoplasia 2010, 12, 183–191. [Google Scholar]
- Gadji, M.; Awe, J.A.; Rodrigues, P.; Kumar, R.; Houston, D.S.; Klewes, L.; Dieye, T.N.; Rego, E.M.; Passetto, R.F.; de Oliveira, F.M.; et al. Profiling three-dimensional nuclear telomeric architecture of myelodysplastic syndromes and acute myeloid leukemia defines patient subgroups. Clin. Cancer Res. 2012, 18, 3293–3304. [Google Scholar] [CrossRef]
- Klewes, L.; Hobsch, C.; Katzir, N.; Rourke, D.; Garini, Y.; Mai, S. Novel automated three-dimensional genome scanning based on the nuclear architecture of telomeres. Cytometry A 2011, 79, 159–166. [Google Scholar]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.; Chuang, A.Y.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytometry A 2005, 67, 144–150. [Google Scholar]
- Mai, S.; Garini, Y. The significance of telomeric aggregates in the interphase nuclei of tumor cells. J. Cell. Biochem. 2006, 97, 904–915. [Google Scholar] [CrossRef]
- Lamm, S.H.; Engel, A.; Joshi, K.P.; Byrd, D.M., 3rd.; Chen, R. Chronic myelogenous leukemia and benzene exposure: A systematic review and meta-analysis of the case-control literature. Chem. Biol. Interact. 2009, 182, 93–97. [Google Scholar] [CrossRef]
- Lichtman, M.A. Is there an entity of chemically induced BCR-ABL-positive chronic myelogenous leukemia? Oncologist 2008, 13, 645–654. [Google Scholar]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J. Clin. Invest. 2010, 120, 2254–2264. [Google Scholar] [CrossRef]
- Druker, B.J. Translation of the philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar] [CrossRef]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Stone, J.; de Lange, T.; Ramsay, G.; Jakobovits, E.; Bishop, J.M.; Varmus, H.; Lee, W. Definition of regions in human C-Myc that are Involved in transformation and nuclear localization. Mol. Cell Biol. 1987, 7, 1697–1709. [Google Scholar]
- Quintas-Cardama, A.; Cortes, J. Molecular biology of BCR-ABL1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef]
- Mitelman, F.; Levan, G.; Nilsson, P.G.; Brandt, L. Non-random karyotypic evolution in chronic myeloid leukemia. Int. J. Cancer 1976, 18, 24–30. [Google Scholar] [CrossRef]
- Lawler, S.D.; O’Malley, F.; Lobb, D.S. Chromosome banding studies in philadelphia chromosome positive myeloid leukaemia. Scand. J. Haematol. 1976, 17, 17–28. [Google Scholar] [CrossRef]
- Oehler, V.G.; Yeung, K.Y.; Choi, Y.E.; Bumgarner, R.E.; Raftery, A.E.; Radich, J.P. The derivation of diagnostic markers of chronic myeloid leukemia progression from microarray data. Blood 2009, 114, 3292–3298. [Google Scholar] [CrossRef]
- Samassekou, O.; Ntwari, A.; Hebert, J.; Yan, J. Individual telomere lengths in chronic myeloid leukemia. Neoplasia 2009, 11, 1146–1154. [Google Scholar]
- Brummendorf, T.H.; Holyoake, T.L.; Rufer, N.; Barnett, M.J.; Schulzer, M.; Eaves, C.J.; Eaves, A.C.; Lansdorp, P.M. Prognostic implications of differences in telomere length between normal and malignant cells from patients with chronic myeloid leukemia measured by flow cytometry. Blood 2000, 95, 1883–1890. [Google Scholar]
- Brummendorf, T.H.; Ersoz, I.; Hartmann, U.; Bartolovic, K.; Balabanov, S.; Wahl, A.; Paschka, P.; Kreil, S.; Lahaye, T.; Berger, U.; et al. Telomere length in peripheral blood granulocytes reflects response to treatment with imatinib in patients with chronic myeloid leukemia. Blood 2003, 101, 375–376. [Google Scholar] [CrossRef]
- Drummond, M.; Lennard, A.; Brummendorf, T.; Holyoake, T. Telomere shortening correlates with prognostic score at diagnosis and proceeds rapidly during progression of chronic myeloid leukemia. Leuk. Lymphoma 2004, 45, 1775–1781. [Google Scholar] [CrossRef]
- Boultwood, J.; Fidler, C.; Shepherd, P.; Watkins, F.; Snowball, J.; Haynes, S.; Kusec, R.; Gaiger, A.; Littlewood, T.J.; Peniket, A.J.; et al. Telomere length shortening is associated with disease evolution in chronic myelogenous leukemia. Am. J. Hematol. 1999, 61, 5–9. [Google Scholar] [CrossRef]
- Keller, G.; Brassat, U.; Braig, M.; Heim, D.; Wege, H.; Brummendorf, T.H. Telomeres and telomerase in chronic myeloid leukaemia: impact for pathogenesis, disease progression and targeted therapy. Hematol. Oncol. 2009, 27, 123–129. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef]
- Campbell, L.J.; Fidler, C.; Eagleton, H.; Peniket, A.; Kusec, R.; Gal, S.; Littlewood, T.J.; Wainscoat, J.S.; Boultwood, J. hTERT, the catalytic component of telomerase, is downregulated in the haematopoietic stem cells of patients with chronic myeloid leukaemia. Leukemia 2006, 20, 671–679. [Google Scholar] [CrossRef]
- Lobetti-Bodoni, C.; Ferrero, D.; Genuardi, E.; Passera, R.; Bernocco, E.; Sia, D.; Grignani, G.; Crisa, E.; Monitillo, L.; Rocci, A.; et al. Telomere loss in philadelphia-negative hematopoiesis after successful treatment of chronic myeloid leukemia: Evidence for premature aging of the myeloid compartment. Mech. Ageing Dev. 2012, 133, 479–488. [Google Scholar] [CrossRef]
- Samassekou, O.; Li, H.; Hébert, J.; Ntwari, A.; Wang, H.; Cliche, C.G.; Bouchard, E.; Huang, S.; Yan, J. Chromosome-arm-specific long telomeres: A New clonal event in primary chronic myelogenous leukemia cells. Neoplasia 2011, 13, 550–560. [Google Scholar]
- Martens, U.M.; Zijlmans, J.M.; Poon, S.S.; Dragowska, W.; Yui, J.; Chavez, E.A.; Ward, R.K.; Lansdorp, P.M. Short telomeres on human chromosome 17p. Nat. Genet. 1998, 18, 76–80. [Google Scholar]
- Perner, S.; Bruderlein, S.; Hasel, C.; Waibel, I.; Holdenried, A.; Ciloglu, N.; Chopurian, H.; Nielsen, K.V.; Plesch, A.; Hogel, J.; et al. Quantifying telomere lengths of human individual chromosome arms by centromere-calibrated fluorescence in situ hybridization and digital imaging. Am. J. Pathol. 2003, 163, 1751–1756. [Google Scholar] [CrossRef]
- Mitelman, F. The cytogenetic scenario of chronic myeloid leukemia. Leuk. Lymphoma 1993, 11, 11–15. [Google Scholar] [CrossRef]
- Krejci, K.; Stentoft, J.; Koch, J. Molecular cytogenetics investigation of the telomeres in a case of philadelphia positive B-all with a single telomere expansion. Neoplasia 1999, 1, 492–497. [Google Scholar]
- Baur, J.A.; Zou, Y.; Shay, J.W.; Wright, W.E. Telomere position effect in human cells. Science 2001, 292, 2075–2077. [Google Scholar] [CrossRef]
- Koering, C.E.; Pollice, A.; Zibella, M.P.; Bauwens, S.; Puisieux, A.; Brunori, M.; Brun, C.; Martins, L.; Sabatier, L.; Pulitzer, J.F.; et al. Human telomeric position effect is determined by chromosomal context and telomeric chromatin integrity. EMBO Rep. 2002, 3, 1055–1061. [Google Scholar] [CrossRef]
- Samassekou, O.; Malina, A.; Hebert, J.; Yan, J. Presence of alternative lengthening of telomeres associated circular extrachromosome telomere repeats in primary leukemia cells of chronic myeloid leukemia. J. Hematol. Oncol. 2013. [Google Scholar] [CrossRef]
- Broccoli, D.; Smogorzewska, A.; Chong, L.; de Lange, T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 1997, 17, 231–235. [Google Scholar] [CrossRef]
- Ohyashiki, K.; Ohyashiki, J.H.; Iwama, H.; Hayashi, S.; Shay, J.W.; Toyama, K. Telomerase activity and cytogenetic changes in chronic myeloid leukemia with disease progression. Leukemia 1997, 11, 190–194. [Google Scholar]
- Engelhardt, M.; Mackenzie, K.; Drullinsky, P.; Silver, R.T.; Moore, M.A. Telomerase activity and telomere length in acute and chronic leukemia, pre- and post-ex vivo culture. Cancer Res. 2000, 60, 610–617. [Google Scholar]
- Ohyashiki, K.; Iwama, H.; Tauchi, T.; Shimamoto, T.; Hayashi, S.; Ando, K.; Kawakubo, K.; Ohyashiki, J.H. Telomere dynamics and genetic instability in disease progression of chronic myeloid leukemia. Leuk. Lymphoma 2000, 40, 49–56. [Google Scholar] [CrossRef]
- Uziel, O.; Fenig, E.; Nordenberg, J.; Beery, E.; Reshef, H.; Sandbank, J.; Birenbaum, M.; Bakhanashvili, M.; Yerushalmi, R.; Luria, D.; et al. Imatinib mesylate (Gleevec) downregulates telomerase activity and inhibits proliferation in telomerase-expressing cell lines. Br. J. Cancer 2005, 92, 1881–1891. [Google Scholar] [CrossRef]
- Yamada, O.; Kawauchi, K.; Akiyama, M.; Ozaki, K.; Motoji, T.; Adachi, T.; Aikawa, E. Leukemic cells with increased telomerase activity exhibit resistance to imatinib. Leuk. Lymphoma 2008, 49, 1168–1177. [Google Scholar] [CrossRef]
- Deville, L.; Hillion, J.; Pendino, F.; Samy, M.; Nguyen, E.; Segal-Bendirdjian, E. hTERT promotes imatinib resistance in chronic myeloid leukemia cells: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 711–719. [Google Scholar] [CrossRef]
- Chai, J.H.; Zhang, Y.; Tan, W.H.; Chng, W.J.; Li, B.; Wang, X. Regulation of hTERT by BCR-ABL at multiple levels in K562 cells. BMC Cancer 2011. [Google Scholar] [CrossRef]
- Yuan, Z.; Mei, H.D. Inhibition of telomerase activity with hTERT Antisense increases the effect of CDDP-induced apoptosis in myeloid leukemia. Hematol. J. 2002, 3, 201–205. [Google Scholar] [CrossRef]
- Lau, L.M.; Dagg, R.A.; Henson, J.D.; Au, A.Y.; Royds, J.A.; Reddel, R.R. Detection of alternative lengthening of telomeres by telomere quantitative PCR. Nucleic Acids Res. 2013, 41, e34. [Google Scholar] [CrossRef]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous T-loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef]
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of T-circles. EMBO J. 2009, 28, 799–809. [Google Scholar] [CrossRef]
- Fasching, C.L.; Neumann, A.A.; Muntoni, A.; Yeager, T.R.; Reddel, R.R. DNA damage induces alternative lengthening of telomeres (ALT) associated promyelocytic leukemia bodies that preferentially associate with linear telomeric DNA. Cancer Res. 2007, 67, 7072–7077. [Google Scholar] [CrossRef]
- Morrish, T.A.; Greider, C.W. Short telomeres initiate telomere recombination in primary and tumor cells. PLoS Genet. 2009, 5, e1000357. [Google Scholar] [CrossRef]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef]
- Chen, Q.; Ijpma, A.; Greider, C.W. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol. Cell. Biol. 2001, 21, 1819–1827. [Google Scholar] [CrossRef]
- Gonzalo, S.; Jaco, I.; Fraga, M.F.; Chen, T.; Li, E.; Esteller, M.; Blasco, M.A. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat. Cell Biol. 2006, 8, 416–424. [Google Scholar] [CrossRef]
- Garcia-Cao, M.; O’Sullivan, R.; Peters, A.H.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2004, 36, 94–99. [Google Scholar] [CrossRef]
- Benetti, R.; Garcia-Cao, M.; Blasco, M.A. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007, 39, 243–250. [Google Scholar] [CrossRef]
- Benetti, R.; Gonzalo, S.; Jaco, I.; Schotta, G.; Klatt, P.; Jenuwein, T.; Blasco, M.A. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J. Cell Biol. 2007, 178, 925–936. [Google Scholar] [CrossRef]
- Broccoli, D.; Young, J.W.; de Lange, T. Telomerase activity in normal and malignant hematopoietic cells. Proc. Natl. Acad. Sci. USA 1995, 92, 9082–9086. [Google Scholar] [CrossRef]
- Cerone, M.A.; Autexier, C.; Londono-Vallejo, J.A.; Bacchetti, S. A human cell line that maintains telomeres in the absence of telomerase and of key markers of ALT. Oncogene 2005, 24, 7893–7901. [Google Scholar] [CrossRef]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: Implications for telomere-based therapies. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef]
- Knecht, H.; Kongruttanachok, N.; Sawan, B.; Brossard, J.; Prevost, S.; Turcotte, E.; Lichtensztejn, Z.; Lichtensztejn, D.; Mai, S. Three-dimensional telomere signatures of hodgkin- and reed-sternberg cells at diagnosis identify patients with poor response to conventional chemotherapy. Transl. Oncol. 2012, 5, 269–277. [Google Scholar]
- Samassekou, O.; Hebert, J.; Mai, S.; Yan, J. Nuclear remodeling of telomeres in chronic myeloid leukemia. Genes Chromosomes Cancer 2013, 52, 495–502. [Google Scholar] [CrossRef]
- Oliveira, F.M.; Gadji, M.; Simões, B.P.; Rego, E.M.; Falcão, R.P.; Mai, S. Three-dimensional nuclear telomeric organization (3D) of chronic myeloid leukemia patients (CML) predicts accelerated phase and blast crisis. In Proceedings of the Ninth AACR-Japanese Cancer Association Joint Conference: Breakthroughs in Basic and Translational Cancer Research, Maui, Hawaii, HI, USA, 21–25 February 2012; p. 47.
- De Lange, T. Human telomeres are attached to the nuclear matrix. EMBO J. 1992, 11, 717–724. [Google Scholar]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef]
- Pampalona, J.; Soler, D.; Genesca, A.; Tusell, L. Telomere dysfunction and chromosome structure modulate the contribution of individual chromosomes in abnormal nuclear morphologies. Mutat. Res. 2010, 683, 16–22. [Google Scholar] [CrossRef]
- Pandita, T.K.; Hunt, C.R.; Sharma, G.G.; Yang, Q. Regulation of telomere movement by telomere chromatin structure. Cell Mol. Life Sci. 2007, 64, 131–138. [Google Scholar] [CrossRef]
- Ramirez, M.J.; Surralles, J. Laser confocal microscopy analysis of human interphase nuclei by three-dimensional fish reveals dynamic perinucleolar clustering of telomeres. Cytogenet. Genome Res. 2008, 122, 237–242. [Google Scholar] [CrossRef]
- Marella, N.V.; Seifert, B.; Nagarajan, P.; Sinha, S.; Berezney, R. Chromosomal rearrangements during human epidermal keratinocyte differentiation. J. Cell. Physiol. 2009, 221, 139–146. [Google Scholar] [CrossRef]
- Solovei, I.; Kreysing, M.; Lanctot, C.; Kosem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef]
- De Vos, W.H.; Hoebe, R.A.; Joss, G.H.; Haffmans, W.; Baatout, S.; van Oostveldt, P.; Manders, E.M. Controlled light exposure microscopy reveals dynamic telomere microterritories throughout the cell cycle. Cytometry A 2009, 75, 428–439. [Google Scholar]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Samassekou, O. Dynamic Length Changes of Telomeres and Their Nuclear Organization in Chronic Myeloid Leukemia. Cancers 2013, 5, 1086-1102. https://doi.org/10.3390/cancers5031086
Samassekou O. Dynamic Length Changes of Telomeres and Their Nuclear Organization in Chronic Myeloid Leukemia. Cancers. 2013; 5(3):1086-1102. https://doi.org/10.3390/cancers5031086
Chicago/Turabian StyleSamassekou, Oumar. 2013. "Dynamic Length Changes of Telomeres and Their Nuclear Organization in Chronic Myeloid Leukemia" Cancers 5, no. 3: 1086-1102. https://doi.org/10.3390/cancers5031086