Novel Systemic Therapies in Advanced Liposarcoma: A Review of Recent Clinical Trial Results

Abstract
:1. Introduction
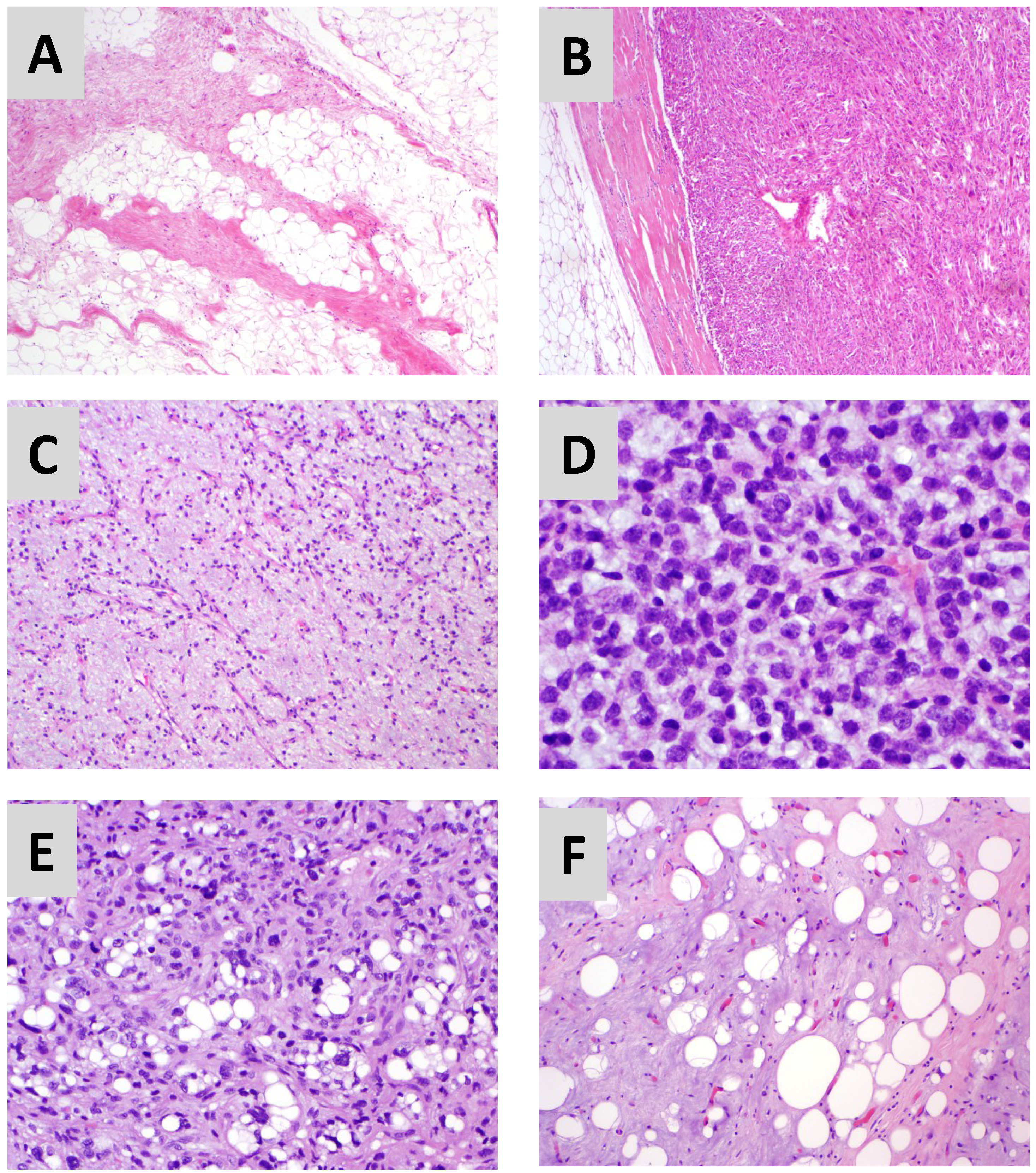
2. Three Distinct Liposarcoma Subtypes

{kind=link}
| Liposarcoma Histologic Subtype | Genetic and/or Molecular Aberration | Histologic Features | Anatomic Site | Clinical Behavior | Response to Current Therapy |
|---|---|---|---|---|---|
| Well differentiated (WD) | 12q13-15 amplification (MDM2, CDK4, etc.) | Adipocytes of varying size, prominent fibrous stroma | Retroperitoneum >extremities, paratesticular areas, trunk | Locoregional recurrence | Poor |
| Dedifferentiated (DD) | same as WD | Highly-cellular portion (5 or more mitoses/10 HPF) next to WD portion | same as WD | Locoregional recurrence and distant metastasis (10–15%) | Low |
| Myxoid/Round Cell (MRC) | Translocation (t12;16)(q13;p11) or (t12;22)(q13;q12) leading to FUS-CHOP/DDIT3 or EWS-CHOP/DDITS fusion protein | Abundant extracellular myxoid material; sparse cellular portion w/mature adipocytes, immature lipoblasts, round cells (>5% of tumor) | Proximal lower extremities | Distant metastasis (10–20%) to visceral organ sites, bone, and fat bearing areas | High |
| Pleomorphic | Complex | Highly cellular resembling MFH; pleomorphic lipoblasts; occassional multinucleated cells | Lower extremities >retroperitoneum; mediastinum | Distant metastasis (30–50%) | Low |
3. Conventional Cytotoxic Chemotherapy
4. Novel Systemic Therapies in Liposarcoma
| Novel Therapy | Mechanism of Action | Liposarcoma Histologic Subtype | Study Type/Clinical Trial Phase | References^ (n = liposarcoma pts) |
|---|---|---|---|---|
| Trabectedin | Binding of DNA minor groove; direct interaction w/FUS-CHOP | MRC | Phase II, Retrospective, and Neoadjuvant | Garcia-Cabonero, 2004 (10); Yovine, 2004 (6); Le Cesne, 2005 (10); Grosso, 2007 (51 *); Grosso, 2009 (32 *); Demetri, 2009 (93); Gronchi, 2012 (23 *), Samuels, 2013 (233) |
| Eribulin | Microtubule inhibitor | DD | Phase II | Schoffski, 2011 (37) |
| RG7112 | MDM2 antagonist | WD/DD | Phase I (Neoadjuvant) | Ray-Coquard, 2012 (20 *) |
| Flavopiridol | pan-CDK inhibitor, including CDK4 | WD/DD | Phase I | Luke, 2012 (16) |
| PD 0332991 | CDK4/6 inhibitor | WD/DD | Phase I | Schwartz, 2012 (7) |
| Troglitazone, Rosiglitazone, Efatutazone | PPAR-gamma agonist | all | Phase I, II | Debrock, 2003 (12 *); Pishvaian, 2012 (5) |
| Nelfinavir | SREBP-1 inhibitor | WD/DD | Phase I | Pan, 2012 (20 *) |
| Pazopanib, Sorafenib, Sunitinib | Tyrosine kinase receptor inhibitor | all | Phase II | Sleijfer, 2009 (19); von Mehren, 2012 (10); Tariq Mahmood, 2011 (17) |
4.1. Marine-Derived Compounds—Trabectedin and Eribulin
4.2. MDM2 Antagonists
4.3. CDK4 Antagonists
4.4. PPAR-Gamma Agonists
4.6. Tyrosine Kinase Receptor Inhibitors
4.7. Immunotherapy
4.8. Other Potential Novel Systemic Therapies from Preclinical Studies
5. Discussion/Conclusions
References
- Clark, M.A.; Fisher, C.; Judson, I.; Thomas, J.M. Soft-tissue sarcomas in adults. N. Engl. J. Med. 2005, 353, 701–711. [Google Scholar] [CrossRef]
- Dodd, L.G. Update on liposarcoma: A review for cytopathologists. Diagn. Cytopathol. 2012, 40, 1122–1131. [Google Scholar] [CrossRef]
- Hoffman, A.; Lazar, A.J.; Pollock, R.E.; Lev, D. New frontiers in the treatment of liposarcoma, a therapeutically resistant malignant cohort. Drug Resist. Updat. 2011, 14, 52–66. [Google Scholar] [CrossRef]
- Conyers, R.; Young, S.; Thomas, D.M. Liposarcoma: Molecular genetics and therapeutics. Sarcoma 2011. [Google Scholar] [CrossRef]
- Singer, S.; Antonescu, C.R.; Riedel, E.; Brennan, M.F. Histologic subtype and margin of resection predict pattern of recurrence and survival for retroperitoneal liposarcoma. Ann. Surg. 2003, 238, 358–370. [Google Scholar]
- Lahat, G.; Anaya, D.A.; Wang, X.; Tuvin, D.; Lev, D.; Pollock, R.E. Resectable well-differentiated versus dedifferentiated liposarcomas: Two different diseases possibly requiring different treatment approaches. Ann. Surg. Oncol. 2008, 15, 1585–1593. [Google Scholar] [CrossRef]
- Ghadimi, M.P.; Al-Zaid, T.; Madewell, J.; Peng, T.; Colombo, C.; Hoffman, A.; Creighton, C.J.; Zhang, Y.; Zhang, A.; Lazar, A.J.; et al. Diagnosis, management, and outcome of patients with dedifferentiated liposarcoma systemic metastasis. Ann. Surg. Oncol. 2011, 18, 3762–3770. [Google Scholar] [CrossRef]
- Fiore, M.; Grosso, F.; Lo Vullo, S.; Pennacchioli, E.; Stacchiotti, S.; Ferrari, A.; Collini, P.; Lozza, L.; Mariani, L.; Casali, P.G.; et al. Myxoid/round cell and pleomorphic liposarcomas: Prognostic factors and survival in a series of patients treated at a single institution. Cancer 2007, 109, 2522–2531. [Google Scholar] [CrossRef]
- De Vreeze, R.S.; de Jong, D.; Tielen, I.H.; Ruijter, H.J.; Nederlof, P.M.; Haas, R.L.; van Coevorden, F. Primary retroperitoneal myxoid/round cell liposarcoma is a nonexisting disease: An immunohistochemical and molecular biological analysis. Mod. Pathol. 2009, 22, 223–231. [Google Scholar] [CrossRef]
- Schwab, J.H.; Boland, P.; Guo, T.; Brennan, M.F.; Singer, S.; Healey, J.H.; Antonescu, C.R. Skeletal metastases in myxoid liposarcoma: An unusual pattern of distant spread. Ann. Surg. Oncol. 2007, 14, 1507–1514. [Google Scholar] [CrossRef]
- Guadagnolo, B.A.; Zagars, G.K.; Ballo, M.T.; Patel, S.R.; Lewis, V.O.; Benjamin, R.S.; Pollock, R.E. Excellent local control rates and distinctive patterns of failure in myxoid liposarcoma treated with conservation surgery and radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 760–765. [Google Scholar] [CrossRef]
- Asano, N.; Susa, M.; Hosaka, S.; Nakayama, R.; Kobayashi, E.; Takeuchi, K.; Horiuchi, K.; Suzuki, Y.; Anazawa, U.; Mukai, M.; et al. Metastatic patterns of myxoid/round cell liposarcoma: A review of a 25-year experience. Sarcoma 2012, 2012, 345161. [Google Scholar]
- De Vreeze, R.; de Jong, D.; Nederlof, P.; Ruijter, H.J.; Boerrigter, L.; Haas, R.; van Coevorden, F. Multifocal myxoid liposarcoma—Metastasis or second primary tumor? A molecular biological analysis. J. Mol. Diagn. 2010, 12, 238–243. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Elahi, A.; Healey, J.H.; Brennan, M.F.; Lui, M.Y.; Lewis, J.; Jhanwar, S.C.; Woodruff, J.M.; Ladanyi, M. Monoclonality of multifocal myxoid liposarcoma: Confirmation by analysis of TLS-CHOP or EWS-CHOP rearrangements. Clin. Cancer Res. 2000, 6, 2788–2793. [Google Scholar]
- Patel, S.R.; Burgess, M.A.; Plager, C.; Papadopoulos, N.E.; Linke, K.A.; Benjamin, R.S. Myxoid liposarcoma. Experience with chemotherapy. Cancer 1994, 74, 1265–1269. [Google Scholar] [CrossRef]
- Jones, R.L.; Fisher, C.; Al-Muderis, O.; Judson, I.R. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur. J. Cancer 2005, 41, 2853–2860. [Google Scholar] [CrossRef]
- Miettinen, M.; Enzinger, F.M. Epithelioid variant of pleomorphic liposarcoma: A study of 12 cases of a distinctive variant of high-grade liposarcoma. Mod. Pathol. 1999, 12, 722–728. [Google Scholar]
- Hornick, J.L.; Bosenberg, M.W.; Mentzel, T.; McMenamin, M.E.; Oliveira, A.M.; Fletcher, C.D. Pleomorphic liposarcoma: Clinicopathologic analysis of 57 cases. Am. J. Surg. Pathol. 2004, 28, 1257–1267. [Google Scholar] [CrossRef]
- Ghadimi, M.P.; Liu, P.; Peng, T.; Bolshakov, S.; Young, E.D.; Torres, K.E.; Colombo, C.; Hoffman, A.; Broccoli, D.; Hornick, J.L.; et al. Pleomorphic liposarcoma: Clinical observations and molecular variables. Cancer 2011, 117, 5359–5369. [Google Scholar] [CrossRef]
- Coindre, J.M.; Mariani, O.; Chibon, F.; Mairal, A.; de Saint Aubain Somerhausen, N.; Favre-Guillevin, E.; Bui, N.B.; Stoeckle, E.; Hostein, I.; Aurias, A. Most malignant fibrous histiocytomas developed in the retroperitoneum are dedifferentiated liposarcomas: A review of 25 cases initially diagnosed as malignant fibrous histiocytoma. Mod. Pathol. 2003, 16, 256–262. [Google Scholar] [CrossRef]
- De Vreeze, R.S.; de Jong, D.; Nederlof, P.M.; Ariaens, A.; Tielen, I.H.; Frenken, L.; Haas, R.L.; van Coevorden, F. Added Value of Molecular Biological Analysis in Diagnosis and Clinical Management of Liposarcoma: A 30-Year Single-Institution Experience. Ann. Surg. Oncol. 2010, 17, 686–693. [Google Scholar] [CrossRef]
- Penel, N.; van Glabbeke, M.; Marreaud, S.; Ouali, M.; Blay, J.Y.; Hohenberger, P. Testing new regimens in patients with advanced soft tissue sarcoma: Analysis of publications from the last 10 years. Ann. Oncol. 2011, 22, 1266–1272. [Google Scholar] [CrossRef]
- Borden, E.C.; Amato, D.A.; Rosenbaum, C.; Enterline, H.T.; Shiraki, M.J.; Creech, R.H.; Lerner, H.J.; Carbone, P.P. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J. Clin. Oncol. 1987, 5, 840–850. [Google Scholar]
- Bramwell, V.H.; Anderson, D.; Charette, M.L. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft-tissue sarcoma: A meta-analysis and clinical practice guideline. Sarcoma 2000, 4, 103–112. [Google Scholar] [CrossRef]
- Edmonson, J.H.; Ryan, L.M.; Blum, R.H.; Brooks, J.S.; Shiraki, M.; Frytak, S.; Parkinson, D.R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J. Clin. Oncol. 1993, 11, 1269–1275. [Google Scholar]
- Santoro, A.; Tursz, T.; Mouridsen, H.; Verweij, J.; Steward, W.; Somers, R.; Buesa, J.; Casali, P.; Spooner, D.; Rankin, E.; et al. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: A randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J. Clin. Oncol. 1995, 13, 1537–1545. [Google Scholar]
- Le Cesne, A.; Judson, I.; Crowther, D.; Rodenhuis, S.; Keizer, H.J.; van Hoesel, Q.; Blay, J.Y.; Frisch, J.; van Glabbeke, M.; Hermans, C.; et al. Randomized phase III study comparing conventional-dose doxorubicin plus ifosfamide versus high-dose doxorubicin plus ifosfamide plus recombinant human granulocyte-macrophage colony-stimulating factor in advanced soft tissue sarcomas: A trial of the European Organization for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group. J. Clin. Oncol. 2000, 18, 2676–2684. [Google Scholar]
- Van Oosterom, A.T.; Mouridsen, H.T.; Nielsen, O.S.; Dombernowsky, P.; Krzemieniecki, K.; Judson, I.; Svancarova, L.; Spooner, D.; Hermans, C.; van Glabbeke, M.; et al. EORTC Soft Tissue and Bone Sarcoma Group. Results of randomised studies of the EORTC Soft Tissue and Bone Sarcoma Group (STBSG) with two different ifosfamide regimens in first- and second-line chemotherapy in advanced soft tissue sarcoma patients. Eur. J. Cancer 2002, 38, 2397–2406. [Google Scholar]
- Buesa, J.M.; Mouridsen, H.T.; van Oosterom, A.T.; Verweij, J.; Wagener, T.; Steward, W.; Poveda, A.; Vestlev, P.M.; Thomas, D.; Sylvester, R. High-dose DTIC in advanced soft-tissue sarcomas in the adult. A phase II study of the E.O.R.T.C. Soft Tissue and Bone Sarcoma Group. Ann. Oncol. 1991, 2, 307–309. [Google Scholar]
- Antman, K.; Crowley, J.; Balcerzak, S.P.; Rivkin, S.E.; Weiss, G.R.; Elias, A.; Natale, R.B.; Cooper, R.M.; Barlogie, B.; Trump, D.L.; et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J. Clin. Oncol. 1993, 11, 1276–1285. [Google Scholar]
- Maki, R.G.; Wathen, J.K.; Patel, S.R.; Priebat, D.A.; Okuno, S.H.; Samuels, B.; Fanucchi, M.; Harmon, D.C.; Schuetze, S.M.; Reinke, D.; Thall, P.F.; et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: Results of sarcoma alliance for research through collaboration study 002. J. Clin. Oncol. 2007, 25, 2755–2763. [Google Scholar] [CrossRef]
- Katz, D.; Boonsirikamchai, P.; Choi, H.; Lazar, A.J.; Wang, W.L.; Xiao, L.; Park, M.S.; Ravi, V.; Benjamin, R.S.; Araujo, D.M. Efficacy of first-line doxorubicin and ifosfamide in myxoid liposarcoma. Clin. Sarcoma Res. 2012, 2, 2. [Google Scholar]
- Italiano, A.; Toulmonde, M.; Cioffi, A.; Penel, N.; Isambert, N.; Bompas, E.; Duffaud, F.; Patrikidou, A.; Lortal, B.; Le Cesne, A.; et al. Advanced well-differentiated/dedifferentiated liposarcomas: Role of chemotherapy and survival. Ann. Oncol. 2012, 23, 1601–1607. [Google Scholar] [CrossRef]
- Italiano, A.; Garbay, D.; Cioffi, A.; Maki, R.G.; Bui, B. Advanced pleomorphic liposarcomas: Clinical outcome and impact of chemotherapy. Ann. Oncol. 2012, 23, 2205–2206. [Google Scholar]
- Jelić, S.; Kovcin, V.; Milanović, N.; Babović, N.; Kreacić, M.; Ristović, Z.; Vlajić, M.; Filipović-Ljesković, I. Randomised study of high-dose epirubicin versus high-dose epirubicin-cisplatin chemotherapy for advanced soft tissue sarcoma. Eur. J. Cancer 1997, 33, 220–225. [Google Scholar]
- Hartmann, J.T.; Oechsle, K.; Huober, J.; Jakob, A.; Azemar, M.; Horger, M.; Kanz, L.; Bokemeyer, C. An open label, non-comparative phase II study of gemcitabine as salvage treatment for patients with pretreated adult type soft tissue sarcoma. Invest. New Drugs 2006, 24, 249–253. [Google Scholar] [CrossRef]
- Reichardt, P.; Oechsle, K.; Pink, D.; Bokemeyer, C.; Schneller, F.; Issels, R.; Kanz, L.; Hartmann, J.T. An open label, non-comparative phase II study of topotecan as salvage treatment for patients with soft tissue sarcoma. Invest. New Drugs 2003, 21, 481–486. [Google Scholar] [CrossRef]
- Van Glabbeke, M.; Verweij, J.; Judson, I.; Nielsen, O.S. EORTC Soft Tissue and Bone Sarcoma Group. Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur. J. Cancer 2002, 38, 543–549. [Google Scholar]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar]
- Forni, C.; Minuzzo, M.; Virdis, E.; Tamborini, E.; Simone, M.; Tavecchio, M.; Erba, E.; Grosso, F.; Gronchi, A.; Aman, P.; et al. Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol. Cancer Ther. 2009, 8, 449–457. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Supko, J.G.; Manola, J.; Seiden, M.V.; Harmon, D.; Ryan, D.P.; Quigley, M.T.; Merriam, P.; Canniff, J.; Goss, G.; et al. Phase II and pharmacokinetic study of ecteinascidin 743 in patients with progressive sarcomas of soft tissues refractory to chemotherapy. J. Clin. Oncol. 2004, 22, 1480–1490. [Google Scholar] [CrossRef]
- Yovine, A.; Riofrio, M.; Blay, J.Y.; Brain, E.; Alexandre, J.; Kahatt, C.; Taamma, A.; Jimeno, J.; Martin, C.; Salhi, Y.; et al. Phase II study of ecteinascidin-743 in advanced pretreated soft tissue sarcoma patients. J. Clin. Oncol. 2004, 22, 890–899. [Google Scholar] [CrossRef]
- Le Cesne, A.; Blay, J.Y.; Judson, I.; van Oosterom, A.; Verweij, J.; Radford, J.; Lorigan, P.; Rodenhuis, S.; Ray-Coquard, I.; Bonvalot, S.; et al. Phase II study of ET-743 in advanced soft tissue sarcomas: A European Organisation for the Research and Treatment of Cancer (EORTC) soft tissue and bone sarcoma group trial. J. Clin. Oncol. 2005, 23, 576–584. [Google Scholar]
- Demetri, G.D.; Chawla, S.P.; von Mehren, M.; Ritch, P.; Baker, L.H.; Blay, J.Y.; Hande, K.R.; Keohan, M.L.; Samuels, B.L.; Schuetze, S.; et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: Results of a randomized phase II study of two different schedules. J. Clin. Oncol. 2009, 27, 4188–4196. [Google Scholar] [CrossRef]
- Samuels, B.L.; Chawla, S.; Patel, S.; von Mehren, M.; Hamm, J.; Kaiser, P.E.; Schuetze, S.; Li, J.; Aymes, A.; Demetri, G.D. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: Results of a worldwide expanded access program study. Ann. Oncol. 2013. [Google Scholar] [CrossRef]
- Grosso, F.; Jones, R.L.; Demetri, G.D.; Judson, I.R.; Blay, J.Y.; Le Cesne, A.; Sanfilippo, R.; Casieri, P.; Collini, P.; Dileo, P.; et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: A retrospective study. Lancet Oncol. 2007, 8, 595–602. [Google Scholar]
- Grosso, F.; Sanfilippo, R.; Virdis, E.; Piovesan, C.; Collini, P.; Dileo, P.; Morosi, C.; Tercero, J.C.; Jimeno, J.; D’Incalci, M.; et al. Trabectedin in myxoid liposarcomas (MLS): A long-term analysis of a single-institution series. Ann. Oncol. 2009, 20, 1439–1444. [Google Scholar] [CrossRef]
- Gronchi, A.; Bui, B.N.; Bonvalot, S.; Pilotti, S.; Ferrari, S.; Hohenberger, P.; Hohl, R.J.; Demetri, G.D.; Le Cesne, A.; Lardelli, P.; et al. Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann. Oncol. 2012, 23, 771–776. [Google Scholar] [CrossRef]
- Casali, P.G.; Sanfilippo, R.; D’Incalci, M. Trabectedin therapy for sarcomas. Curr. Opin. Oncol. 2010, 22, 342–346. [Google Scholar] [CrossRef]
- Schöffski, P.; Ray-Coquard, I.L.; Cioffi, A.; Bui, N.B.; Bauer, S.; Hartmann, J.T.; Krarup-Hansen, A.; Grünwald, V.; Sciot, R.; Dumez, H.; et al. European Organisation for Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group (STBSG). Activity of eribulin mesylate in patients with soft-tissue sarcoma: A phase 2 study in four independent histological subtypes. Lancet Oncol. 2011, 12, 1045–1052. [Google Scholar]
- Müller, C.R.; Paulsen, E.B.; Noordhuis, P.; Pedeutour, F.; Saeter, G.; Myklebost, O. Potential for treatment of liposarcomas with the MDM2 antagonist Nutlin-3A. Int. J. Cancer 2007, 121, 199–205. [Google Scholar] [CrossRef]
- Singer, S.; Socci, N.D.; Ambrosini, G.; Sambol, E.; Decarolis, P.; Wu, Y.; O’Connor, R.; Maki, R.; Viale, A.; Sander, C.; et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res. 2007, 67, 6626–6636. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar]
- Luke, J.J.; D’Adamo, D.R.; Dickson, M.A.; Keohan, M.L.; Carvajal, R.D.; Maki, R.G.; de Stanchina, E.; Musi, E.; Singer, S.; Schwartz, G.K. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: Preclinical investigations and results of a phase I dose-escalation clinical trial. Clin. Cancer Res. 2012, 18, 2638–2647. [Google Scholar] [CrossRef]
- Schwartz, G.K.; LoRusso, P.M.; Dickson, M.A.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; Courtney, R.; O’Dwyer, P.J. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br. J. Cancer 2011, 104, 1862–1868. [Google Scholar]
- Tontonoz, P.; Singer, S.; Forman, B.M.; Sarraf, P.; Fletcher, J.A.; Fletcher, C.D.; Brun, R.P.; Mueller, E.; Altiok, S.; Oppenheim, H.; et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 237–241. [Google Scholar] [CrossRef]
- Demetri, G.D.; Fletcher, C.D.; Mueller, E.; Sarraf, P.; Naujoks, R.; Campbell, N.; Spiegelman, B.M.; Singer, S. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc. Natl. Acad. Sci. USA 1999, 96, 3951–3956. [Google Scholar]
- Debrock, G.; Vanhentenrijk, V.; Sciot, R.; Debiec-Rychter, M.; Oyen, R.; van Oosterom, A. A phase II trial with rosiglitazone in liposarcoma patients. Br. J. Cancer 2003, 89, 1409–1412. [Google Scholar]
- Pishvaian, M.J.; Marshall, J.L.; Wagner, A.J.; Hwang, J.J.; Malik, S.; Cotarla, I.; Deeken, J.F.; He, A.R.; Daniel, H.; Halim, A.B.; et al. A phase 1 study of efatutazone, an oral peroxisome proliferator-activated receptor gamma agonist, administered to patients with advanced malignancies. Cancer 2012, 118, 5403–5413. [Google Scholar] [CrossRef]
- Wagner, K.D.; Benchetrit, M.; Bianchini, L.; Michiels, J.F.; Wagner, N. Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) is highly expressed in liposarcoma and promotes migration and proliferation. J. Pathol. 2011, 224, 575–588. [Google Scholar] [CrossRef]
- Chow, W.A.; Guo, S.; Valdes-Albini, F. Nelfinavir induces liposarcoma apoptosis and cell cycle arrest by upregulating sterol regulatory element binding protein-1. Anticancer Drugs 2006, 17, 891–903. [Google Scholar] [CrossRef]
- Guan, M.; Fousek, K.; Jiang, C.; Guo, S.; Synold, T.; Xi, B.; Shih, C.C.; Chow, W.A. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin. Cancer Res. 2011, 17, 1796–1806. [Google Scholar] [CrossRef]
- Pan, J.; Mott, M.; Xi, B.; Hepner, E.; Guan, M.; Fousek, K.; Magnusson, R.; Tinsley, R.; Valdes, F.; Frankel, P.; et al. Phase I study of nelfinavir in liposarcoma. Cancer Chemother. Pharmacol. 2012, 70, 791–799. [Google Scholar] [CrossRef]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schöffski, P.; Collin, F.; Pandite, L.; Marreaud, S.; de Brauwer, A.; et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J. Clin. Oncol. 2009, 27, 3126–3132. [Google Scholar] [CrossRef]
- Von Mehren, M.; Rankin, C.; Goldblum, J.R.; Demetri, G.D.; Bramwell, V.; Ryan, C.W.; Borden, E. Phase 2 Southwest Oncology Group-directed intergroup trial (S0505) of sorafenib in advanced soft tissue sarcomas. Cancer 2012, 118, 770–776. [Google Scholar] [CrossRef]
- Maki, R.G.; D’Adamo, D.R.; Keohan, M.L.; Saulle, M.; Schuetze, S.M.; Undevia, S.D.; Livingston, M.B.; Cooney, M.M.; Hensley, M.L.; Mita, M.M.; et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin. Oncol. 2009, 27, 3133–3140. [Google Scholar]
- Tariq Mahmood, S.; Agresta, S.; Vigil, C.E.; Zhao, X.; Han, G.; D’Amato, G.; Calitri, C.E.; Dean, M.; Garrett, C.; Schell, M.J.; et al. Phase II study of sunitinib malate, a multitargeted tyrosine kinase inhibitor in patients with relapsed or refractory soft tissue sarcomas. Focus on three prevalent histologies: Leiomyosarcoma, liposarcoma and malignant fibrous histiocytoma. Int. J. Cancer 2011, 129, 1963–1969. [Google Scholar]
- George, S.; Merriam, P.; Maki, R.G.; van den Abbeele, A.D.; Yap, J.T.; Akhurst, T.; Harmon, D.C.; Bhuchar, G.; O’Mara, M.M.; D’Adamo, D.R.; et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J. Clin. Oncol. 2009, 27, 3154–3160. [Google Scholar]
- Peng, T.; Zhang, P.; Liu, J.; Nguyen, T.; Bolshakov, S.; Belousov, R.; Young, E.D.; Wang, X.; Brewer, K.; López-Terrada, D.H.; et al. An experimental model for the study of well-differentiated and dedifferentiated liposarcoma; deregulation of targetable tyrosine kinase receptors. Lab. Invest. 2011, 91, 392–403. [Google Scholar]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; Davidson, N.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Pollack, S.M.; Loggers, E.T.; Rodler, E.T.; Yee, C.; Jones, R.L. Immune-based therapies for sarcoma. Sarcoma 2011, 2011, 438940. [Google Scholar]
- Tseng, W.W.; Demicco, E.G.; Lazar, A.J.; Lev, D.C.; Pollock, R.E. Lymphocyte composition and distribution in inflammatory, well-differentiated retroperitoneal liposarcoma: Clues to a potential adaptive immune response and therapeutic implications. Am. J. Surg. Pathol. 2012, 36, 941–944. [Google Scholar] [CrossRef]
- Pollack, S.M.; Jungbluth, A.A.; Hoch, B.L.; Farrar, E.A.; Bleakley, M.; Schneider, D.J.; Loggers, E.T.; Rodler, E.; Eary, J.F.; Conrad, E.U. NY-ESO-1 is a ubiquitous immunotherapeutic target antigen for patients with myxoid/round cell liposarcoma. Cancer 2012, 118, 4564–4570. [Google Scholar]
- Hemminger, J.A.; Ewart Toland, A.; Scharschmidt, T.J.; Mayerson, J.L.; Kraybill, W.G.; Guttridge, D.C.; Iwenofu, O.H. The cancer-testis antigen NY-ESO-1 is highly expressed in myxoid and round cell subset of liposarcomas. Mod. Pathol. 2013, 26, 282–288. [Google Scholar]
- Lai, J.P.; Robbins, P.F.; Raffeld, M.; Aung, P.P.; Tsokos, M.; Rosenberg, S.A.; Miettinen, M.M.; Lee, C.C. NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: Significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod. Pathol. 2012, 25, 854–858. [Google Scholar]
- Pollack, S. Personal communication, Fred Hutchinson Cancer Center: Seattle, WA, USA, 2013.
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [Green Version]
- Gutierrez, A.; Snyder, E.L.; Marino-Enriquez, A.; Zhang, Y.X.; Sioletic, S.; Kozakewich, E.; Grebliunaite, R.; Ou, W.B.; Sicinska, E.; Raut, C.P.; Demetri, G.D.; et al. Aberrant AKT activation drives well-differentiated liposarcoma. Proc. Natl. Acad. Sci. USA 2011, 108, 16386–16391. [Google Scholar]
- Smith, K.B.; Tran, L.M.; Tam, B.M.; Shurell, E.M.; Li, Y.; Braas, D.; Tap, W.D.; Christofk, H.R.; Dry, S.M.; Eilber, F.C.; et al. Novel Dedifferentiated Liposarcoma Xenograft Models Reveal PTEN Down-Regulation as a Malignant Signature and Response to PI3K Pathway Inhibition. Am. J. Pathol. 2013, 182, 1400–1411. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar]
- Pedeutour, F.; Maire, G.; Pierron, A.; Thomas, D.M.; Garsed, D.W.; Bianchini, L.; Duranton-Tanneur, V.; Cortes-Maurel, A.; Italiano, A.; Squire, J.A.; et al. A newly characterized human well-differentiated liposarcoma cell line contains amplifications of the 12q12-21 and 10p11-14 regions. Virchows Arch. 2012, 461, 67–78. [Google Scholar] [CrossRef]
- Ariizumi, T.; Ogose, A.; Kawashima, H.; Hotta, T.; Li, G.; Xu, Y.; Hirose, T.; Endo, N. Establishment and characterization of a novel dedifferentiated liposarcoma cell line, NDDLS-1. Pathol. Int. 2011, 61, 461–468. [Google Scholar] [CrossRef]
- Uboldi, S.; Bernasconi, S.; Romano, M.; Marchini, S.; Fuso Nerini, I.; Damia, G.; Ganzinelli, M.; Marangon, E.; Sala, F.; Clivio, L.; et al. Characterization of a new trabectedin-resistant myxoid liposarcoma cell line that shows collateral sensitivity to methylating agents. Int. J. Cancer 2012, 131, 59–69. [Google Scholar]
- Stratford, E.W.; Castro, R.; Daffinrud, J.; Skårn, M.; Lauvrak, S.; Munthe, E.; Myklebost, O. Characterization of liposarcoma cell lines for preclinical and biological studies. Sarcoma 2012, 2012, 148614. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tseng, W.W.; Somaiah, N.; Lazar, A.J.; Lev, D.C.; Pollock, R.E. Novel Systemic Therapies in Advanced Liposarcoma: A Review of Recent Clinical Trial Results. Cancers 2013, 5, 529-549. https://doi.org/10.3390/cancers5020529
Tseng WW, Somaiah N, Lazar AJ, Lev DC, Pollock RE. Novel Systemic Therapies in Advanced Liposarcoma: A Review of Recent Clinical Trial Results. Cancers. 2013; 5(2):529-549. https://doi.org/10.3390/cancers5020529
Chicago/Turabian StyleTseng, William W., Neeta Somaiah, Alexander J. Lazar, Dina C. Lev, and Raphael E. Pollock. 2013. "Novel Systemic Therapies in Advanced Liposarcoma: A Review of Recent Clinical Trial Results" Cancers 5, no. 2: 529-549. https://doi.org/10.3390/cancers5020529