Annotating Cancer Variants and Anti-Cancer Therapeutics in Reactome

Abstract
:1. Introduction
2. Results and Discussion
2.1. Annotation of Cancer-Perturbed Pathways
2.1.1. Extension of Protein Modifications to Accommodate Annotation of Changes in Amino Acid Sequence

2.1.2. Associating Disease Attributes with Physical Entities and Events
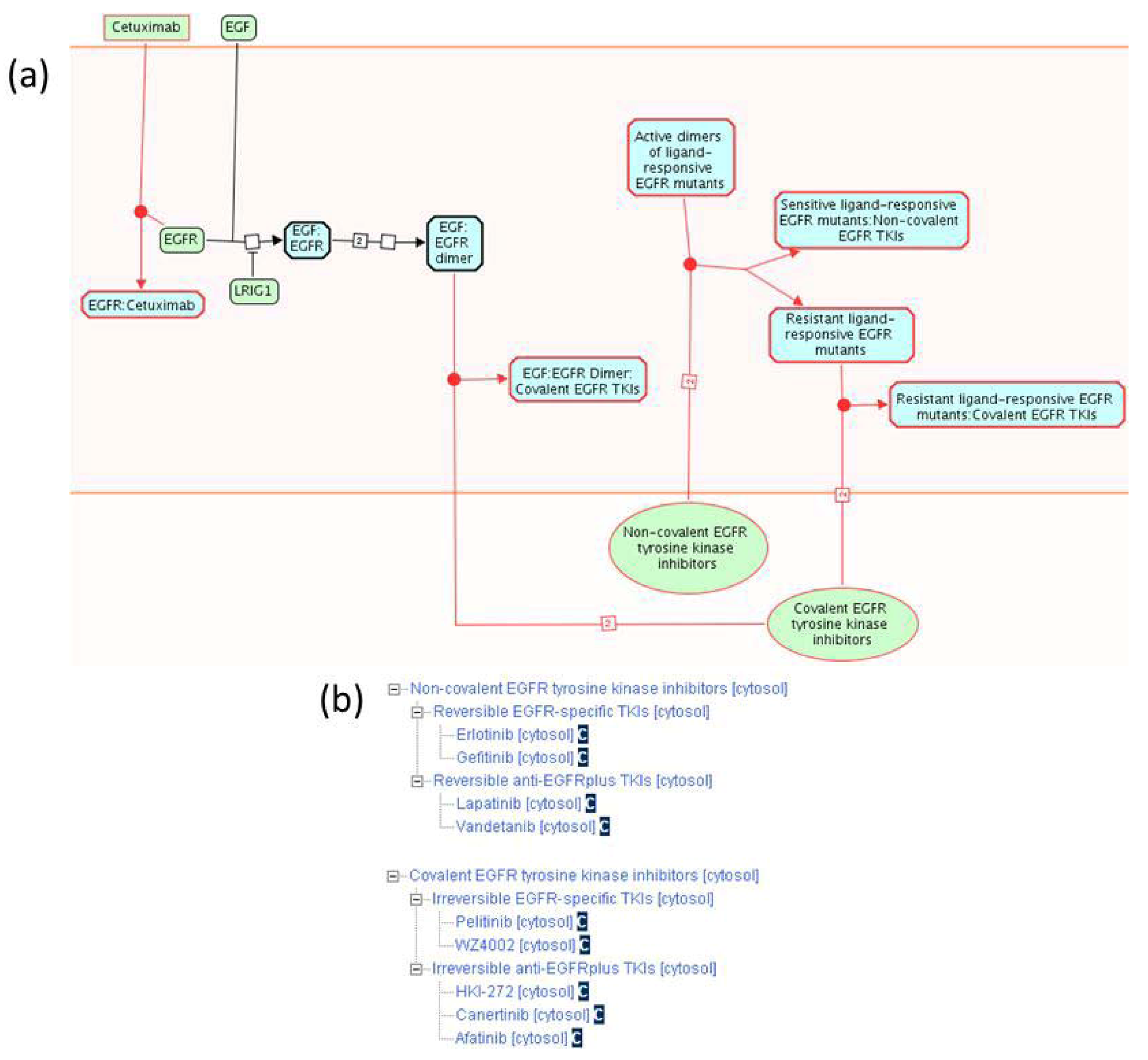
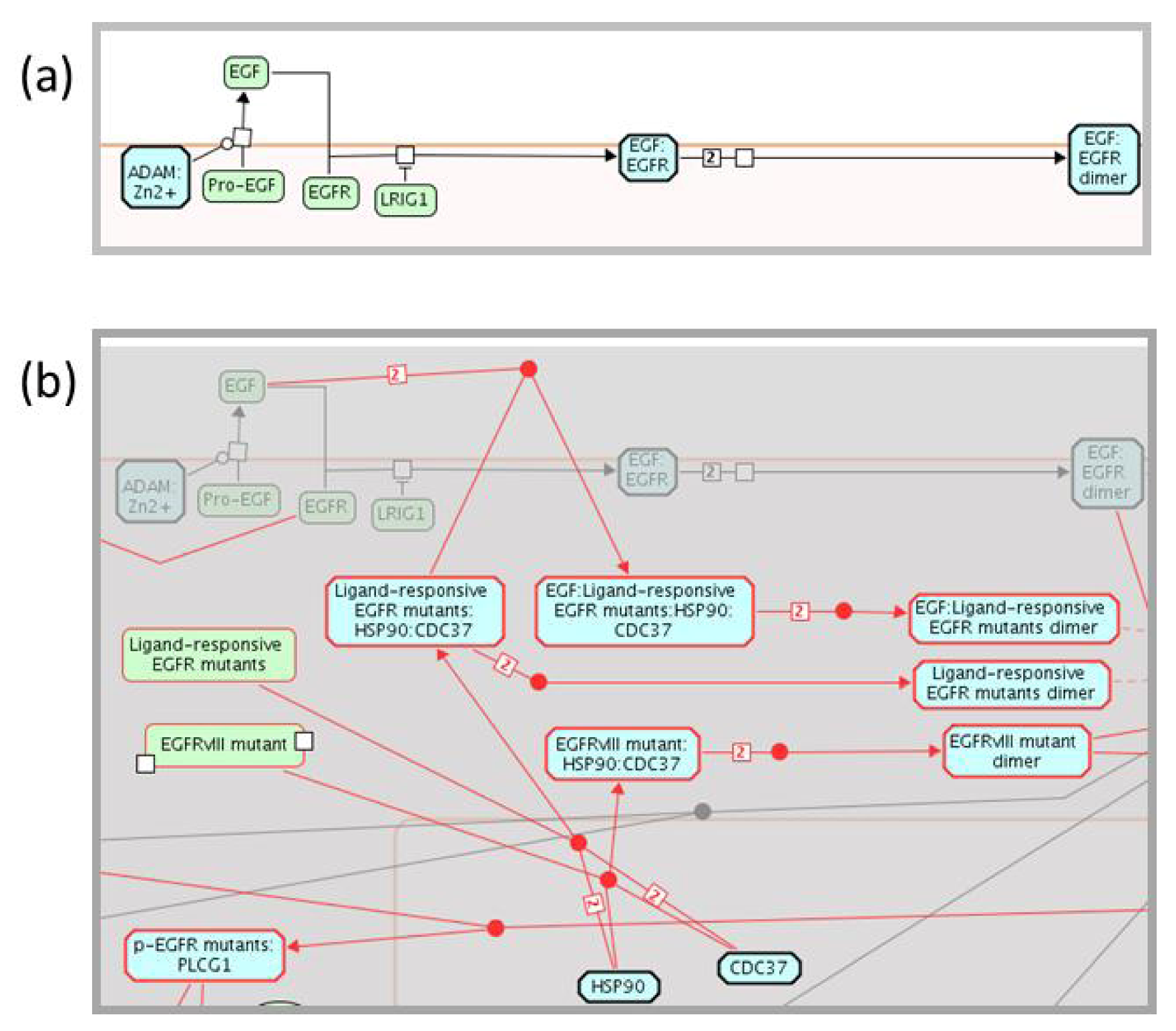
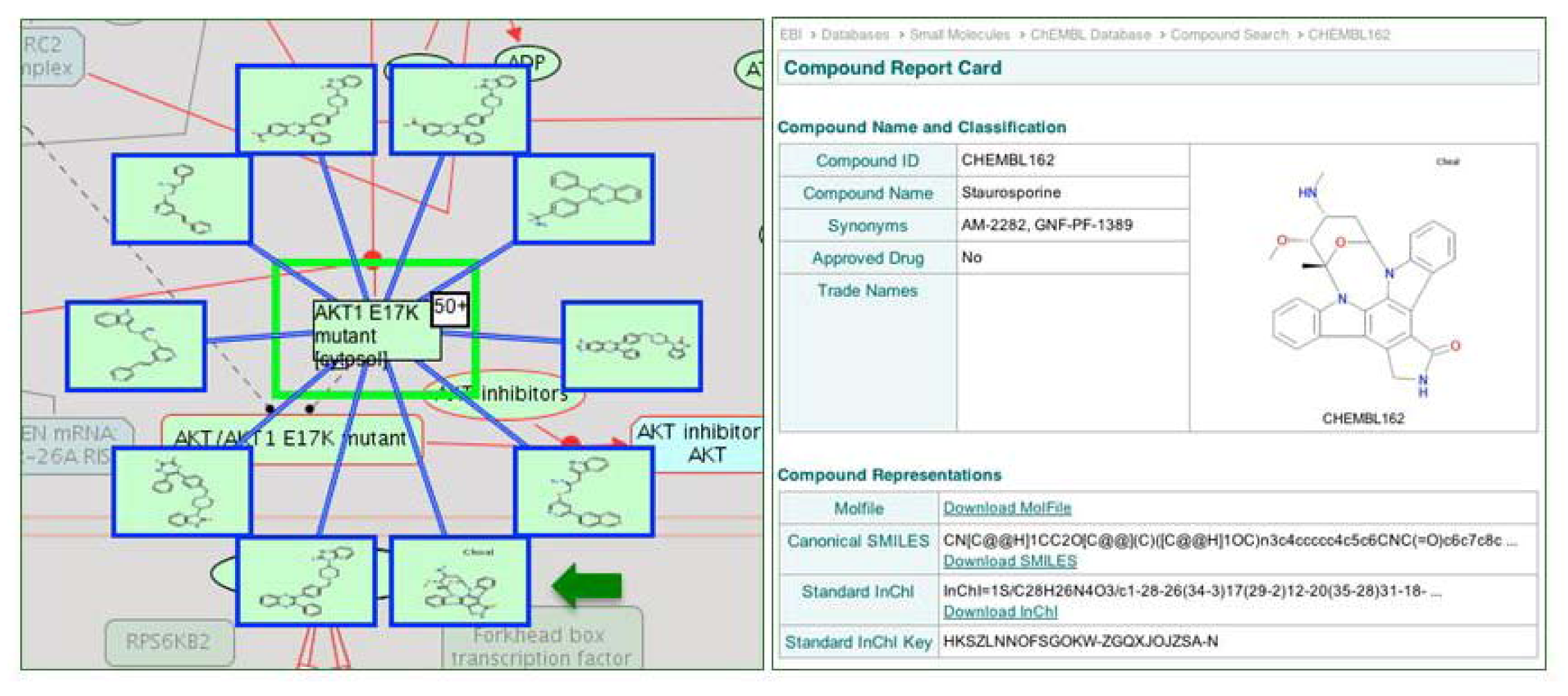
2.1.3. Mode of Action and Specificity of Anti-Cancer Therapeutics

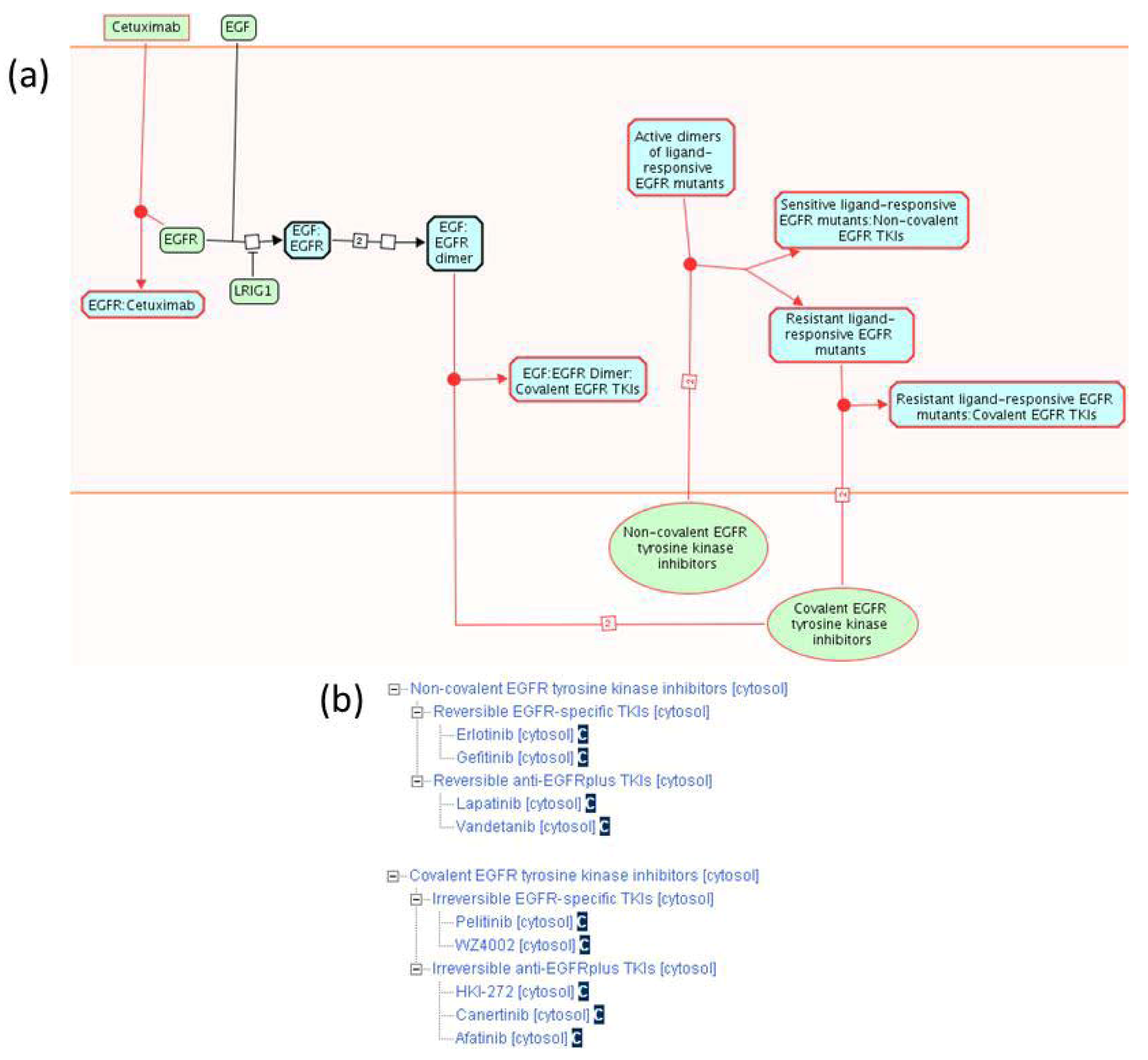

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Variant | COSMIC Identifier(s) | Mutation Type | Disease | Reactome Pathway Name |
|---|---|---|---|---|
| EGFR A289D mutant | 21685 | Missense | Glioblastoma | Signaling by EGFR in Cancer |
| EGFR A289T mutant | 21686 | Missense | Glioblastoma, oligodendroglioma | Signaling by EGFR in Cancer |
| EGFR A289V mutant | 21687 | Missense | Glioblastoma | Signaling by EGFR in Cancer |
| EGFR D770_N771insNPG mutant | Insertion | Lung cancer | Signaling by EGFR in Cancer | |
| EGFR D770_N771insNPH mutant | 48920 | Insertion | Lung cancer | Signaling by EGFR in Cancer |
| EGFR E746_A750del mutant | 6223, 129800, 6225 | Deletion | Breast, head and neck, kidney, lung, ovarian, salivary gland and thyroid cancer | Signaling by EGFR in Cancer |
| EGFR E746_A750del; T790M mutant | Deletion; Missense | Lung cancer | Signaling by EGFR in Cancer | |
| EGFR E746_S752delinsV mutant | 18492, 18426, 12384, 85797 | Deletion | Lung cancer | Signaling by EGFR in Cancer |
| EGFR E746_T751delinsA mutant | 20845, 12678, 13549 | Deletion | Head and neck, lung cancer | Signaling by EGFR in Cancer |
| EGFR G598V mutant | 34167, 21690 | Missense | Glioblastoma | Signaling by EGFR in Cancer |
| EGFR G719A mutant | 6239, 13448 | Missense | Lung cancer | Signaling by EGFR in Cancer |
| EGFR G719C mutant | 6253, 20881 | Missense | Lung cancer | Signaling by EGFR in Cancer |
| EGFR G719S mutant | 6252, 13983 | Missense | Colorectal, lung cancer | Signaling by EGFR in Cancer |
| EGFR L747_A750delinsP mutant | 13562, 12382, 12422 | Deletion | Head and neck, lung cancer | Signaling by EGFR in Cancer |
| EGFR L747_P753delinsS mutant | 13564, 12370 | Deletion | Head and neck, lung, prostate cancer | Signaling by EGFR in Cancer |
| EGFR L747_S752del mutant | 13984, 6255 | Deletion | Lung cancer | Signaling by EGFR in Cancer |
| EGFR L747_T751del mutant | 24432, 12369, 6254, 23571 | Deletion | Lung cancer | Signaling by EGFR in Cancer |
| EGFR L747_T751delinsP mutant | 24573, 12383, 22944 | Deletion | Lung cancer | Signaling by EGFR in Cancer |
| EGFR L858R mutant | 6224, 12979 | Missense | Breast, lung, ovarian, stomach, thymus and thyroid cancer, mesothelioma | Signaling by EGFR in Cancer |
| EGFR L858R;T790M mutant | Missense; Missense | Lung cancer | Signaling by EGFR in Cancer | |
| EGFR L861Q mutant | 6213, 13173 | Missense | Lung cancer, glioblastoma | Signaling by EGFR in Cancer |
| EGFR M766_A767insASV mutant | Insertion | Lung cancer | Signaling by EGFR in Cancer | |
| EGFR R108K mutant | 21683, 34166 | Missense | Glioblastoma | Signaling by EGFR in Cancer |
| EGFR T263P mutant | 21684 | Missense | Glioblastoma | Signaling by EGFR in Cancer |
| EGFR V738_K739insKIPVAI mutant | Insertion | Lung cancer | Signaling by EGFR in Cancer | |
| EGFRvIII mutant | 21351 | Deletion | Lung cancer, glioblastoma | Signaling by EGFR in Cancer |
| BCR-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| CNTRL-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| CPSF6-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| CUX1-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| FGFR1 K656E mutant | 35673 | Missense | Glioblastoma | Signaling by FGFR in Disease |
| FGFR1 N546K mutant | 19176 | Missense | Glioblastoma, hypochondroplasia | Signaling by FGFR in Disease |
| FGFR1 P252S mutant | Missense | Melanoma | Signaling by FGFR in Disease | |
| FGFR1 P252T mutant | 12834 | Missense | Lung cancer | Signaling by FGFR in Disease |
| FGFR1 R576W mutant | 19177 | Missense | Glioblastoma | Signaling by FGFR in Disease |
| FGFR1OP-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| FGFR1OP2-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| FGFR1c P252R mutant | Missense | Pfeiffer syndrome | Signaling by FGFR in Disease | |
| FGFR2 K660E mutant | 36909 | Missense | Endometrial cancer | Signaling by FGFR in Disease |
| FGFR2 K660M mutant | 49175 | Missense | Cervical cancer | Signaling by FGFR in Disease |
| FGFR2 K660N mutant | 49173 | Missense | Endometrial cancer, Crouzon syndrome | Signaling by FGFR in Disease |
| FGFR2 L764fs*4STOP mutant | Frameshift | Endometrial cancer | Signaling by FGFR in Disease | |
| FGFR2 N549H mutant | Missense | Crouzon syndrome | Signaling by FGFR in Disease | |
| FGFR2 N549K mutant | 36912, 36902 | Missense | Endometrial cancer | Signaling by FGFR in Disease |
| FGFR2 S267P mutant | Missense | Stomach cancer, Crouzon syndrome | Signaling by FGFR in Disease | |
| FGFR2 W290C mutant | 41286 | Missense | Lung cancer, Pfeiffer syndrome | Signaling by FGFR in Disease |
| FGFR2b P253R mutant | 49170 | Missense | Endometrial cancer, acrocephalosyndactylia | Signaling by FGFR in Disease |
| FGFR2b S252W mutant | 36903, 41289 | Missense | Endometrial, ovarian cancer, acrocephalosyndactylia, craniosynostosis | Signaling by FGFR in Disease |
| FGFR2b S373C mutant | 36905 | Missense | Endometrial cancer | Signaling by FGFR in Disease |
| FGFR2b Y376C mutant | 36904, 41290 | Missense | Endometrial, ovarian cancer | Signaling by FGFR in Disease |
| FGFR2c A314D mutant | 49171 | Missense | Endometrial cancer | Signaling by FGFR in Disease |
| FGFR2c A314S mutant | Missense | Bone development disease | Signaling by FGFR in Disease | |
| FGFR2c A315S mutant | Missense | Syndactyly | Signaling by FGFR in Disease | |
| FGFR2c A315T mutant | 30777 | Missense | Endometrial cancer | Signaling by FGFR in Disease |
| FGFR2c P253R mutant | 49170 | Missense | Endometrial cancer, acrocephalosyndactylia | Signaling by FGFR in Disease |
| FGFR2c S252W mutant | 41289, 36903 | Missense | Endometrial, ovarian cancer, acrocephalosyndactylia, craniosynostosis | Signaling by FGFR in Disease |
| FGFR2c S372C mutant | Missense | Beare-Stevenson cutis gyrata syndrome | Signaling by FGFR in Disease | |
| FGFR2c W290G mutant | Missense | Crouzon syndrome, Pfeiffer syndrome | Signaling by FGFR in Disease | |
| FGFR2c Y375C mutant | Missense | Beare-Stevenson cutis gyrata syndrome | Signaling by FGFR in Disease | |
| FGFR3 795fs*139STOP mutant | Frameshift | Multiple myeloma, thanatophoric dysplasia | Signaling by FGFR in Disease | |
| FGFR3 A391E mutant | 721 | Missense | Bladder cancer, Crouzon syndrome | Signaling by FGFR in Disease |
| FGFR3 G370C mutant | 716, 35897 | Missense | Bladder cancer, thanatophoric dysplasia | Signaling by FGFR in Disease |
| FGFR3 G380R mutant | 24842, 24812 | Missense | Bladder cancer, multiple myeloma, achondroplasia | Signaling by FGFR in Disease |
| FGFR3 G382D mutant | 727 | Missense | Multiple myeloma | Signaling by FGFR in Disease |
| FGFR3 K650E mutant | 719, 35899 | Missense | Bladder, testicular cancer, multiple myeloma, thanatophoric dysplasia | Signaling by FGFR in Disease |
| FGFR3 K650M mutant | 720, 85791 | Missense | Bladder, testicular cancer, multiple myeloma, thanatophoric dysplasia | Signaling by FGFR in Disease |
| FGFR3 K650N mutant | Missense | Bladder, testicular cancer, hypochondroplasia | Signaling by FGFR in Disease | |
| FGFR3 K650Q mutant | 726 | Missense | Bladder cancer, hypochondroplasia | Signaling by FGFR in Disease |
| FGFR3 K650T mutant | 731 | Missense | Bladder, testicular cancer, hypochondroplasia | Signaling by FGFR in Disease |
| FGFR3 R248C mutant | 714, 35896 | Missense | Bladder cancer, multiple myeloma, thanatophoric dysplasia | Signaling by FGFR in Disease |
| FGFR3 S371C mutant | 17461, 35898 | Missense | Bladder cancer, thanatophoric dysplasia | Signaling by FGFR in Disease |
| FGFR3 Y373C mutant | 718, 29428 | Missense | Bladder cancer, multiple myeloma, thanatophoric dysplasia | Signaling by FGFR in Disease |
| FGFR3b G697C mutant | 24802 | Missense | Head and neck cancer | Signaling by FGFR in Disease |
| FGFR3b S249C mutant | 715, 29427 | Missense | Bladder, cervical, head and neck, prostate cancer, thanatophoric dysplasia | Signaling by FGFR in Disease |
| FGFR3c P250R mutant | Missense | Acrocephalosyndactylia, craniosynostosis | Signaling by FGFR in Disease | |
| FGFR4 N535D mutant | Missense | Rhabdomyosarcoma | Signaling by FGFR in Disease | |
| FGFR4 N535K mutant | Missense | Rhabdomyosarcoma | Signaling by FGFR in Disease | |
| FGFR4 V550E mutant | Missense | Rhabdomyosarcoma | Signaling by FGFR in Disease | |
| FGFR4 V550L mutant | Missense | Rhabdomyosarcoma | Signaling by FGFR in Disease | |
| FGFR4 Y367C mutant | Missense | Breast cancer | Signaling by FGFR in Disease | |
| LRRFIP1-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| MYO18A-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| TRIM24-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| ZMYM2-FGFR1 fusion mutant | Translocation | Myeloid leukemia | Signaling by FGFR in Disease | |
| IDH1 R132C mutant | 28747, 41294 | Missense | Glioblastoma | The citric acid (TCA) cycle and respiratory electron transport |
| IDH1 R132H mutant | 28746, 41291 | Missense | Glioblastoma | The citric acid (TCA) cycle and respiratory electron transport |
| IDH1 R132L mutant | 28750 | Missense | Glioblastoma | The citric acid (TCA) cycle and respiratory electron transport |
| IDH1 R132S mutant | 28748 | Missense | Glioblastoma | The citric acid (TCA) cycle and respiratory electron transport |
| PIK3CA E542K mutant | 760, 29329 | Missense | Bladder, breast, cervical, colorectal, endometrial, esophageal, gallbladder, head and neck, kidney, liver, lung, ovarian, penis, pharynx, pituitary, skin sweat gland, stomach, thyroid cancer, glioblastoma, lymphocytic leukemia | PI3K/AKT Signaling in Cancer |
| PIK3CA E542Q mutant | 17442 | Missense | Breast, colorectal, endometrial, lung cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA E542V mutant | 762 | Missense | Breast, colorectal, endometrial, ovarian cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA E545A mutant | 12458 | Missense | Breast, colorectal, endometrial, esophageal, lung, ovarian, prostate, thyroid cancer, glioblastoma, hepatoblastoma, synovial sarcoma | PI3K/AKT Signaling in Cancer |
| PIK3CA E545G mutant | 764 | Missense | Bladder, breast, colorectal, endometrial, head and neck, larynx, pituitary, stomach cancer, myeloid leukemia, non-Hodgkin lymphoma, retinoblastoma | PI3K/AKT Signaling in Cancer |
| PIK3CA E545K mutant | 763, 29328 | Missense | Bladder, breast. cervical, colorectal, endometrial, esophageal, gallbladder, head and neck, kidney, lung, ovarian, pancreatic, penis, pharynx, skin, stomach, sweat gland, thyroid cancer, melanoma, glioblastoma, medulloblastoma, myeloma, pituitary adenoma | PI3K/AKT Signaling in Cancer |
| PIK3CA E545Q mutant | 27133 | Missense | Bladder, breast, colorectal, esophageal, head and neck, ovarian, thyroid cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA E545V mutant | 144201 | Missense | Ovarian cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA H1047L mutant | 776, 30744 | Missense | Bladder, breast, colorectal, endometrial, head and neck, liver, lung, ovarian, pharynx, thyroid cancer, glioblastoma, non-Hodgkin lymphoma, pituitary adenoma | PI3K/AKT Signaling in Cancer |
| PIK3CA H1047R mutant | 775, 29325 | Missense | Bladder, breast, cervical, colorectal, endometrial, gallbladder, head and neck, liver, lung, ovarian, pancreatic, pharynx, prostate, stomach, thyroid cancer, glioblastoma, medulloblastoma, melanoma, meningioma, neuroectodermal tumor, non-Hodgkin lymphoma, pituitary adenoma | PI3K/AKT Signaling in Cancer |
| PIK3CA H1047Y mutant | 774, 29326 | Missense | Breast, colorectal, endometrial, lung, ovarian cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3CA M1043I mutant | 773, 29313, 94984 | Missense | Bladder, breast, cervical, colorectal, endometrial, lung, ovarian, thyroid cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3CA M1043T mutant | 12463 | Missense | Ovarian, stomach cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3CA M1043V mutant | 12591, 30743 | Missense | Breast, colorectal, endometrial, head and neck, lung, ovarian, pharynx cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3CA Q546E mutant | 6147 | Missense | Breast, cervical, colorectal, endometrial cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA Q546H mutant | 24712, 30740 | Missense | Cervical, colorectal, endometrial cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA Q546K mutant | 766, 30738 | Missense | Breast, colorectal, endometrial, lung, ovarian, stomach cancer, lymphocytic leukemia | PI3K/AKT Signaling in Cancer |
| PIK3CA Q546L mutant | 25041, 85754 | Missense | Breast, colorectal, gallbladder, head and neck cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA Q546P mutant | 767 | Missense | Breast, colorectal, endometrial, ovarian cancer, glioma | PI3K/AKT Signaling in Cancer |
| PIK3CA Q546R mutant | 12459, 30739 | Missense | Breast. colorectal, endometrial, prostate, stomach cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA R38C mutant | 744 | Missense | Colorectal cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA R38G mutant | 40945 | Missense | Glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3CA R38H mutant | 745, 49022 | Missense | Breast, colorectal, endometrial cancer | PI3K/AKT Signaling in Cancer |
| PIK3CA R38S mutant | 87310 | Missense | Stomach cancer | PI3K/AKT Signaling in Cancer |
| PIK3R1 D560H mutant | 125378 | Missense | Pharynx cancer | PI3K/AKT Signaling in Cancer |
| PIK3R1 D560Y mutant | 335765 | Missense | Glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3R1 G376R mutant | 35827, 132923 | Missense | Endometrial cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3R1 H450_E451del mutant | 39296 | Deletion | Endometrial cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3R1 K459del mutant | 87216 | Deletion | Endometrial cancer | PI3K/AKT Signaling in Cancer |
| PIK3R1 N564D mutant | 42912 | Missense | Colorectal, endometrial cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3R1 N564K mutant | 35808 | Missense | Glioblastoma | PI3K/AKT Signaling in Cancer |
| PIK3R1 R574_T576del mutant | 87219 | Deletion | Endometrial cancer | PI3K/AKT Signaling in Cancer |
| PIK3R1 R574I mutant | 85927 | Missense | Colorectal cancer | PI3K/AKT Signaling in Cancer |
| PIK3R1 R574T mutant | 87544 | Missense | Bladder, breast cancer | PI3K/AKT Signaling in Cancer |
| PIK3R1 Y463_L466del mutant | 87228 | Deletion | Endometrial cancer | PI3K/AKT Signaling in Cancer |
| AKT1 E17K mutant | 33765, 34142 | Missense | Breast, colorectal, ovarian cancer | PI3K/AKT Signaling in Cancer |
| PTEN R130G mutant | 5219 | Missense | Endometrial, lung, ovarian cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN R130Q mutant | 5033 | Missense | Breast, colorectal, endometrial, ovarian, thyroid cancer, glioma, histiocytoma | PI3K/AKT Signaling in Cancer |
| PTEN R130L mutant | 5216 | Missense | Breast, endometrial cancer, Cowden syndrome | PI3K/AKT Signaling in Cancer |
| PTEN C124S mutant | 5224, 5271 | Missense | Endometrial, thyroid cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN C124R mutant | Missense | Thyroid adenoma, Cowden syndrome | PI3K/AKT Signaling in Cancer | |
| PTEN R173H mutant | 5039 | Missense | Endometrial, ovarian cancer, glioma | PI3K/AKT Signaling in Cancer |
| PTEN R173C mutant | 5089, 24682 | Missense | Endometrial cancer, glioblastoma, lymphocytic leukemia, melanoma | PI3K/AKT Signaling in Cancer |
| PTEN R173P mutant | 12735 | Missense | Testicular cancer | PI3K/AKT Signaling in Cancer |
| PTEN S170N mutant | 5045 | Missense | Endometrial cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN S170R mutant | Missense | Bannayan-Riley-Ruvalcaba syndrome | PI3K/AKT Signaling in Cancer | |
| PTEN H123Y mutant | 5078 | Missense | Endometrial cancer | PI3K/AKT Signaling in Cancer |
| PTEN G129E mutant | 28917 | Missense | Endometrial cancer | PI3K/AKT Signaling in Cancer |
| PTEN G129R mutant | 5092 | Missense | Thyroid cancer, glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN H93Y mutant | 5043 | Missense | Endometrial cancer, glioma, medulloblastoma | PI3K/AKT Signaling in Cancer |
| PTEN H93A mutant | Missense | Cancer | PI3K/AKT Signaling in Cancer | |
| PTEN H93R mutant | 5060 | Missense | Glioblastoma, autism spectrum disorders | PI3K/AKT Signaling in Cancer |
| PTEN H93D mutant | 5283 | Missense | Endometrial cancer | PI3K/AKT Signaling in Cancer |
| PTEN H93Q mutant | 5186 | Missense | Glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN R130P mutant | 5277 | Missense | Breast, endometrial, glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN C124F mutant | 13578 | Missense | Lung cancer | PI3K/AKT Signaling in Cancer |
| PTEN C124Y mutant | 5140 | Missense | Stomach cancer | PI3K/AKT Signaling in Cancer |
| PTEN S170I mutant | 5218 | Missense | Glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN S170G mutant | 5063 | Missense | Glioblastoma | PI3K/AKT Signaling in Cancer |
| PTEN G129V mutant | 5276 | Missense | Endometrial cancer | PI3K/AKT Signaling in Cancer |
| PTEN R130* mutant | 21342, 5152 | Nonsense | Cervical, colorectal, endometrial, lung, ovarian, prostate, thyroid cancer, glioblastoma, medulloblastoma, leimyosarcoma | PI3K/AKT Signaling in Cancer |
| PTEN R233* mutant | 5154, 21343 | Nonsense | Cervical, colorectal, endometrial, lung, ovarian cancer, glioblastoma, histiocytoma, lymphocytic leukemia, | PI3K/AKT Signaling in Cancer |
| PTEN R335* mutant | 5775, 5151 | Nonsense | Head and neck, stomach cancer, glioblastoma, melanoma, Burkitt lymphoma, lymphocytic leukemia | PI3K/AKT Signaling in Cancer |
| Anti-Cancer Therapeutic | Reference Molecule Identifier | Specificity | Reactome Pathway Name |
|---|---|---|---|
| 17-AAG | ChEBIa: 64153 | HSP90 | Signaling by EGFR in Cancer |
| 17-DMAG | ChEBI:65324 | HSP90 | Signaling by EGFR in Cancer |
| Afatinib | ChEBI:61390 | EGFR, ERBB2 | Signaling by EGFR in Cancer |
| Canertinib | ChEBI:61399 | Pan-ERBB | Signaling by EGFR in Cancer |
| Cetuximab | Recombinant antibody | EGFR | Signaling by EGFR in Cancer |
| Erlotinib | ChEBI:114785 | EGFR | Signaling by EGFR in Cancer |
| Gefitinib | ChEBI:49668 | EGFR | Signaling by EGFR in Cancer |
| Geldanamycin | ChEBI:5292 | HSP90 | Signaling by EGFR in Cancer |
| HKI-272 | ChEBI:61390 | EGFR, ERBB2 | Signaling by EGFR in Cancer |
| Herbimycin A | ChEBI:5674 | HSP90 | Signaling by EGFR in Cancer |
| Lapatinib | ChEBI:49603 | EGFR, ERBB2 | Signaling by EGFR in Cancer |
| Pelitinib | ChEBI:38927 | EGFR | Signaling by EGFR in Cancer |
| Vandetanib | ChEBI:49960 | EGFR, VEGFR | Signaling by EGFR in Cancer |
| WZ4002 | ChEBI:61400 | EGFR | Signaling by EGFR in Cancer |
| IPI-504 | Pending | HSP90 | Signaling by EGFR in Cancer |
| AZ 2171 | ChEBI:556867 | FGFR, PDGFR, VEGFR. KIT | Signaling by FGFR in Disease |
| Brivanib | ChEBI:443041 | FGFR, VEGFR | Signaling by FGFR in Disease |
| Brivanib alaninate | ChEBI:270995 | FGFR, VEGFR | Signaling by FGFR in Disease |
| Dovitinib | ChEBI:594834 | FGFR, FLT3, VEGFR, PDGFR, KIT, CSFR | Signaling by FGFR in Disease |
| E3810 | Pending | FGFR, VEGFR | Signaling by FGFR in Disease |
| E7080 | ChEBI:816009 | FGFR VEGFR, PDGFR | Signaling by FGFR in Disease |
| Masitinib | ChEBI:63450 | FGFR3, PDGFR, KIT | Signaling by FGFR in Disease |
| GP369 | Recombinant antibody | FGFR2b | Signaling by FGFR in Disease |
| Midostaurin | ChEBI:63452 | FGFR, FLT3, PDGFR, VEGFR, KIT, PKCA | Signaling by FGFR in Disease |
| PD173074 | ChEBI:63448 | Pan-FGFR | Signaling by FGFR in Disease |
| AZD4547 | ChEBI:63453 | Pan-FGFR | Signaling by FGFR in Disease |
| BGJ398 | ChEBI:63451 | Pan-FGFR | Signaling by FGFR in Disease |
| SU5402 | ChEBI:63449 | FGFR, VEGFR | Signaling by FGFR in Disease |
| GSK1059615 | Pending | Pan-PI3K | PI3K/AKT Signaling in Cancer |
| BEZ235 | Pending | PI3K Class I, mTOR | PI3K/AKT Signaling in Cancer |
| BGT226 | Pending | PI3K Class I, mTOR | PI3K/AKT Signaling in Cancer |
| BKM120 | Pending | PI3K Class I | PI3K/AKT Signaling in Cancer |
| XL765 | Pending | PI3K Class I, mTOR | PI3K/AKT Signaling in Cancer |
| XL147 | Pending | PI3K Class I | PI3K/AKT Signaling in Cancer |
| GDC0941 | ChEBI:65326 | PI3K Class I | PI3K/AKT Signaling in Cancer |
| PX-866 | ChEBI:65345 | PIK3CA, PIK3CD, PIK3CG | PI3K/AKT Signaling in Cancer |
| LY294002 | ChEBI:65329 | Pan-PI3K | PI3K/AKT Signaling in Cancer |
| wortmannin | ChEBI:52289 | Pan-PI3K | PI3K/AKT Signaling in Cancer |
| Perifosine | ChEBI:428891 | AKT | PI3K/AKT Signaling in Cancer |
| MK2206 | ChEBI:716367 | AKT | PI3K/AKT Signaling in Cancer |
| Triciribine | ChEBI:65310 | AKT | PI3K/AKT Signaling in Cancer |
2.2. Other Disease Pathways in Reactome
2.3. Enhancing the Reactome Pathway Browser for Display of Disease Variants
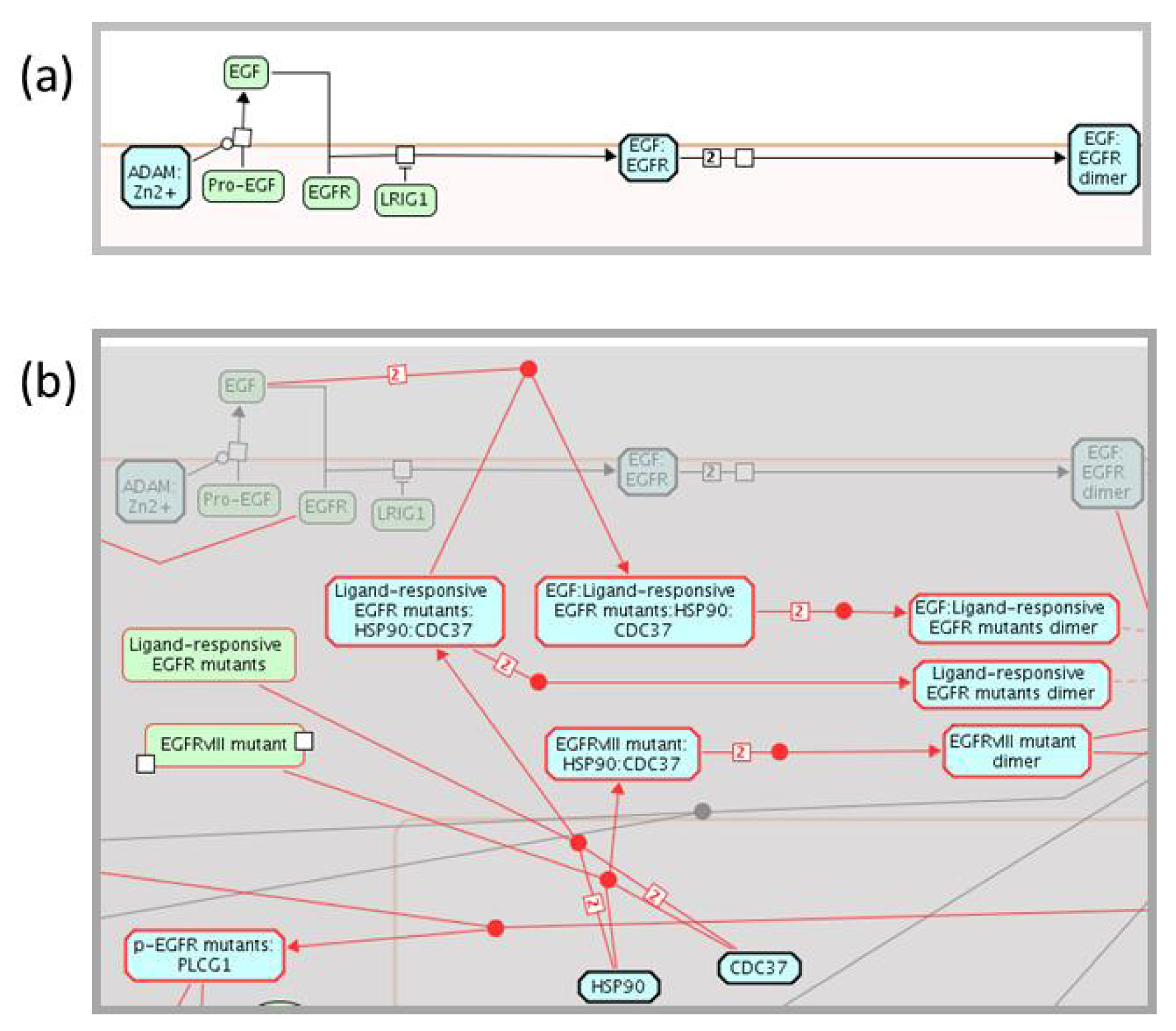
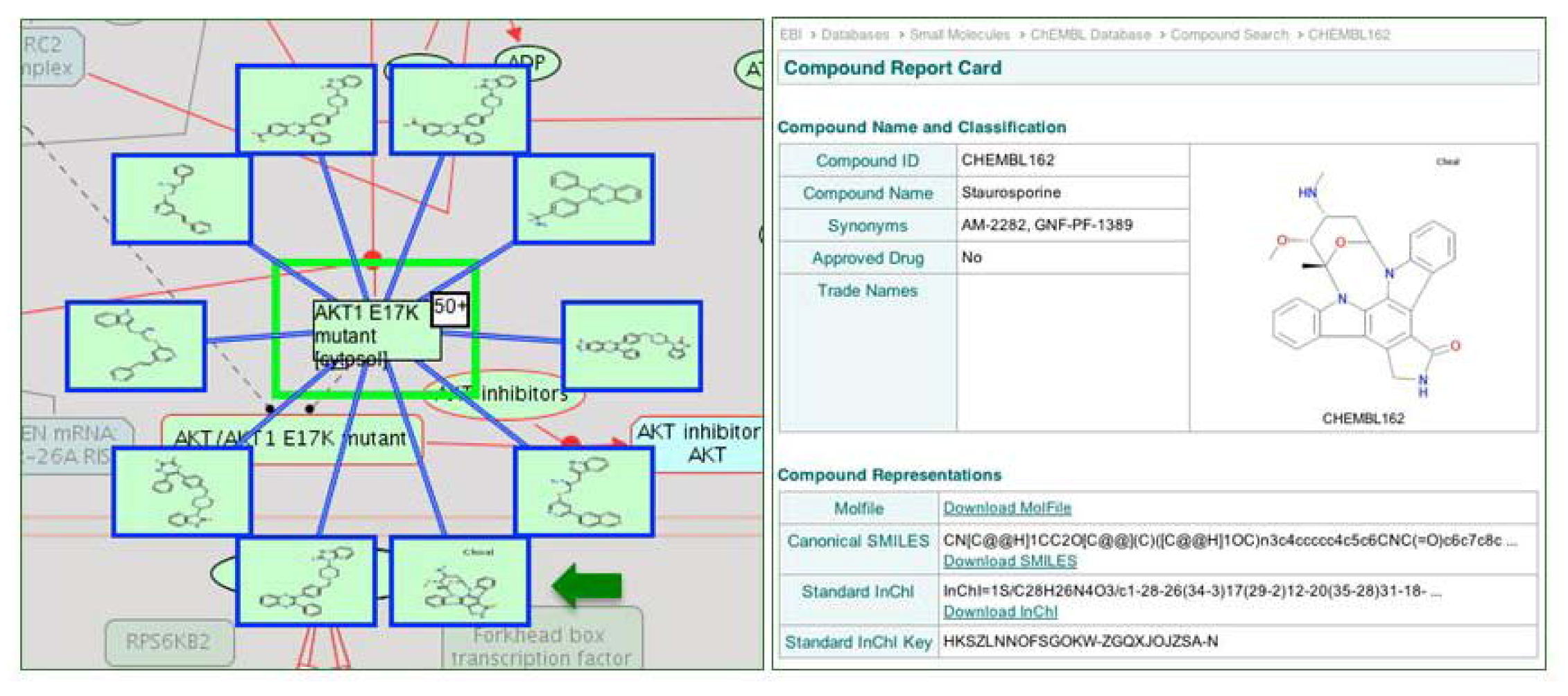
2.4. Reactome Cancer-Perturbed Pathways Support Pathway Visualization and Analysis
3. Experimental Section
4. Conclusions
Acknowledgments
References
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; et al. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar]
- Hockenbery, D.; Nunez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar]
- Lew, D.J.; Dulic, V.; Reed, S.I. Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell 1991, 66, 1197–1206. [Google Scholar] [CrossRef]
- Motokura, T.; Bloom, T.; Kim, H.G.; Juppner, H.; Ruderman, J.V.; Kronenberg, H.M.; Arnold, A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature 1991, 350, 512–515. [Google Scholar]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar]
- Matthews, L.; Gopinath, G.; Gillespie, M.; Caudy, M.; Croft, D.; de Bono, B.; Garapati, P.; Hemish, J.; Hermjakob, H.; Jassal, B.; et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009, 37, D619–D622. [Google Scholar]
- Vastrik, I.; D’Eustachio, P.; Schmidt, E.; Gopinath, G.; Croft, D.; de Bono, B.; Gillespie, M.; Jassal, B.; Lewis, S.; Matthews, L.; et al. Reactome: A knowledge base of biologic pathways and processes. Genome Biol. 2007, 8, R39. [Google Scholar] [CrossRef]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.R.; Wu, G.R.; Matthews, L.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33, D428–D432. [Google Scholar]
- Gillespie, M.; Shamovsky, V.; D’Eustachio, P. Human and chicken TLR pathways: Manual curation and computer-based orthology analysis. Mamm. Genome 2010, 22, 130–138. [Google Scholar]
- Jassal, B.; Jupe, S.; Caudy, M.; Birney, E.; Stein, L.; Hermjakob, H.; D’Eustachio, P. The systematic annotation of the three main GPCR families in Reactome. Database (Oxford) 2010, 2010, aq018. [Google Scholar]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2011, 39, D52–D57. [Google Scholar]
- Flicek, P.; Aken, B.L.; Ballester, B.; Beal, K.; Bragin, E.; Brent, S.; Chen, Y.; Clapham, P.; Coates, G.; Fairley, S.; et al. Ensembl’s 10th year. Nucleic Acids Res. 2010, 38, D557–D562. [Google Scholar] [CrossRef]
- Consortium, T.U. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012, 40, D71–D75. [Google Scholar] [CrossRef]
- Fujita, P.A.; Rhead, B.; Zweig, A.S.; Hinrichs, A.S.; Karolchik, D.; Cline, M.S.; Goldman, M.; Barber, G.P.; Clawson, H.; Coelho, A.; et al. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res. 2011, 39, D876–D882. [Google Scholar] [CrossRef]
- De Matos, P.; Alcantara, R.; Dekker, A.; Ennis, M.; Hastings, J.; Haug, K.; Spiteri, I.; Turner, S.; Steinbeck, C. Chemical Entities of Biological Interest: An update. Nucleic Acids Res. 2010, 38, D249–D254. [Google Scholar]
- Consortium, G.O. The Gene Ontology in 2010: Extensions and refinements. Nucleic Acids Res. 2010, 38, D331–D335. [Google Scholar] [CrossRef]
- Reactome-Signaling by EGFR. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_9417/ (accessed on 20 September 2012).
- Reactome-Signaling by FGFR. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_9470/ (accessed on 20 September 2012).
- Reactome-Signaling by NOTCH. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_299/ (accessed on 20 September 2012).
- Reactome-PIP3 Activates AKT Signaling. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_75829/ (accessed on 20 September 2012).
- Reactome-RAF/MAP Kinase Cascade. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_634/ (accessed on 20 September 2012).
- Reactome-Apoptosis. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_578/ (accessed on 20 September 2012).
- Reactome-Cell Cycle Checkpoints. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_1538/ (accessed on 20 September 2012).
- Reactome-Mitotic G1-G1/S phases. Available online: http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=REACT_21267/ (accessed on 20 September 2012).
- Sherrill, J.M.; Kyte, J. Activation of epidermal growth factor receptor by epidermal growth factor. Biochemistry 1996, 35, 5705–5718. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef]
- Furdui, C.M.; Lew, E.D.; Schlessinger, J.; Anderson, K.S. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol. Cell 2006, 21, 711–717. [Google Scholar]
- Hart, K.C.; Robertson, S.C.; Donoghue, D.J. Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Mol. Biol. Cell 2001, 12, 931–942. [Google Scholar]
- Mohammadi, M.; Dikic, I.; Sorokin, A.; Burgess, W.H.; Jaye, M.; Schlessinger, J. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol. Cell. Biol. 1996, 16, 977–989. [Google Scholar]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Schlessinger, J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004, 306, 1506–1507. [Google Scholar]
- Ong, S.H.; Hadari, Y.R.; Gotoh, N.; Guy, G.R.; Schlessinger, J.; Lax, I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 6074–6079. [Google Scholar]
- Rodrigues, G.A.; Falasca, M.; Zhang, Z.; Ong, S.H.; Schlessinger, J. A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol. Cell. Biol. 2000, 20, 1448–1459. [Google Scholar]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Burke, J.E.; Vadas, O.; Berndt, A.; Finegan, T.; Perisic, O.; Williams, R.L. Dynamics of the phosphoinositide 3-kinase p110delta interaction with p85alpha and membranes reveals aspects of regulation distinct from p110alpha. Structure 2011, 19, 1127–1137. [Google Scholar]
- Mandelker, D.; Gabelli, S.B.; Schmidt-Kittler, O.; Zhu, J.; Cheong, I.; Huang, C.H.; Kinzler, K.W.; Vogelstein, B.; Amzel, L.M. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA 2009, 106, 16996–17001. [Google Scholar]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Scheid, M.P.; Marignani, P.A.; Woodgett, J.R. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol. Cell. Biol. 2002, 22, 6247–6260. [Google Scholar]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Greulich, H.; Chen, T.H.; Feng, W.; Janne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar]
- Fernandes, H.; Cohen, S.; Bishayee, S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: A study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J. Biol. Chem. 2001, 276, 5375–5383. [Google Scholar]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62–ra93. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar]
- Cappellen, D.; de Oliveira, C.; Ricol, D.; de Medina, S.; Bourdin, J.; Sastre-Garau, X.; Chopin, D.; Thiery, J.P.; Radvanyi, F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat. Genet. 1999, 23, 18–20. [Google Scholar]
- Neilson, K.M.; Friesel, R. Ligand-independent activation of fibroblast growth factor receptors by point mutations in the extracellular, transmembrane, and kinase domains. J. Biol. Chem. 1996, 271, 25049–25057. [Google Scholar] [CrossRef]
- Sun, M.; Hillmann, P.; Hofmann, B.T.; Hart, J.R.; Vogt, P.K. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 15547–15552. [Google Scholar]
- Jaiswal, B.S.; Janakiraman, V.; Kljavin, N.M.; Chaudhuri, S.; Stern, H.M.; Wang, W.; Kan, Z.; Dbouk, H.A.; Peters, B.A.; Waring, P.; et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 2009, 16, 463–474. [Google Scholar] [CrossRef]
- Huang, C.H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Gabelli, S.B.; Amzel, L.M. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007, 318, 1744–1748. [Google Scholar]
- Miled, N.; Yan, Y.; Hon, W.C.; Perisic, O.; Zvelebil, M.; Inbar, Y.; Schneidman-Duhovny, D.; Wolfson, H.J.; Backer, J.M.; Williams, R.L. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007, 317, 239–242. [Google Scholar] [CrossRef]
- Zhao, J.J.; Liu, Z.; Wang, L.; Shin, E.; Loda, M.F.; Roberts, T.M. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2005, 102, 18443–18448. [Google Scholar]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar]
- Han, S.Y.; Kato, H.; Kato, S.; Suzuki, T.; Shibata, H.; Ishii, S.; Shiiba, K.; Matsuno, S.; Kanamaru, R.; Ishioka, C. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 2000, 60, 3147–3151. [Google Scholar]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar]
- Greulich, H.; Pollock, P.M. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol. Med. 2011, 17, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar]
- Montecchi-Palazzi, L.; Beavis, R.; Binz, P.A.; Chalkley, R.J.; Cottrell, J.; Creasy, D.; Shofstahl, J.; Seymour, S.L.; Garavelli, J.S. The PSI-MOD community standard for representation of protein modification data. Nat. Biotechnol. 2008, 26, 864–866. [Google Scholar] [CrossRef]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Urick, M.E.; Rudd, M.L.; Godwin, A.K.; Sgroi, D.; Merino, M.; Bell, D.W. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011, 71, 4061–4067. [Google Scholar]
- Demiroglu, A.; Steer, E.J.; Heath, C.; Taylor, K.; Bentley, M.; Allen, S.L.; Koduru, P.; Brody, J.P.; Hawson, G.; Rodwell, R.; et al. The t(8;22) in chronic myeloid leukemia fuses BCR to FGFR1: Transforming activity and specific inhibition of FGFR1 fusion proteins. Blood 2001, 98, 3778–3783. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Schriml, L.M.; Arze, C.; Nadendla, S.; Chang, Y.W.; Mazaitis, M.; Felix, V.; Feng, G.; Kibbe, W.A. Disease Ontology: A backbone for disease semantic integration. Nucleic Acids Res. 2012, 40, D940–D946. [Google Scholar]
- De Coronado, S.; Wright, L.W.; Fragoso, G.; Haber, M.W.; Hahn-Dantona, E.A.; Hartel, F.W.; Quan, S.L.; Safran, T.; Thomas, N.; Whiteman, L. The NCI Thesaurus quality assurance life cycle. J. Biomed. Inform. 2009, 42, 530–539. [Google Scholar]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef]
- Bai, A.; Meetze, K.; Vo, N.Y.; Kollipara, S.; Mazsa, E.K.; Winston, W.M.; Weiler, S.; Poling, L.L.; Chen, T.; Ismail, N.S.; et al. GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res. 2010, 70, 7630–7639. [Google Scholar]
- Le Novere, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.I.; Wimalaratne, S.M.; et al. The Systems Biology Graphical Notation. Nat. Biotechnol. 2009, 27, 735–741. [Google Scholar] [CrossRef]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; Kerssemakers, J.; et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010, 38, D525–D531. [Google Scholar] [CrossRef]
- Overington, J. ChEMBL. An interview with John Overington, team leader, chemogenomics at the European Bioinformatics Institute Outstation of the European Molecular Biology Laboratory (EMBL-EBI). Interview by Wendy A. Warr. J. Comput. Aided Mol. Des. 2009, 23, 195–198. [Google Scholar] [CrossRef]
- Funahashi, A.; Tanimura, N.; Morohashi, M.; Kitano, H. CellDesigner: A process diagram editor for gene-regulatory and biochemical networks. BioSilico 2003, 1, 159–162. [Google Scholar] [CrossRef]
- Killcoyne, S.; Carter, G.W.; Smith, J.; Boyle, J. Cytoscape: A community-based framework for network modeling. Methods Mol. Biol. 2009, 563, 219–239. [Google Scholar]
- Junker, B.H.; Klukas, C.; Schreiber, F. VANTED: A system for advanced data analysis and visualization in the context of biological networks. BMC Bioinformatics 2006, 7, 109. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- International Cancer Genome Consortium. Available online: http://www.icgc.org (accessed on 20 September 2012).
- Caspi, R.; Altman, T.; Dale, J.M.; Dreher, K.; Fulcher, C.A.; Gilham, F.; Kaipa, P.; Karthikeyan, A.S.; Kothari, A.; Krummenacker, M.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010, 38, D473–D479. [Google Scholar]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar]
- Bello, S.M.; Richardson, J.E.; Davis, A.P.; Wiegers, T.C.; Mattingly, C.J.; Dolan, M.E.; Smith, C.L.; Blake, J.A.; Eppig, J.T. Disease model curation improvements at Mouse Genome Informatics. Database (Oxford) 2012, 2012, ar063. [Google Scholar]
- Davis, A.P.; King, B.L.; Mockus, S.; Murphy, C.G.; Saraceni-Richards, C.; Rosenstein, M.; Wiegers, T.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2011. Nucleic Acids Res. 2011, 39, D1067–D1072. [Google Scholar]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar]
- Wu, G.; Feng, X.; Stein, L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010, 11, R53. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Milacic, M.; Haw, R.; Rothfels, K.; Wu, G.; Croft, D.; Hermjakob, H.; D'Eustachio, P.; Stein, L. Annotating Cancer Variants and Anti-Cancer Therapeutics in Reactome. Cancers 2012, 4, 1180-1211. https://doi.org/10.3390/cancers4041180
Milacic M, Haw R, Rothfels K, Wu G, Croft D, Hermjakob H, D'Eustachio P, Stein L. Annotating Cancer Variants and Anti-Cancer Therapeutics in Reactome. Cancers. 2012; 4(4):1180-1211. https://doi.org/10.3390/cancers4041180
Chicago/Turabian StyleMilacic, Marija, Robin Haw, Karen Rothfels, Guanming Wu, David Croft, Henning Hermjakob, Peter D'Eustachio, and Lincoln Stein. 2012. "Annotating Cancer Variants and Anti-Cancer Therapeutics in Reactome" Cancers 4, no. 4: 1180-1211. https://doi.org/10.3390/cancers4041180