A microRNA Link to Glioblastoma Heterogeneity

Abstract
:1. Introduction
2. Glioma Stem Cells (GSCs)
3. Tumor Associated Vasculature
4. GBM Heterogeneity Orchestrated by Non-Physiological Levels of Oxygen, pH, and Metabolites
5. Tumor Hypoxia
6. Identifying Underlying Mechanisms for Diverse Events in Heterogeneous Tumors like GBMs
7. microRNA: Biogenesis and Functions
8. miRNA Targets with Established Role in Gliomagenesis

{kind=link}
| MicroRNA functions | MicroRNAs | Relevant targets | Reference |
|---|---|---|---|
| Glioma stem cells | miR-124 | PTBP1, CDK6, SCP1, LAMC1, ITGB1 | [106,107,108,109,110,111,112,113] |
| miR-137 | CDK6 | ||
| miR-128 | Bmi-1, E2F3A | ||
| miR-7 | EGFR, IRS2 | ||
| miR-425 | |||
| miR-486 | |||
| miR-451 | |||
| miR-34a | Notch1, Notch2 | ||
| miR-326 | Notch | ||
| Pro-agiomiRs | miR-126, | Spred-1, PIK3R2 | [114,115,116,117,118,119,120,121,122,123,124,125,126] |
| miR-17-92 cluster | TSP-1 | ||
| miR-378 | Sufu, Fus-1 | ||
| miR-296 | HGS | ||
| miR-21 | |||
| miR-210 | Ephrin-A3 | ||
| miR-130a | HOXA5, GAX | ||
| miR-125b | MAZ | ||
| miR-101 | EZH2 | ||
| Anti-angiomiRs | miR 221/222 | c-Kit | [127,128] |
| miR-320 | IGF-1 | ||
| Aerobic glycolysis & related signaling pathways | miR-326 | PKM2 | [129,130,131] |
| miR-21 | PI3K/AKT/P53 | ||
| miR-26a, | PI3K/AKT | ||
| miR-221/222 | PI3K/AKT | ||
| miR-451 | LKB1/AMPK | ||
| miR-128 | AKT | ||
| miR-25 | P53 | ||
| mir-32 | P53 | ||
| miR-34a | MYC | ||
| miR-155/143 | CEBPb | ||
| mir-23a/b | MYC | ||
| Hypoxamirs | miR-20b | Sirt1 | [132,133,134,135] |
| miR-210 | ISCU1/2 | ||
| miR-199a | HIF-1α, Sirt1 |
| Glioma de novo pathway (KEGG) | |||
|---|---|---|---|
| S. No. | Gene name | miRNA(s) | |
| 1 | PDGFA | hsa-let-7d | |
| 2 | PDGFB | hsa-miR-146b-3p | |
| 3 | EGFR | hsa-miR-1, hsa-miR-128, hsa-miR-146a, hsa-miR-16, hsa-miR-21, hsamiR-7 | |
| 4 | PDGFRA | hsa-let-7b | |
| 5 | PDGFRB | hsa-miR-224 | |
| 6 | IGF1R | hsa-miR-7, hsa-miR-122, hsa-miR-133b, hsa-miR-138, hsa-miR-145, hsamiR-194 | |
| 7 | GRB2 | hsa-miR-433 | |
| 8 | CALM2, CALM3 | hsa-miR-1 | |
| 9 | CAMK2G | hsa-miR-219-5p | |
| 10 | HRAS | hsa-let-7a, hsa-miR-143, hsa-miR-181a, | |
| 11 | KRAS | hsa-let-7a, hsa-let-7g, hsa-miR-143, hsa-miR-155, hsa-miR-181c, hsa-miR-18a*, hsa-miR-96 | |
| 12 | NRAS | hsa-let-7a, hsa-let-7b, hsa-let-7c, hsa-miR-20a | |
| 13 | PIK3R1 | hsa-miR-29a | |
| 14 | PIK3R2 | hsa-miR-126 | |
| 15 | PTEN | hsa-miR-106b, hsa-miR-141, hsa-miR-17, hsa-miR-18a, hsa-miR-19a, hsa-miR-19b, hsa-miR-20a, hsa-miR-21, hsa-miR-214, hsa-miR-216a, hsa-miR-217, hsa-miR-221, hsa-miR-222, hsa-miR-26a, hsa-miR-494 | |
| 16 | ARAF | hsa-miR-124 | |
| 17 | RAF1 | hsa-miR-125b, hsa-miR-7 | |
| 18 | AKT1 | hsa-miR-125b, hsa-miR-149*, hsa-miR-185, hsa-miR-451 | |
| 19 | MAP2K1 | hsa-miR-34a, hsa-miR-424 | |
| 20 | MAPK1 | hsa-miR-199b-3p | |
| 21 | CDKN2A | hsa-miR-24, hsa-let-7g, hsa-miR-125b | |
| 22 | TP53 | hsa-miR-25, hsa-miR-30d, hsa-miR-612, hsa-miR-125a-5p, hsa-miR-125b, hsa-miR-1285, hsa-miR-15a, hsa-miR-16, hsa-miR-221, hsa-miR-222 | |
| 23 | CDKN1A | hsa-miR-106a, hsa-miR-106b, hsa-miR-125a-5p, hsa-miR-132, hsa-miR-145, hsa-miR-146a, hsa-miR-146b-5p, hsa-miR-17, hsa-miR-182, hsa-miR-208a, hsa-miR-208b, hsa-miR-20a, hsa-miR-20b, hsa-miR-28-5p, hsa-miR-298, hsa-miR-299-5p, hsa-miR-302a, hsa-miR-345, hsa-miR-363, hsa-miR-372, hsa-miR-423-3p, hsa-miR-503, hsa-miR-515-3p, hsa-miR-519d, hsa-miR-519e, hsa-miR-520a-3p, hsa-miR-520b, hsa-miR-520h, hsa-miR-572, hsa-miR-639, hsa-miR-654-3p, hsa-miR-657,hsa-miR-93, hsa-miR-942, hsa-miR-96 | |
| 24 | CDKN2A | hsa-let-7g, hsa-miR-125b, hsa-miR-24 | |
| 25 | CCND1 | hsa-let-7b, hsa-miR-106b, hsa-miR-15a, hsa-miR-15b, hsa-miR-16, hsa-miR-16-1*, hsa-miR-17, hsa-miR-193b, hsa-miR-19a, hsa-miR-20a, hsa-miR-302a, hsa-miR-302c, hsa-miR-34a, hsa-miR-424, hsa-miR-449a, hsa-miR-503 | |
| 26 | CDK4 | hsa-miR-124, hsa-miR-145, hsa-miR-24, hsa-miR-302a, hsa-miR-34a, hsa-miR-34b, hsa-miR-34b*, hsa-miR-34c-5p | |
| 27 | CDK6 | hsa-miR-124, hsa-miR-137, hsa-miR-16, hsa-miR-185, hsa-miR-203, hsa-miR-29a, hsa-miR-29b, hsa-miR-29c, hsa-miR-30a*, hsa-miR-34a, hsa-miR-34b, hsa-miR-34b*, hsa-miR-424, hsa-miR-449a | |
| 28 | RB1 | hsa-miR-106a, hsa-miR-106b, hsa-miR-17, hsa-miR-20a, hsa-miR-23b, hsa-miR-26a, hsa-miR-335, hsa-miR-675 | |
| 29 | E2F1 | hsa-let-7a, hsa-miR-106a, hsa-miR-106b, hsa-miR-126, hsa-miR-149*, hsa-miR-17, hsa-miR-20a, hsa-miR-21, hsa-miR-223, hsa-miR-23b, hsa-miR-330-3p, hsa-miR-34a, hsa-miR-93, hsa-miR-98 | |
| TGF beta Signaling pathway (KEGG) | |||
| S. No. | Gene name | miRNA(s) | |
| 1 | BMP7 | hsa-miR-22, hsa-miR-342-3p | |
| 2 | THBS1 | hsa-let-7a, hsa-let-7b, hsa-miR-1, hsa-miR-17, hsa-miR-20a, hsa-miR-30a*, hsa-miR-92a, hsa-miR-98 | |
| 3 | TGFB1 | hsa-miR-24 | |
| 4 | TGFB2 | hsa-miR-141 | |
| 5 | TGFB3 | hsa-miR-29a | |
| 6 | LEFTY1, LEFTY2 | hsa-miR-302a, hsa-miR-302d | |
| 7 | BMPR1B | hsa-miR-125b | |
| 8 | BMPR2 | hsa-miR-129-5p, hsa-miR-17, hsa-miR-19a, hsa-miR-19b, hsa-miR-20a, hsa-miR21, hsa-miR-92a | |
| 9 | TGFBR1 | hsa-let-7c, hsa-miR-128, hsa-miR-204 | |
| 10 | TGFBR2 | hsa-miR-17, hsa-miR-18a, hsa-miR-19a, hsa-miR-19b, hsa-miR-204, hsa-miR-20a, hsa-miR-21, hsa-miR-302b, hsa-miR-372, hsa-miR-590-5p, hsa-miR-92a | |
| 11 | ACVR1 | hsa-miR-197 | |
| 12 | ACVR2A | hsa-miR-16 | |
| 13 | ACVR1C | hsa-miR-122, hsa-miR-147, hsa-miR-22, hsa-miR-376a, hsa-miR376a*, hsa-miR-376b, hsa-miR-376c, hsa-miR-412 | |
| 14 | SMAD1 | hsa-miR-155, hsa-miR-26a | |
| 15 | SMAD2 | hsa-miR-155 | |
| 16 | SMAD5 | hsa-miR-155 | |
| 17 | RHOA | hsa-miR-122, hsa-miR-155, hsa-miR-185, hsa-miR-31 | |
| 18 | ROCK1 | hsa-miR-146a, hsa-miR-584 | |
| 19 | ROCK2 | hsa-miR-138 | |
| 20 | SMAD4 | hsa-miR-17, hsa-miR-18a, hsa-miR-19a, hsa-miR-20a, hsa-miR-26a, hsa-miR-483-3p, hsa-miR-92a | |
| 21 | MAPK1 | hsa-miR-199b-3p | |
| 22 | ID1 | hsa-miR-100, hsa-miR-520h | |
| 23 | ID3 | hsa-miR-520h | |
| 24 | RBL1, RBL2 | hsa-miR-106b, hsa-miR-17, hsa-miR-20a | |
| 25 | E2F5 | hsa-let-7b, hsa-miR-192, hsa-miR-34a | |
| 26 | CREBBP | hsa-miR-324-3p | |
| 27 | EP300 | hsa-miR-182, hsa-miR-194, hsa-miR-200b, hsa-miR-200c, hsa-miR-26b, hsa-miR-374a, hsa-miR-429 | |
| 28 | SP1 | hsa-miR-124, hsa-miR-218, hsa-miR-29b | |
| 29 | MYC | hsa-let-7a, hsa-let-7g, hsa-miR-145, hsa-miR-17, hsa-miR-20a, hsa-miR-21, hsa-miR-24, hsa-miR-26a, hsa-miR-34a, hsa-miR-34b, hsa-miR-34b*, hsa-miR-34c-5p, hsa-miR-378, hsa-miR-98 | |
| Notch signaling pathway (KEGG) | |||
| S. No. | Gene name | miRNA(s) | |
| 1 | DLL1 | hsa-miR-34a | |
| 2 | JAG1 | hsa-miR-200c, hsa-miR-21, hsa-miR-34a | |
| 3 | ADAM17 | hsa-miR-122 | |
| 4 | NOTCH1 | hsa-miR-129-5p, hsa-miR-144, hsa-miR-30a, hsa-miR-326, hsa-miR-34a | |
| 5 | NOTCH2 | hsa-miR-1, hsa-miR-16, hsa-miR-181c, hsa-miR-326, hsa-miR-34a | |
| 6 | NOTCH3 | hsa-miR-206 | |
| 7 | NOTCH4 | hsa-miR-181c | |
| 8 | DVL2 | hsa-miR-324-3p | |
| 9 | NUMB | hsa-miR-31 | |
| 10 | NUMBL | hsa-miR-122 | |
| 11 | PSEN1 | hsa-miR-562 | |
| 12 | CREBBP | hsa-miR-324-3p | |
| 13 | EP300 | hsa-miR-182, hsa-miR-194, hsa-miR-200b, hsa-miR-200c, hsa-miR-26b, hsa-miR374a, hsa-miR-429 | |
| 14 | KAT2B | hsa-miR-106b, hsa-miR-181a, hsa-miR-181b, hsa-miR-19a, hsa-miR-19b, hsa-miR-25, hsa-miR-32, hsa-miR-92a, hsa-miR-93 | |
| 15 | CTBP1 | hsa-miR-137 | |
| 16 | HDAC1 | hsa-miR-449a | |
| 17 | NCOR2 | hsa-miR-10a, hsa-miR-10b | |
| 18 | HES1 | hsa-miR-199b-5p, hsa-miR-23a | |
| VEGF signaling pathway (KEGG) | |||
| S. No. | Gene name | miRNA(s) | |
| 1 | VEGFA | hsa-miR-106a, hsa-miR-106b, hsa-miR-107, hsa-miR-125a-5p, hsa-miR-126, hsa-miR-134, hsa-miR-140-5p, hsa-miR-147, hsa-miR-150, hsa-miR-15a, hsa-miR-15b, hsa-miR-16, hsa-miR-17, hsa-miR-195, hsa-miR-205, hsa-miR-20a, hsa-miR-20b, hsa-miR-29b, hsa-miR-302d, hsa-miR-330-3p, hsa-miR-34a, hsa-miR-34b, hsa-miR-361-5p, hsa-miR-372, hsa-miR-373, hsa-miR-378, hsa-miR-383, hsa-miR-504, hsa-miR-520g, hsa-miR-520h, hsa-miR-93 | |
| 2 | CDC42 | hsa-miR-137, hsa-miR-185, hsa-miR-216a, hsa-miR-224, hsa-miR-330-3p, hsa-miR-608 | |
| 3 | PIK3R1 | hsa-miR-29a | |
| 4 | PIK3R2 | hsa-miR-126 | |
| 5 | MAPK11 | hsa-miR-122 | |
| 6 | MAPK14 | hsa-miR-124, hsa-miR-24 | |
| 7 | AKT1 | hsa-miR-125b, hsa-miR-149*, hsa-miR-185, hsa-miR-451 | |
| 8 | HRAS | hsa-let-7a, hsa-miR-143, hsa-miR-181a | |
| 9 | KRAS | hsa-let-7a, hsa-let-7g, hsa-miR-143, hsa-miR-155, hsa-miR-181c, hsa-miR-18a*, hsa-miR-96 | |
| 10 | NRAS | hsa-let-7a, hsa-let-7b, hsa-let-7c, hsa-miR-20a | |
| 11 | RAF1 | hsa-miR-125b, hsa-miR-7 | |
| 12 | PPP3CA | hsa-miR-145, hsa-miR-30a | |
| 13 | PPP3R1 | hsa-miR-30a | |
| 14 | NFAT5 | hsa-miR-24, hsa-miR-31 | |
| 15 | NFATC2 | hsa-miR-184 | |
| 16 | PTK2 | hsa-miR-193a-3p | |
| 17 | RAC1 | hsa-miR-122, hsa-miR-194 | |
| 18 | CASP9 | hsa-let-7a, hsa-miR-133a | |
| 19 | PTGS2 | hsa-let-7b, hsa-miR-101, hsa-miR-16, hsa-miR-26b | |
| 20 | PLA2G4B | hsa-miR-338-3p | |
| 21 | MAPK1 | hsa-miR-199b-3p | |
| PDGF Signaling Pathway (Biocarta) | |||
| S. No. | Gene name | miRNA(s) | |
| 1 | PDGFA | hsa-let-7d | |
| 2 | PDGFRA | hsa-let-7b, | |
| 3 | GRB2 | hsa-miR-433 | |
| 4 | JAK1 | hsa-miR-17 | |
| 5 | RASA1 | hsa-miR-335 | |
| 6 | STAT1 | hsa-miR-145 | |
| 7 | STAT3 | hsa-miR-125b, hsa-miR-20b | |
| 8 | STAT5A | hsa-miR-222 | |
| 9 | SRF | hsa-miR-122 | |
| 10 | FOS | hsa-miR-101, hsa-miR-221, hsa-miR-222 | |
| 11 | JUN | hsa-miR-30a | |
| 12 | HRAS | hsa-let-7a, hsa-miR-143, hsa-miR-181a, | |
| 13 | RAF1 | hsa-miR-125b, hsa-miR-7 | |
| 14 | MAP2K1 | hsa-miR-34a, hsa-miR-424 | |
| 15 | MAP3K1 | hsa-miR-192 | |
9. miRNAs in Glioma Stem Cells (GSCs)
10. miRNAs in Tumor Associated Angiogenesis
11. miRNAs in Tumor Metabolism
12. miRNA in Tumor Hypoxia
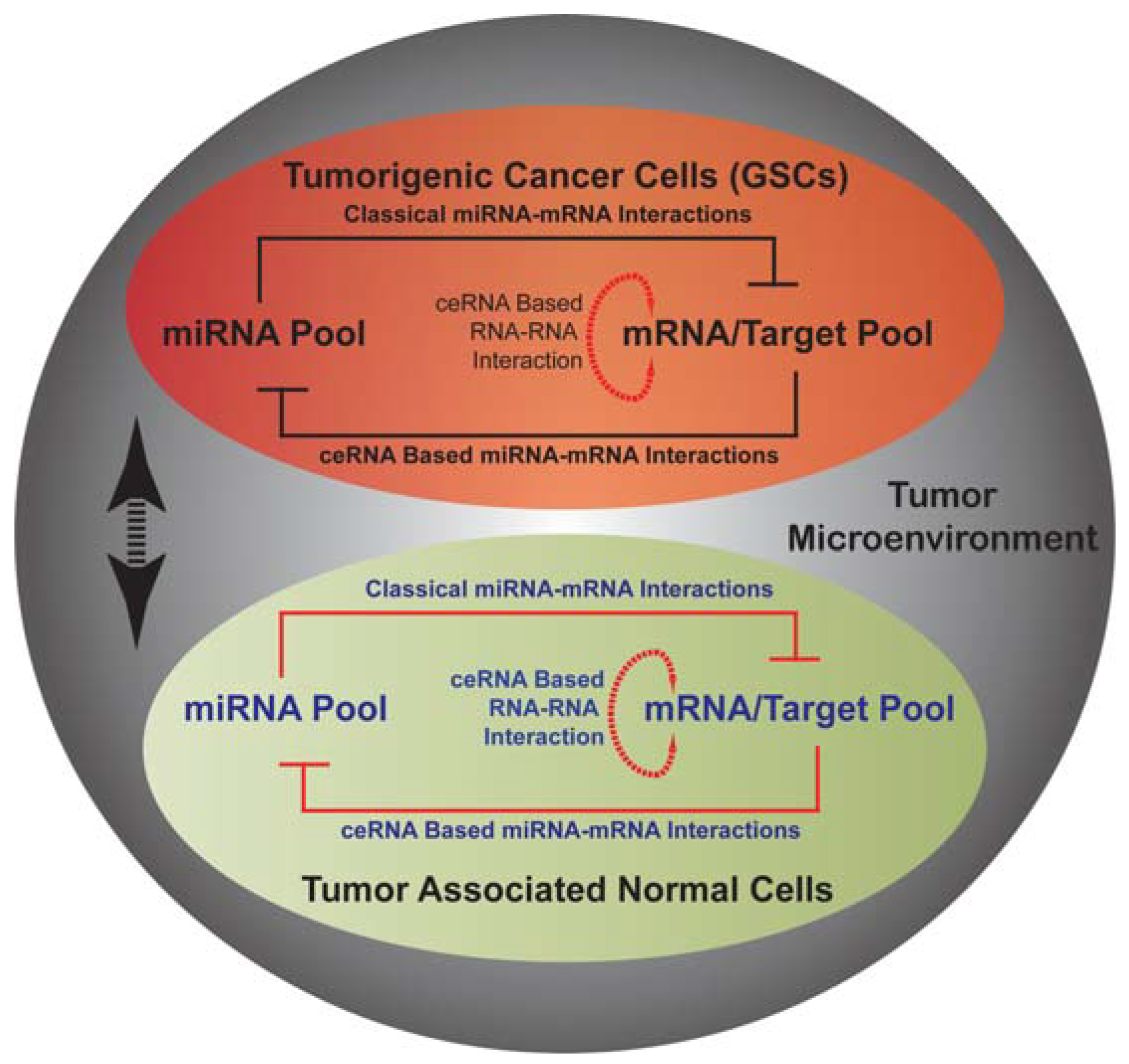
13. miRNA Mediated Networks in Glioma
14. Conclusions
Acknowledgements
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-Year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hjelmeland, A.B.; Lathia, J.D.; Sathornsumetee, S.; Rich, J.N. Twisted tango: Brain tumor neurovascular interactions. Nat. Neurosci. 2011, 14, 1375–1381. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of cd133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Salmaggi, A.; Boiardi, A.; Gelati, M.; Russo, A.; Calatozzolo, C.; Ciusani, E.; Sciacca, F.L.; Ottolina, A.; Parati, E.A.; La Porta, C.; et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia 2006, 54, 850–860. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’Avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and mgmt expression in glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; de Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; de Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in pdgf-induced glioma cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neurooncology 2010, 12, 1113–1125. [Google Scholar]
- Deleyrolle, L.P.; Harding, A.; Cato, K.; Siebzehnrubl, F.A.; Rahman, M.; Azari, H.; Olson, S.; Gabrielli, B.; Osborne, G.; Vescovi, A.; et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 2011, 134, 1331–1343. [Google Scholar] [CrossRef]
- Knizetova, P.; Ehrmann, J.; Hlobilkova, A.; Vancova, I.; Kalita, O.; Kolar, Z.; Bartek, J. Autocrine regulation of glioblastoma cell cycle progression, viability and radioresistance through the vegf-vegfr2 (kdr) interplay. Cell Cycle 2008, 7, 2553–2561. [Google Scholar]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar]
- Assanah, M.C.; Bruce, J.N.; Suzuki, S.O.; Chen, A.; Goldman, J.E.; Canoll, P. PDGF stimulates the massive expansion of glial progenitors in the neonatal forebrain. Glia 2009, 57, 1835–1847. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef]
- Chirasani, S.R.; Sternjak, A.; Wend, P.; Momma, S.; Campos, B.; Herrmann, I.M.; Graf, D.; Mitsiadis, T.; Herold-Mende, C.; Besser, D.; et al. Bone morphogenetic protein-7 release from endogenous neural precursor cells suppresses the tumourigenicity of stem-like glioblastoma cells. Brain 2010, 133, 1961–1972. [Google Scholar] [CrossRef]
- Walzlein, J.H.; Synowitz, M.; Engels, B.; Markovic, D.S.; Gabrusiewicz, K.; Nikolaev, E.; Yoshikawa, K.; Kaminska, B.; Kempermann, G.; Uckert, W.; et al. The antitumorigenic response of neural precursors depends on subventricular proliferation and age. Stem Cells 2008, 26, 2945–2954. [Google Scholar] [CrossRef]
- Sanai, N.; Nguyen, T.; Ihrie, R.A.; Mirzadeh, Z.; Tsai, H.H.; Wong, M.; Gupta, N.; Berger, M.S.; Huang, E.; Garcia-Verdugo, J.M.; et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011, 478, 382–386. [Google Scholar]
- Dwain, I.; Xiangpeng, Y.; Zeng, Z.; Patricia, T.; Joh, S.Y. Neural stem cells—A promising potential therapy for brain tumors. Curr. Stem Cell Res. Ther. 2006, 1, 79–84. [Google Scholar] [CrossRef]
- Mokry, J.; Cizkova, D.; Filip, S.; Ehrmann, J.; Osterreicher, J.; Kolar, Z.; English, D. Nestin expression by newly formed human blood vessels. Stem Cells Dev. 2004, 13, 658–664. [Google Scholar]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Brat, D.J.; van Meir, E.G. Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: A new world of angiogenesis research. Am. J. Pathol. 2001, 158, 789–796. [Google Scholar] [CrossRef]
- Zadeh, G.; Koushan, K.; Baoping, Q.; Shannon, P.; Guha, A. Role of angiopoietin-2 in regulating growth and vascularity of astrocytomas. J. Oncol. 2010, 2010, 659231. [Google Scholar]
- Wesseling, P.; Schlingemann, R.O.; Rietveld, F.J.; Link, M.; Burger, P.C.; Ruiter, D.J. Early and extensive contribution of pericytes/vascular smooth muscle cells to microvascular proliferation in glioblastoma multiforme: An immuno-light and immuno-electron microscopic study. J. Neuropathol. Exp. Neurol. 1995, 54, 304–310. [Google Scholar]
- Ahluwalia, M.S.; Gladson, C.L. Progress on antiangiogenic therapy for patients with malignant glioma. J. Oncol. 2010, 2010, 689018. [Google Scholar]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Takezaki, T.; Hide, T.; Takanaga, H.; Nakamura, H.; Kuratsu, J.; Kondo, T. Essential role of the hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 2011, 102, 1306–1312. [Google Scholar]
- Dong, J.; Grunstein, J.; Tejada, M.; Peale, F.; Frantz, G.; Liang, W.C.; Bai, W.; Yu, L.; Kowalski, J.; Liang, X.; et al. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004, 23, 2800–2810. [Google Scholar]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of ldh-a: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Barker, F.G., 2nd; Davis, R.L.; Chang, S.M.; Prados, M.D. Necrosis as a prognostic factor in glioblastoma multiforme. Cancer 1996, 77, 1161–1166. [Google Scholar] [CrossRef]
- Brat, D.J.; Kaur, B.; van Meir, E.G. Genetic modulation of hypoxia induced gene expression and angiogenesis: Relevance to brain tumors. Front. Biosci. 2003, 8, d100–d116. [Google Scholar] [CrossRef]
- Burger, P.C.; Vollmer, R.T. Histologic factors of prognostic significance in the glioblastoma multiforme. Cancer 1980, 46, 1179–1186. [Google Scholar] [CrossRef]
- Evans, S.M.; Judy, K.D.; Dunphy, I.; Jenkins, W.T.; Nelson, P.T.; Collins, R.; Wileyto, E.P.; Jenkins, K.; Hahn, S.M.; Stevens, C.W.; et al. Comparative measurements of hypoxia in human brain tumors using needle electrodes and EF5 binding. Cancer Res. 2004, 64, 1886–1892. [Google Scholar]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Kim, J.W.; Gao, P.; Dang, C.V. Effects of hypoxia on tumor metabolism. Cancer Metastasis Rev. 2007, 26, 291–298. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar]
- Maslov, S.; Sneppen, K. Specificity and stability in topology of protein networks. Science 2002, 296, 910–913. [Google Scholar] [CrossRef]
- Kim, J.; Chu, J.; Shen, X.; Wang, J.; Orkin, S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell 2008, 132, 1049–1061. [Google Scholar]
- Orkin, S.H.; Wang, J.; Kim, J.; Chu, J.; Rao, S.; Theunissen, T.W.; Shen, X.; Levasseur, D.N. The transcriptional network controlling pluripotency in ES cells. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 195–202. [Google Scholar] [CrossRef]
- Sumazin, P.; Yang, X.; Chiu, H.S.; Chung, W.J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An extensive microrna-mediated network of rna-rna interactions regulates established oncogenic pathways in glioblastoma. Cell 2011, 147, 370–381. [Google Scholar]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. Mirecords: An integrated resource for microrna-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human micrornas are processed from capped, polyadenylated transcripts that can also function as mrnas. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by rna polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar]
- Seitz, H.; Youngson, N.; Lin, S.P.; Dalbert, S.; Paulsen, M.; Bachellerie, J.P.; Ferguson-Smith, A.C.; Cavaille, J. Imprinted microrna genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat. Genet. 2003, 34, 261–262. [Google Scholar]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The mirtron pathway generates microrna-class regulatory RNAs in drosophila. Cell 2007, 130, 89–100. [Google Scholar]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef]
- Zhang, H.; Kolb, F.A.; Jaskiewicz, L.; Westhof, E.; Filipowicz, W. Single processing center models for human Dicer and bacterial RNase III. Cell 2004, 118, 57–68. [Google Scholar] [CrossRef]
- Mourelatos, Z.; Dostie, J.; Paushkin, S.; Sharma, A.; Charroux, B.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Mirnps: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002, 16, 720–728. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Pillai, R.S.; Artus, C.G.; Filipowicz, W. Tethering of human ago proteins to mRNA mimics the miRNA-mediated repression of protein synthesis. RNA 2004, 10, 1518–1525. [Google Scholar]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Shomron, N.; Sandberg, R.; Hornstein, E.; Kitzman, J.; Burge, C.B. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA 2007, 13, 1894–1910. [Google Scholar]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef]
- Zhang, Y.; Dutta, A.; Abounader, R. The role of microRNAs in glioma initiation and progression. Front. Biosci. 2012, 17, 700–712. [Google Scholar] [CrossRef]
- Sana, J.; Hajduch, M.; Michalek, J.; Vyzula, R.; Slaby, O. MicroRNAs and glioblastoma: Roles in core signalling pathways and potential clinical implications. J. Cell. Mol. Med. 2011, 15, 1636–1644. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Han, L.; Zhang, A.; Zhang, C.; Zheng, Y.; Jiang, T.; Pu, P.; Jiang, C.; Kang, C. Downregulation of miR-221/222 sensitizes glioma cells to temozolomide by regulating apoptosis independently of p53 status. Oncol. Rep. 2012, 27, 854–860. [Google Scholar]
- Quintavalle, C.; Garofalo, M.; Zanca, C.; Romano, G.; Iaboni, M.; del Basso de Caro, M.; Martinez-Montero, J.C.; Incoronato, M.; Nuovo, G.; Croce, C.M.; et al. MiR-221/222 overexpession in human glioblastoma increases invasiveness by targeting the protein phosphate ptpmu. Oncogene 2012, 31, 858–868. [Google Scholar]
- Hsu, S.D.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. Mirtarbase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011, 39, D163–D169. [Google Scholar]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar]
- Dong, H.; Luo, L.; Hong, S.; Siu, H.; Xiao, Y.; Jin, L.; Chen, R.; Xiong, M. Integrated analysis of mutations, miRNA and mRNA expression in glioblastoma. BMC Syst. Biol. 2010, 4, 163. [Google Scholar] [CrossRef]
- Gal, H.; Pandi, G.; Kanner, A.A.; Ram, Z.; Lithwick-Yanai, G.; Amariglio, N.; Rechavi, G.; Givol, D. MiR-451 and imatinib mesylate inhibit tumor growth of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2008, 376, 86–90. [Google Scholar]
- Godlewski, J.; Newton, H.B.; Chiocca, E.A.; Lawler, S.E. MicroRNAs and glioblastoma; the stem cell connection. Cell Death Differ. 2010, 17, 221–228. [Google Scholar] [CrossRef]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. MicroRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar]
- Silber, J.; Lim, D.A.; Petritsch, C.; Persson, A.I.; Maunakea, A.K.; Yu, M.; Vandenberg, S.R.; Ginzinger, D.G.; James, C.D.; Costello, J.F.; et al. MiR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008, 6, 14. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, T.; Li, R.; Liu, W.; Chen, Y.; Yan, X.; Gong, Y.; Yin, B.; Qiang, B.; Zhao, J.; et al. MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J. Mol. Med. 2009, 87, 43–51. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. Notch pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; diPierro, C.G.; Li, Y.; Chiocca, E.A.; et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 2009, 29, 15161–15168. [Google Scholar]
- Chen, Y.; Gorski, D.H. Regulation of angiogenesis through a microRNA (miR-130a) that down-regulates antiangiogenic homeobox genes gax and hoxa5. Blood 2008, 111, 1217–1226. [Google Scholar]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar]
- Fasanaro, P.; D’Alessandra, Y.; di Stefano, V.; Melchionna, R.; Romani, S.; Pompilio, G.; Capogrossi, M.C.; Martelli, F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J. Biol. Chem. 2008, 283, 15878–15883. [Google Scholar]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.; Srivastava, D. miR-126 Regulates angiogenic signaling and vascular integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar]
- Hua, Z.; Lv, Q.; Ye, W.; Wong, C.K.; Cai, G.; Gu, D.; Ji, Y.; Zhao, C.; Wang, J.; Yang, B.B.; et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One 2006, 1, e116. [Google Scholar]
- Kuehbacher, A.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Role of dicer and drosha for endothelial microRNA expression and angiogenesis. Circ. Res. 2007, 101, 59–68. [Google Scholar] [CrossRef]
- Kuhnert, F.; Mancuso, M.R.; Hampton, J.; Stankunas, K.; Asano, T.; Chen, C.Z.; Kuo, C.J. Attribution of vascular phenotypes of the murine EGFL7 locus to the microRNA miR-126. Development 2008, 135, 3989–3993. [Google Scholar] [CrossRef]
- Lee, D.Y.; Deng, Z.; Wang, C.H.; Yang, B.B. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc. Natl. Acad. Sci. USA 2007, 104, 20350–20355. [Google Scholar] [CrossRef]
- Pulkkinen, K.; Malm, T.; Turunen, M.; Koistinaho, J.; Yla-Herttuala, S. Hypoxia induces microRNA miR-210 in vitro and in vivo Ephrin-a3 and neuronal pentraxin 1 are potentially regulated by miR-210. FEBS Lett. 2008, 582, 2397–2401. [Google Scholar]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef]
- Wurdinger, T.; Tannous, B.A.; Saydam, O.; Skog, J.; Grau, S.; Soutschek, J.; Weissleder, R.; Breakefield, X.O.; Krichevsky, A.M. miR-296 Regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 2008, 14, 382–393. [Google Scholar] [CrossRef]
- Smits, M.; Wurdinger, T.; van Het Hof, B.; Drexhage, J.A.; Geerts, D.; Wesseling, P.; Noske, D.P.; Vandertop, W.P.; de Vries, H.E.; Reijerkerk, A. Myc-associated zinc finger protein (MAZ) is regulated by miR-125b and mediates VEGF-induced angiogenesis in glioblastoma. FASEB J. 2012. [Google Scholar]
- Smits, M.; Mir, S.E.; Nilsson, R.J.; van der Stoop, P.M.; Niers, J.M.; Marquez, V.E.; Cloos, J.; Breakefield, X.O.; Krichevsky, A.M.; Noske, D.P.; et al. Down-regulation of miR-101 in endothelial cells promotes blood vessel formation through reduced repression of EZH2. PLoS One 2011, 6, e16282. [Google Scholar]
- Poliseno, L.; Tuccoli, A.; Mariani, L.; Evangelista, M.; Citti, L.; Woods, K.; Mercatanti, A.; Hammond, S.; Rainaldi, G. MicroRNAs modulate the angiogenic properties of huvecs. Blood 2006, 108, 3068–3071. [Google Scholar] [CrossRef]
- Wang, X.H.; Qian, R.Z.; Zhang, W.; Chen, S.F.; Jin, H.M.; Hu, R.M. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin. Exp. Pharmacol. Physiol. 2009, 36, 181–188. [Google Scholar] [CrossRef]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; de Lay, M.; van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; de Marzo, A.M.; van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar]
- Olive, V.; Bennett, M.J.; Walker, J.C.; Ma, C.; Jiang, I.; Cordon-Cardo, C.; Li, Q.J.; Lowe, S.W.; Hannon, G.J.; He, L. miR-19 Is a key oncogenic component of miR-17-92. Genes Dev. 2009, 23, 2839–2849. [Google Scholar]
- Kulshreshtha, R.; Ferracin, M.; Negrini, M.; Calin, G.A.; Davuluri, R.V.; Ivan, M. Regulation of microRNA expression: The hypoxic component. Cell Cycle 2007, 6, 1426–1431. [Google Scholar]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef]
- Cascio, S.; D’Andrea, A.; Ferla, R.; Surmacz, E.; Gulotta, E.; Amodeo, V.; Bazan, V.; Gebbia, N.; Russo, A. miR-20b Modulates VEGF expression by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells. J. Cell. Physiol. 2010, 224, 242–249. [Google Scholar]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef]
- Stiles, C.D.; Rowitch, D.H. Glioma stem cells: A midterm exam. Neuron 2008, 58, 832–846. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, J.; Pan, T.; Zhou, J.; Gong, W.; Liu, N.; Fu, Z.; You, Y. MiR-125b is critical for the suppression of human U251 glioma stem cell proliferation. Brain Res. 2010, 1312, 120–126. [Google Scholar]
- Bruggeman, S.W.; Hulsman, D.; Tanger, E.; Buckle, T.; Blom, M.; Zevenhoven, J.; van Tellingen, O.; van Lohuizen, M. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell 2007, 12, 328–341. [Google Scholar]
- Dirks, P. Bmi1 and cell of origin determinants of brain tumor phenotype. Cancer Cell 2007, 12, 295–297. [Google Scholar] [CrossRef]
- Jin, X.; Yin, J.; Kim, S.H.; Sohn, Y.W.; Beck, S.; Lim, Y.C.; Nam, D.H.; Choi, Y.J.; Kim, H. EGFR-AKT-SMAD signaling promotes formation of glioma stem-like cells and tumor angiogenesis by ID3-driven cytokine induction. Cancer Res. 2011, 71, 7125–7134. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar]
- Shih, A.H.; Holland, E.C. Notch signaling enhances nestin expression in gliomas. Neoplasia 2006, 8, 1072–1082. [Google Scholar]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar]
- Zhang, X.P.; Zheng, G.; Zou, L.; Liu, H.L.; Hou, L.H.; Zhou, P.; Yin, D.D.; Zheng, Q.J.; Liang, L.; Zhang, S.Z.; et al. Notch activation promotes cell proliferation and the formation of neural stem cell-like colonies in human glioma cells. Mol. Cell. Biochem. 2008, 307, 101–108. [Google Scholar]
- Cao, Y.; Nagesh, V.; Hamstra, D.; Tsien, C.I.; Ross, B.D.; Chenevert, T.L.; Junck, L.; Lawrence, T.S. The extent and severity of vascular leakage as evidence of tumor aggressiveness in high-grade gliomas. Cancer Res. 2006, 66, 8912–8917. [Google Scholar]
- Rutten, E.H.; Doesburg, W.H.; Slooff, J.L. Histologic factors in the grading and prognosis of astrocytoma grade I-IV. J. Neurooncol. 1992, 13, 223–230. [Google Scholar]
- Wang, S.; Olson, E.N. Angiomirs—Key regulators of angiogenesis. Curr. Opin. Genet. Dev. 2009, 19, 205–211. [Google Scholar] [CrossRef]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neurooncology 2010, 12, 1102–1112. [Google Scholar]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009, 23, 1327–1337. [Google Scholar] [CrossRef]
- Nan, Y.; Han, L.; Zhang, A.; Wang, G.; Jia, Z.; Yang, Y.; Yue, X.; Pu, P.; Zhong, Y.; Kang, C. MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res. 2010, 1359, 14–21. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Zhang, J.; Sun, G.; Luo, H.; Kang, C.; Pu, P.; Jiang, T.; Liu, N.; You, Y. MicroRNAs involved in the EGFR/PTEN/AKT pathway in gliomas. J. Neurooncol. 2012, 106, 217–224. [Google Scholar] [CrossRef]
- Zhang, J.; Han, L.; Ge, Y.; Zhou, X.; Zhang, A.; Zhang, C.; Zhong, Y.; You, Y.; Pu, P.; Kang, C. miR-221/222 Promote malignant progression of glioma through activation of the Akt pathway. Int. J. Oncol. 2010, 36, 913–920. [Google Scholar]
- Zhou, X.; Ren, Y.; Moore, L.; Mei, M.; You, Y.; Xu, P.; Wang, B.; Wang, G.; Jia, Z.; Pu, P.; et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of pten status. Lab. Invest. 2010, 90, 144–155. [Google Scholar] [CrossRef]
- Papagiannakopoulos, T.; Shapiro, A.; Kosik, K.S. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008, 68, 8164–8172. [Google Scholar] [CrossRef]
- Suh, S.S.; Yoo, J.Y.; Nuovo, G.J.; Jeon, Y.J.; Kim, S.; Lee, T.J.; Kim, T.; Bakacs, A.; Alder, H.; Kaur, B.; et al. MicroRNAs/TP53 feedback circuitry in glioblastoma multiforme. Proc. Natl. Acad. Sci. USA 2012, 109, 5316–5321. [Google Scholar]
- Luan, S.; Sun, L.; Huang, F. MicroRNA-34a: A novel tumor suppressor in p53-mutant glioma cell line U251. Arch. Med. Res. 2010, 41, 67–74. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, L.F.; Zhang, H.W.; Hu, S.; Lu, M.H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.D.; et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar]
- Chan, S.Y.; Loscalzo, J. MicroRNA-210: A unique and pleiotropic hypoxamir. Cell Cycle 2010, 9, 1072–1083. [Google Scholar] [CrossRef]
- Lages, E.; Guttin, A.; El Atifi, M.; Ramus, C.; Ipas, H.; Dupre, I.; Rolland, D.; Salon, C.; Godfraind, C.; deFraipont, F.; et al. MicroRNA and target protein patterns reveal physiopathological features of glioma subtypes. PLoS One 2011, 6, e20600. [Google Scholar]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Sumazin, P.; Yang, X.; Chiu, H.S.; Chung, W.J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 2011, 147, 370–381. [Google Scholar]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; di Cunto, F.; et al. Coding-independent regulation of the tumor suppressor pten by competing endogenous mRNAs. Cell 2011, 147, 344–357. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Singh, S.K.; Vartanian, A.; Burrell, K.; Zadeh, G. A microRNA Link to Glioblastoma Heterogeneity. Cancers 2012, 4, 846-872. https://doi.org/10.3390/cancers4030846
Singh SK, Vartanian A, Burrell K, Zadeh G. A microRNA Link to Glioblastoma Heterogeneity. Cancers. 2012; 4(3):846-872. https://doi.org/10.3390/cancers4030846
Chicago/Turabian StyleSingh, Sanjay K., Alenoush Vartanian, Kelly Burrell, and Gelareh Zadeh. 2012. "A microRNA Link to Glioblastoma Heterogeneity" Cancers 4, no. 3: 846-872. https://doi.org/10.3390/cancers4030846