A New Player in the Development of TRAIL Based Therapies for Hepatocarcinoma Treatment: ATM Kinase

Abstract
:1. Role of Apoptosis in Cancer
2. Role of TRAIL Signalling in HCC Development and Therapy

{kind=link}
| TRAIL | Description | Company | Reference/Phase |
|---|---|---|---|
| Apo2L/TRAIL (AMG 951) | Soluble TRAIL activates TRAIL-R1 and TRAIL-R2 (DR4 and DR5) | Amgen/Genentech | Phase I/II Ongoing trial active, not recruiting. |
| HGS-ETR1 (Mapatumumab) | Humanized anti-TRAIL-R1 (DR4) agonistic mAb | Human Genome Science | Completed [13,14] More trials currently active or recruiting. |
| HGS-ETR2 (Lexatumumab) | Humanized anti-TRAIL-R2 (DR5) agonistic mAb | Human Genome Science | Phase I advanced solid tumors. Completed [12,15], More trials currently active or recruiting. |
| HGS-TR2J | Humanized anti-TRAIL-R2 (DR5) agonistic mAb | Human Genome Science | Phase I No ongoing trials [16]. |
| TRA-8 (CS-1008; Tigatuzumab) | Humanized anti-TRAIL-R2 (DR5) agonistic mAb | Daiiki Sankyo Inc. | More trials currently recruiting [17,18]. |
| Conatumumab (AMG 655) | Humanized anti-TRAIL-R2 (DR5) agonistic mAb | Amgen/Takeda | More trials currently active [19,20]. |
| Apomab | Humanized anti-TRAIL-R2 (DR5) agonistic mAb | Genentech | No ongoing trials [21]. |
| LBY135 | Chimeric anti-TRAIL-R2 (DR5) agonistic mAb | Novartis | Phase I/II: advanced solid tumors No ongoing trials [22]. |
3. Molecular Mechanisms That Trigger TRAIL Resistance in HCC
3.1. Role of the Expression of the Different TRAIL Receptors
3.2. c-FLIP Proteins
3.3. Bcl-2 Family
3.4. IAPs
3.5. NF-kappaB
3.6. Tyrosine Kinases
3.7. STAT Proteins
3.8. PI3K/Akt Pathway
4. Development of TRAIL-Based Combined Therapeutic Approaches in HCC
| Type of Combined Therapy | Agent | Mechanism of Action | References |
|---|---|---|---|
| DNA Damage drugs | 5-FU | downregulation of FLIP, upregulation of TRAIL receptors | [27] |
| Cisplatin | downregulation of FLIP, upregulation of TRAIL receptors | [60] | |
| Etoposide | upregulation of Bax, increased release of cytochrome c and DIABLO | [61,62] | |
| Inhibitors of target molecules | HDAC inhibitors (SAHA, valproic acid) | downregulation of FLIP, upregulation of TRAIL receptors | [63] |
| protesome inhibitors (bortezomib) | downregulation of FLIP,upregulation of TRAIL receptors, suppression of Akt pathway | [64] | |
| Cyclooxygenase (COX)-2 inhibitors (NS398 and CAY10404) | up-regulation of TRAIL receptors, down-regulation of both survivin and AKT signaling | [65] | |
| ABT-263 | inhibition of the Bcl-2 family | [66] | |
| Kinase inhibitor | Genistein (isoflavone, tyrosine kinase inhibitor) | increasd cleavage of Bid, suppression of p38 MAPK signaling | [67,68] |
| Quercitin (flavonoid, inhibitor of I-kappaB kinase) | downregulation of FLIP, upregulation of TRAIL receptors | [69] | |
| Flavopiridol (cyclin-dependent kinase) | upregulation of TRAIL receptors, down-regulation of survivin, FLIP and Bcl-xL | [70] | |
| Sorafenib (multi-kinase inhibitor) | downregulation of STAT3 phosphorylation, down-regulation of Mcl-1 | [71] | |
| JNK inhibitor (AS601245, SP600125) | enhancer of caspase-8 activity and the downstream recruitment of the mitochondrial machinery | [72] | |
| Glycogen synthase kinase-3 inhibitors (lithium and SB-415286) | enhancer of caspase-8 activity and the downstream recruitment of the mitochondrial machinery | [73] | |
| Casein kinase 2 (emodin) | upregulation of TRAIL receptors | [74] | |
| Janus kinase 2 inhibitor (AG490) | inhibition of STAT3, XIAP and survivin | [75] | |
| Src-kinase inhibitor (PP2) | inhibition of caspase-8 activity | [54] | |
| Natural Compound and Synthetic Drugs | Capsaicin | upregulation of TRAIL receptors | [76] |
| Flavonoid and flavonoid-like chemical compound (Wogonin, 5, 7-dimethoxyflavone) | downregulation of FLIP, upregulation of TRAIL receptors | [77] | |
| Parthenolide | inhibition of STAT3,upregulation of TRAIL receptors | [78] | |
| Butein | NF-kappaB inactivation, upregulation of TRAIL receptors | [79] | |
| beta-Ionone | upregulation of TRAIL receptors | [80] | |
| Synthetic cannabinoid | upregulation of TRAIL receptors | [81] | |
| 2-Phenyl-4-quinolone | upregulation of TRAIL receptors | [82] | |
| 8-Chloroadenosine | upregulation of TRAIL receptors | [83] | |
| Quinacrine | downregulation of MCL-1, upregulation of TRAIL receptors | [84] | |
| Curcumin | ROS-mediated upregulation of TRAIL Receptors | [85] | |
| J7, a methyl jasmonate derivative | ROS-mediated upregulation of TRAIL Receptors | [86] | |
| Guggulsterone | ROS-mediated upregulation of TRAIL Receptors | [87] | |
| Peroxiredoxin I | ROS-mediated upregulation of TRAIL Receptors | [88] | |
| Sulforaphane | ROS-mediated upregulation of TRAIL Receptors | [89] | |
| Interferon-alpha | downregulation of Bcl-2, upregulation of TRAIL receptors and of Caspase-8, NFkappaB inhibition | [52] | |
| Celecoxib | downregulation of FLIP | [90] | |
| Melittin | activation of CaMKII-TAK1-JNK/p38, inhibition of IkappaBalpha kinase-NFkappaB. | [91] |
4.1. DNA Damage Drugs
4.2. Inhibitors of Target Molecules, Kinase Inhibitors, Natural Compounds and Synthetic Drugs
4.3. siRNA Based Approaches
| Identifier | Cancer | TRAIL | Combined Treatment | Phase |
|---|---|---|---|---|
| NCT00712855 | HCC | mapatumumab | sorafenib | I |
| NCT01258608 | HCC | mapatumumab | sorafenib | II |
| NCT01033240 | Advanced HCCLiver CancerHepatic CancerLiver Neoplasms | tigatuzumab (CS-1008) | sorafenib | II |
| NCT00819169 | CRC NSCLCLocally AdvancedMetastatic Cancer Ovarian CancerPancreatic CancerSarcoma Solid Tumors | conatumumab (AMG655) | ganitumab (AMG 479) | II |
| NCT01327612 | Advanced Solid TumorsCRCLocally AdvancedLymphomaMetastatic CancerNSCLC Solid Tumor | conatumumab (AMG655) | FOLFOX6 ganitumab (AMG 479) bevacizumab | II |
| siRNAs Target | Reference |
|---|---|
| Gli2 | [97] |
| COX2 | [98] |
| DNA methyltransferases (DNMTs) | [99] |
| hTERT | [100] |
| Notch-1 | [101] |
| Caveolin | [102] |
| XIAP | [103] |
| Survivin | [96] |
| Mcl-1 | [104] |
5. ATM Kinase: A Novel Player in the Development of TRAIL Based Approaches for Cancer Therapy
5.1. ATM Kinase: An Essential Guarantee for Genomic Stability
5.2. Role of ATM in Liver Homeostasis and Carcinogenesis
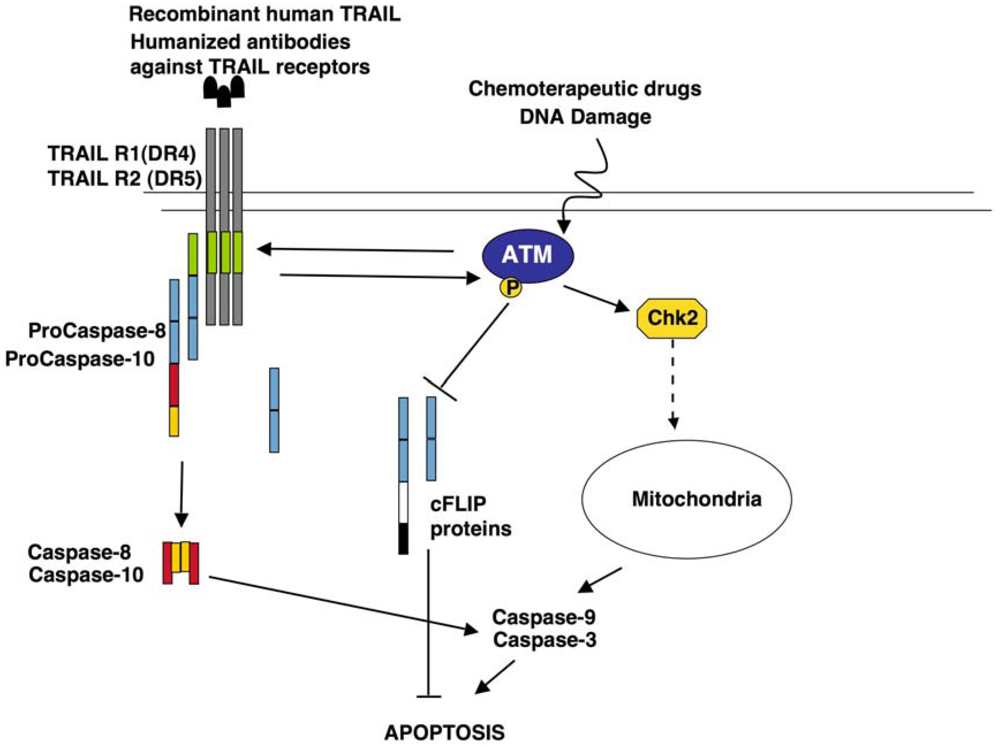
5.3. Role of ATM in TRAIL Signalling
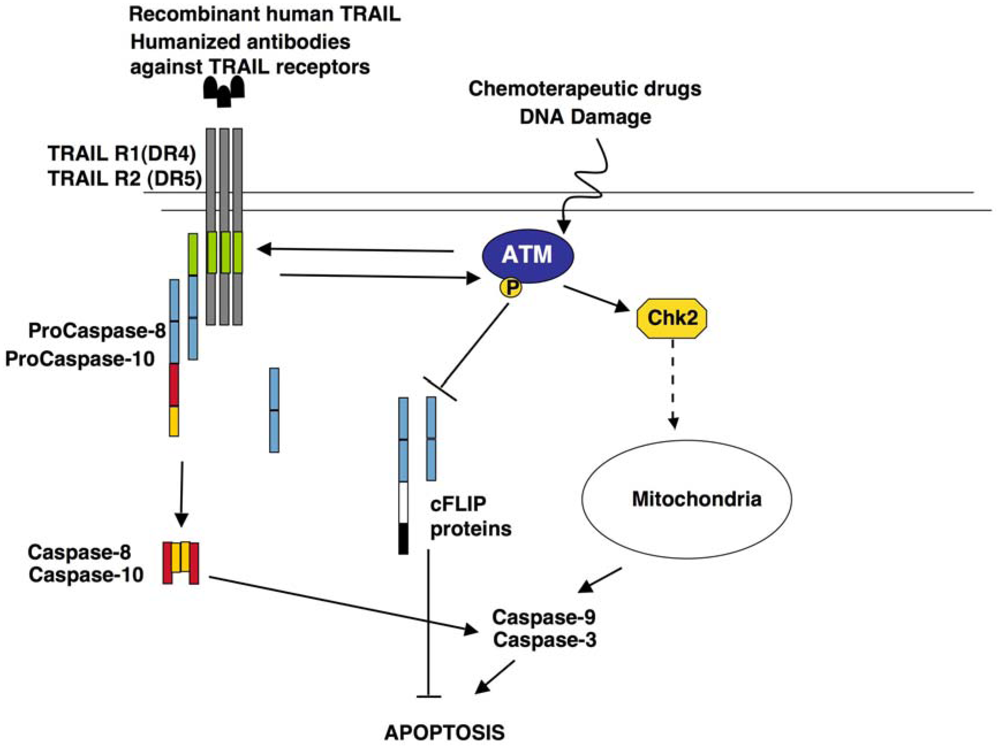
6. Conclusions
Acknowledgments
References
- Ashkenazi, A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov. 2008, 7, 1001–1012. [Google Scholar]
- Fabregat, I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J. Gastroenterol. 2009, 15, 513–520. [Google Scholar]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999, 59, 2770–2775. [Google Scholar]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002, 168, 1356–1361. [Google Scholar]
- Grosse-Wilde, A.; Voloshanenko, O.; Bailly, Y.; Longton, G.M.; Schaefer, U.; Csernok, A.I.; Schütz, G.; Greiner, E.F.; Kemp, C.J.; Walczak, H. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J. Clin. Invest. 2008, 118, 100–110. [Google Scholar]
- Finnberg, N.; Klein-Szanto, A.J.; El-Deiry, W.S. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J. Clin. Invest. 2008, 111–123. [Google Scholar]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar]
- Jo, M.; Kim, T.H.; Seol, D.W.; Esplen, J.E.; Dorko, K.; Billiar, T.R.; Strom, S.C. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 2000, 6, 564–567. [Google Scholar]
- Lawrence, D.; Shahrokh, Z.; Marsters, S.; Achilles, K.; Shih, D.; Mounho, B.; Hillan, K.; Totpal, K.; DeForge, L.; Schow, P.; et al. Differential hepatocyte toxicity of recombinant Apo2l/TRAIL versions. Nat. Med. 2001, 7, 383–385. [Google Scholar]
- Volkmann, X.; Fischer, U.; Bahr, M.J.; Ott, M.; Lehner, F.; MacFarlane, M.; Cohen, G.M.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 2007, 46, 1498–1508. [Google Scholar]
- Plummer, R.; Attard, G.; Pacey, S.; Li, L.; Razak, A.; Perrett, R.; Barrett, M.; Judson, I.; Kaye, S.; Fox, N.L.; et al. Phase I and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin. Cancer Res. 2007, 13, 6187–6194. [Google Scholar]
- Tolcher, A.; Mita, M.; Meropol, N.J.; von Mehren, M.; Patnaik, A.; Padavic, K.; Hill, M.; Mays, T.; McCoy, T.; Fox, N.L.; et al. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J. Clin. Oncol. 2007, 25, 1390–1395. [Google Scholar]
- Trarbach, T.; Moehler, M.; Heinemann, V.; Köhne, C.H.; Przyborek, M.; Schulz, C.; Sneller, V.; Gallant, G.; Kanzler, S. Phase II trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with refractory colorectal cancer. Br. J. Cancer 2010, 102, 506–512. [Google Scholar] [CrossRef]
- Wakelee, H.A.; Patnaik, A.; Sikic, B.I.; Mita, M.; Fox, N.L; Miceli, R.; Ullrich, S.J.; Fisher, G.A.; Tolcher, A.W. Phase I and pharmacokinetic study of lexatumumab (HGS-ETR2) given every 2 weeks in patients with advanced solid tumors. Ann. Oncol. 2010, 21, 376–381. [Google Scholar]
- Human Genome Sciences Initiates Clinical Development of New Drug for the Treatment of Cancer. Available online: http://www.hgsi.com/latest/human-genome-sciences-initiates-clinical-development-of-new-drug-for-the-treatment-of-c-3.html/ (accessed on 15 March 2012).
- Forero-Torres, A.; Shah, J.; Wood, T.; Posey, J.; Carlisle, R.; Copigneaux, C.; Luo, F.R.; Wojtowicz-Praga, S.; Percent, I.; Saleh, M. Phase I trial of weekly tigatuzumab, an agonistic humanized monoclonal antibody targeting death receptor 5 (DR5). Cancer Biother. Radiopharm. 2010, 25, 13–19. [Google Scholar]
- Chen, K.F.; Chen, H.L.; Liu, C.Y.; Tai, W.T.; Ichikawa, K.; Chen, P.J.; Cheng, A.L. Dovitinib sensitizes hepatocellular carcinoma cells to TRAIL and tigatuzumab, a novel anti-DR5 antibody, through SHP-1-dependent inhibition of STAT3. Biochem. Pharmacol. 2012, 83, 769–777. [Google Scholar]
- Herbst, R.S.; Kurzrock, R.; Hong, D.S.; Valdivieso, M.; Hsu, C.P.; Goyal, L.; Juan, G.; Hwang, Y.C.; Wong, S.; Hill, J.S.; et al. A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 5883–5891. [Google Scholar]
- Doi, T.; Murakami, H.; Ohtsu, A.; Fuse, N.; Yoshino, T.; Yamamoto, N.; Boku, N.; Onozawa, Y.; Hsu, C.P.; Gorski, K.S.; et al. Phase 1 study of conatumumab, a pro-apoptotic death receptor 5 agonist antibody, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2011, 68, 733–741. [Google Scholar] [CrossRef]
- Camidge, D.R. Apomab: An agonist monoclonal antibody directed against death receptor 5/TRAIL-receptor 2 for use in the treatment of solid tumors. Expert. Opin. Biol. Ther. 2008, 8, 1167–1176. [Google Scholar]
- Mesothelioma Empowerment. Available online: http://www.mesothel.com/asbestos-cancer/mesothelioma/clinical-trials/lby135.htm/ (accessed on 15 March 2012).
- Zhang, L.; Fang, B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005, 12, 228–237. [Google Scholar]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2l/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar]
- Yang, A.; Wilson, N.S.; Ashkenazi, A. Proapoptotic DR4 and DR5 signaling in cancer cells: Toward clinical translation. Curr. Opin. Cell Biol. 2010, 22, 837–844. [Google Scholar]
- Du, J.; Liang, X.; Liu, Y.; Qu, Z.; Gao, L.; Han, L.; Liu, S.; Cui, M.; Shi, Y.; Zhang, Z.; et al. Hepatitis B virus core protein inhibits trail-induced apoptosis of hepatocytes by blocking DR5 expression. Cell Death Differ. 2009, 16, 219–229. [Google Scholar]
- Yamanaka, T.; Shiraki, K.; Sugimoto, K.; Ito, T.; Fujikawa, K.; Ito, M.; Takase, K.; Moriyama, M.; Nakano, T.; Suzuki, A. Chemotherapeutic agents augment TRAIL-induced apoptosis in human hepatocellular carcinoma cell lines. Hepatology 2000, 32, 482–490. [Google Scholar]
- Shin, E.C.; Seong, Y.R.; Kim, C.H.; Kim, H.; Ahn, Y.S.; Kim, K.; Kim, S.J.; Hong, S.S.; Park, J.H. Human hepatocellular carcinoma cells resist to TRAIL-induced apoptosis, and the resistance is abolished by cisplatin. Exp. Mol. Med. 2002, 34, 114–122. [Google Scholar]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar]
- Kriegl, L.; Jung, A.; Engel, J.; Jackstadt, R.; Gerbes, A.L.; Gallmeier, E.; Reiche, J.A.; Hermeking, H.; Rizzani, A.; Bruns, C.J.; et al. Expression, cellular distribution, and prognostic relevance of trail receptors in hepatocellular carcinoma. Clin. Cancer Res. 2010, 16, 5529–5538. [Google Scholar]
- Peter, M.E. The flip side of FLIP. Biochem. J. 2004, 382, e1–e3. [Google Scholar]
- Budd, R.C.; Yeh, W.C.; Tschopp, J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol. 2006, 6, 196–204. [Google Scholar]
- Safa, A.R.; Pollok, K.E. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers 2011, 3, 1639–1671. [Google Scholar]
- Sharp, D.A.; Lawrence, D.A.; Ashkenazi, A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2005, 280, 19401–19409. [Google Scholar]
- Okano, H.; Shiraki, K.; Inoue, H.; Kawakita, T.; Yamanaka, T.; Deguchi, M.; Sugimoto, K.; Sakai, T.; Ohmori, S.; Fujikawa, K.; et al. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab. Invest. 2003, 83, 1033–1043. [Google Scholar]
- Kelly, P.N.; Strasser, A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011, 18, 1414–1424. [Google Scholar]
- Fulda, S.; Meyer, E.; Debatin, K.M. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002, 21, 2283–2294. [Google Scholar]
- Mott, J.L.; Gores, G.J. Piercing the armor of hepatobiliary cancer: Bcl-2 homology domain 3 (BH3) mimetics and cell death. Hepatology 2007, 46, 906–911. [Google Scholar]
- Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S.L.; Takahashi, H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, 55–61. [Google Scholar]
- Sieghart, W.; Losert, D.; Strommer, S.; Cejka, D.; Schmid, K.; Rasoul-Rockenschaub, S.; Bodingbauer, M.; Crevenna, R.; Monia, B.P.; Peck-Radosavljevic, M.; et al. Mcl-1 overexpression in hepatocellular carcinoma: A potential target for antisense therapy. J. Hepatol. 2006, 44, 151–157. [Google Scholar]
- LeBlanc, H.; Lawrence, D.; Varfolomeev, E.; Totpal, K.; Morlan, J.; Schow, P.; Fong, S.; Schwall, R.; Sinicropi, D.; Ashkenazi, A. Tumor-cell resistance to death receptor-induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat. Med. 2002, 8, 274–281. [Google Scholar]
- Beerheide, W.; Tan, Y.J.; Teng, E.; Ting, A.E.; Jedpiyawongse, A.; Srivatanakul, P. Downregulation of proapoptotic proteins Bax and Bcl-x(s) in p53 overexpressing hepatocellular carcinomas. Biochem. Biophys. Res. Commun. 2000, 273, 54–61. [Google Scholar]
- LaCasse, E.C.; Mahoney, D.J.; Cheung, H.H.; Plenchette, S.; Baird, S.; Korneluk, R.G. Iap-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [Google Scholar]
- Cummins, J.M.; Kohli, M.; Rago, C.; Kinzler, K.W.; Vogelstein, B.; Bunz, F. X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res. 2004, 64, 3006–3008. [Google Scholar]
- Shi, Y.H.; Ding, W.X.; Zhou, J.; He, J.Y.; Xu, Y.; Gambotto, A.A.; Rabinowich, H.; Fan, J.; Yin, X.M. Expression of X-linked inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes metastasis and tumor recurrence. Hepatology 2008, 48, 497–507. [Google Scholar]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar]
- Ito, T.; Shiraki, K.; Sugimot, K.; Yamanaka, T.; Fujikawa, K.; Ito, M.; Takase, K.; Moriyama, M.; Kawano, H.; Hayashida, M.; et al. Survivin promotes cell proliferation in human hepatocellular carcinoma. Hepatology 2000, 31, 1080–1085. [Google Scholar]
- Ye, C.P.; Qiu, C.Z.; Huang, Z.X.; Su, Q.C.; Zhuang, W.; Wu, R.L.; Li, X.F. Relationship between survivin expression and recurrence, and prognosis in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 6264–6268. [Google Scholar]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar]
- Braeuer, S.J.; Büneker, C.; Mohr, A.; Zwacka, R.M. Constitutively activated nuclear factor-kappaB, but not induced NF-kappaB, leads to TRAIL resistance by up-regulation of X-linked inhibitor of apoptosis protein in human cancer cells. Mol. Cancer Res. 2006, 4, 715–728. [Google Scholar] [CrossRef]
- Kim, Y.S.; Schwabe, R.F.; Qian, T.; Lemasters, J.J.; Brenner, D.A. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology 2002, 36, 1498–1508. [Google Scholar]
- Shigeno, M.; Nakao, K.; Ichikawa, T.; Suzuki, K.; Kawakami, A.; Abiru, S.; Miyazoe, S.; Nakagawa, Y.; Ishikawa, H.; Hamasaki, K.; et al. Interferon-alpha sensitizes human hepatoma cells to TRAIL-induced apoptosis through DR5 upregulation and NF-kappaB inactivation. Oncogene 2003, 22, 1653–1662. [Google Scholar]
- Breuhahn, K.; Longerich, T.; Schirmacher, P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006, 25, 3787–3800. [Google Scholar]
- de Toni, E.N.; Kuntzen, C.; Gerbes, A.L.; Thasler, W.E.; Sonuc, N.; Mucha, S.R.; Camaj, P.; Bruns, C.; Göke, B.; Eichhorst, S.T. P60-c-src suppresses apoptosis through inhibition of caspase 8 activation in hepatoma cells, but not in primary hepatocytes. J. Hepatol. 2007, 46, 682–691. [Google Scholar]
- Cursi, S.; Rufini, A.; Stagni, V.; Condo, I.; Matafora, V.; Bachi, A.; Bonifazi, A.P.; Coppola, L.; Superti-Furga, G.; Testi, R.; et al. Src kinase phosphorylates Caspase-8 on Tyr380: A novel mechanism of apoptosis suppression. EMBO J. 2006, 25, 1895–1905. [Google Scholar]
- He, G.; Karin, M. NF-κB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar]
- Kusaba, M.; Nakao, K.; Goto, T.; Nishimura, D.; Kawashimo, H.; Shibata, H.; Motoyoshi, Y.; Taura, N.; Ichikawa, T.; Hamasaki, K.; et al. Abrogation of constitutive STAT3 activity sensitizes human hepatoma cells to TRAIL-mediated apoptosis. J. Hepatol. 2007, 47, 546–555. [Google Scholar]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest. 2004, 113, 1774–1783. [Google Scholar]
- Koehler, B.C.; Urbanik, T.; Vick, B.; Boger, R.J.; Heeger, S.; Galle, P.R.; Schuchmann, M.; Schulze-Bergkamen, H. TRAIL-induced apoptosis of hepatocellular carcinoma cells is augmented by targeted therapies. World J. Gastroenterol. 2009, 15, 5924–5935. [Google Scholar]
- Zhang, B.; Shan, H.; Li, D.; Li, Z.R.; Zhu, K.S.; Jiang, Z.B.; Huang, M.S. Cisplatin sensitizes human hepatocellular carcinoma cells, but not hepatocytes and mesenchymal stem cells, to TRAIL within a therapeutic window partially depending on the upregulation of DR5. Oncol. Rep. 2011, 25, 461–468. [Google Scholar]
- Miao, L.; Yi, P.; Wang, Y.; Wu, M. Etoposide upregulates Bax-enhancing tumour necrosis factor-related apoptosis inducing ligand-mediated apoptosis in the human hepatocellular carcinoma cell line QGY-7703. Eur. J. Biochem. 2003, 270, 2721–2731. [Google Scholar]
- Ganten, T.M.; Koschny, R.; Sykora, J.; Schulze-Bergkamen, H.; Buchler, P.; Haas, T.L.; Schader, M.B.; Untergasser, A.; Stremmel, W.; Walczak, H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. 2006, 12, 2640–2646. [Google Scholar]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Emanuele, S.; Angileri, L.; di Fazio, P.; Santulli, A.; Vento, R.; Tesoriere, G. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to trail-induced apoptosis by TRAIL-disc activation. Eur. J. Cancer 2009, 45, 2425–2438. [Google Scholar]
- Ganten, T.M.; Koschny, R.; Haas, T.L.; Sykora, J.; Li-Weber, M.; Herzer, K.; Walczak, H. Proteasome inhibition sensitizes hepatocellular carcinoma cells, but not human hepatocytes, to TRAIL. Hepatology 2005, 42, 588–597. [Google Scholar] [CrossRef]
- Yamanaka, Y.; Shiraki, K.; Inoue, T.; Miyashita, K.; Fuke, H.; Yamaguchi, Y.; Yamamoto, N.; Ito, K.; Sugimoto, K.; Nakano, T. COX-2 inhibitors sensitize human hepatocellular carcinoma cells to TRAIL-induced apoptosis. 2006, 18, 41–47. [Google Scholar]
- Wang, G.; Zhan, Y.; Wang, H.; Li, W. ABT-263 sensitizes TRAIL-resistant hepatocarcinoma cells by downregulating the Bcl-2 family of anti-apoptotic protein. Cancer Chemother. Pharmacol. 2012, 69, 799–805. [Google Scholar]
- Jin, C.Y.; Park, C.; Kim, G.Y.; Lee, S.J.; Kim, W.J.; Choi, Y.H. Genistein enhances TRAIL-induced apoptosis through inhibition of p38 MAPK signaling in human hepatocellular carcinoma Hep3B cells. Chem. Biol. Interact. 2009, 180, 143–150. [Google Scholar]
- J Jin, C.Y.; Park, C.; Moon, S.K.; Kim, G.Y.; Kwon, T.K.; Lee, S.J.; Kim, W.J.; Choi, Y.H. Genistein sensitizes human hepatocellular carcinoma cells to TRAIL-mediated apoptosis by enhancing bid cleavage. Anticancer Drugs 2009, 20, 713–722. [Google Scholar]
- Kim, J.Y.; Kim, E.H.; Park, S.S.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Quercetin sensitizes human hepatoma cells to TRAIL-induced apoptosis via Sp1-mediated DR5 up-regulation and proteasome-mediated c-FLIPS down-regulation. J. Cell Biochem. 2008, 105, 1386–1398. [Google Scholar]
- Miyashita, K.; Shiraki, K.; Fuke, H.; Inoue, T.; Yamanaka, Y.; Yamaguchi, Y.; Yamamoto, N.; Ito, K.; Sugimoto, K.; Nakano, T. The cyclin-dependent kinase inhibitor flavopiridol sensitizes human hepatocellular carcinoma cells to TRAIL-induced apoptosis. Int. J. Mol. Med. 2006, 18, 249–256. [Google Scholar]
- Chen, K.F.; Tai, W.T.; Liu, T.H.; Huang, H.P.; Lin, Y.C.; Shiau, C.W.; Li, P.K.; Chen, P.J.; Cheng, A.L. Sorafenib overcomes trail resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin. Cancer Res. 2010, 16, 5189–5199. [Google Scholar]
- Mucha, S.R.; Rizzani, A.; Gerbes, A.L.; Camaj, P.; Thasler, W.E.; Bruns, C.J.; Eichhorst, S.T.; Gallmeier, E.; Kolligs, F.T.; Göke, B.; et al. JNK inhibition sensitises hepatocellular carcinoma cells but not normal hepatocytes to the TNF-related apoptosis-inducing ligand. Gut 2009, 58, 688–698. [Google Scholar]
- Beurel, E.; Blivet-Van Eggelpoël, M.J.; Kornprobst, M.; Moritz, S.; Delelo, R.; Paye, F.; Housset, C.; Desbois-Mouthon, C. Glycogen synthase kinase-3 inhibitors augment TRAIL-induced apoptotic death in human hepatoma cells. Biochem. Pharmacol. 2009, 77, 54–65. [Google Scholar]
- Kim, H.R.; Kim, K.; Lee, K.H.; Kim, S.; Kim, J. Inhibition of casein kinase 2 enhances the death ligand- and natural kiler cell-induced hepatocellular carcinoma cell death. Clin. Exp. Immunol. 2008, 152, 336–344. [Google Scholar]
- Fuke, H.; Shiraki, K.; Sugimoto, K.; Tanaka, J.; Beppu, T.; Yoneda, K.; Yamamoto, N.; Ito, K.; Masuya, M.; Takei, Y. Jak inhibitor induces S phase cell-cycle arrest and augments TRAIL-induced apoptosis in human hepatocellular carcinoma cells. Biochem. Biophys Res. Commun. 2007, 363, 738–744. [Google Scholar]
- Moon, D.O.; Kang, C.H.; Kang, S.H.; Choi, Y.H.; Hyun, J.W.; Chang, W.Y.; Kang, H.K.; Koh, Y.S.; Maeng, Y.H.; Kim, Y.R.; et al. Capsaicin sensitizes TRAIL-induced apoptosis through Sp1-mediated DR5 up-regulation: Involvement of Ca2+ influx. Toxicol. Appl. Pharmacol. 2012, 259, 87–95. [Google Scholar]
- Yang, J.F.; Cao, J.G.; Tian, L.; Liu, F. 5, 7-Dimethoxyflavone sensitizes TRAIL-induced apoptosis through DR5 upregulation in hepatocellular carcinoma cells. Cancer Chemother. Pharmacol. 2012, 69, 195–206. [Google Scholar]
- Carlisi, D.; D’Anneo, A.; Angileri, L.; Lauricella, M.; Emanuele, S.; Santulli, A.; Vento, R.; Tesoriere, G. Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3 activation. J. Cell Physiol. 2011, 226, 1632–1641. [Google Scholar]
- Moon, D.O.; Kim, M.O.; Choi, Y.H.; Kim, G.Y. Butein sensitizes human hepatoma cells to TRAIL-induced apoptosis via extracellular signal-regulated kinase/Sp1-dependent DR5 upregulation and NF-kappaB inactivation. Mol. Cancer Ther. 2010, 9, 1583–1595. [Google Scholar]
- Kim, M.O.; Moon, D.O.; Kang, C.H.; Kwon, T.K.; Choi, Y.H.; Kim, G.Y. Beta-ionone enhances TRAIL-induced apoptosis in hepatocellular carcinoma cells through Sp1-dependent upregulation of DR5 and downregulation of NF-kappaB activity. Mol. Cancer Ther. 2010, 9, 833–843. [Google Scholar]
- Pellerito, O.; Calvaruso, G.; Portanova, P.; de Blasio, A.; Santulli, A.; Vento, R.; Tesoriere, G.; Giuliano, M. The synthetic cannabinoid WIN 55,212-2 sensitizes hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating p8/CCAAT/enhancer binding protein homologous protein (CHOP)/death receptor 5 (DR5) axis. Mol. Pharmacol. 2010, 77, 854–863. [Google Scholar]
- Tsai, A.C.; Pan, S.L.; Sun, H.L.; Wang, C.Y.; Peng, C.Y.; Wang, S.W.; Chang, Y.L.; Kuo, S.C.; Lee, K.H.; Teng, C.M. Chm-1, a new vascular targeting agent, induces apoptosis of human umbilical vein endothelial cells via p53-mediated death receptor 5 up-regulation. J. Biol. Chem. 2010, 285, 5497–5506. [Google Scholar]
- Sun, M.; Zhang, J.; Liu, S.; Liu, Y.; Zheng, D. Sp1 is involved in 8-chloro-adenosine-upregulated death receptor 5 expression in human hepatoma cells. Oncol. Rep. 2008, 19, 177–185. [Google Scholar]
- Wang, W.; Gallant, J.N.; Katz, S.I.; Dolloff, N.G.; Smith, C.D.; Abdulghani, J.; Allen, J.E.; Dicker, D.T.; Hong, B.; Navaraj, A.; et al. Quinacrine sensitizes hepatocellular carcinoma cells to TRAIL and chemotherapeutic agents. Cancer Biol. Ther. 2011, 12, 229–238. [Google Scholar]
- Jung, E.M.; Lim, J.H.; Lee, T.J.; Park, J.W.; Choi, K.S.; Kwon, T.K. Curcumin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through reactive oxygen species-mediated upregulation of death receptor 5 (DR5). Carcinogenesis 2005, 26, 1905–1913. [Google Scholar]
- Park, C.; Jin, C.Y.; Hwang, H.J.; Kim, G.Y.; Jung, J.H.; Kim, W.J.; Yoo, Y.H.; Choi, Y.H. J7, a methyl jasmonate derivative, enhances TRAIL-mediated apoptosis through up-regulation of reactive oxygen species generation in human hepatoma HepG2 cells. Toxicol. In Vitro 2011, 26, 86–93. [Google Scholar]
- Moon, D.O.; Park, S.Y.; Choi, Y.H.; Ahn, J.S.; Kim, G.Y. Guggulsterone sensitizes hepatoma cells to TRAIL-induced apoptosis through the induction of CHOP-dependent DR5: Involvement of ROS-dependent ER-stress. Biochem. Pharmacol. 2011, 82, 1641–1650. [Google Scholar]
- Song, I.S.; Kim, S.U.; Oh, N.S.; Kim, J.; Yu, D.Y.; Huang, S.M.; Kim, J.M.; Lee, D.S.; Kim, N.S. Peroxiredoxin I contributes to TRAIL resistance through suppression of redox-sensitive caspase activation in human hepatoma cells. Carcinogenesis 2009, 30, 1106–1114. [Google Scholar]
- Kim, H.; Kim, E.H.; Eom, Y.W.; Kim, W.H.; Kwon, T.K.; Lee, S.J.; Choi, K.S. Sulforaphane sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-resistant hepatoma cells to TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of DR5. Cancer Res. 2006, 66, 1740–1750. [Google Scholar]
- Lu, G.; Liu, Y.; Ji, B.; Wei, F.; Hao, C.; Wang, G. Synergistic effect of celecoxib on TRAIL-induced apoptosis in hepatocellular carcinoma cells. Cancer Invest. 2010, 28, 629–634. [Google Scholar]
- Wang, C.; Chen, T.; Zhang, N.; Yang, M.; Li, B.; Lü, X.; Cao, X.; Ling, C. Melittin, a major component of bee venom, sensitizes human hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating CaMKII-TAK1-JNK/p38 and inhibiting IkappaBalpha kinase-NFkappaB. J. Biol. Chem. 2009, 284, 3804–3813. [Google Scholar]
- Schuchmann, M.; Schulze-Bergkamen, H.; Fleischer, B.; Schattenberg, J.M.; Siebler, J.; Weinmann, A.; Teufel, A.; Worns, M.; Fischer, T.; Strand, S.; et al. Histone deacetylase inhibition by valproic acid down-regulates c-FLIP/CASH and sensitizes hepatoma cells towards CD95- and TRAIL receptor-mediated apoptosis and chemotherapy. Oncol. Rep. 2006, 15, 227–230. [Google Scholar]
- Dzieran, J.; Beck, J.F.; Sonnemann, J. Differential responsiveness of human hepatoma cells versus normal hepatocytes to TRAIL in combination with either histone deacetylase inhibitors or conventional cytostatics. Cancer Sci. 2008, 99, 1685–1692. [Google Scholar]
- Pathil, A.; Armeanu, S.; Venturelli, S.; Mascagni, P.; Weiss, T.S.; Gregor, M.; Lauer, U.M.; Bitzer, M. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology 2006, 43, 425–434. [Google Scholar]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar]
- Nakao, K.; Hamasaki, K.; Ichikawa, T.; Arima, K.; Eguchi, K.; Ishii, N. Survivin downregulation by siRNA sensitizes human hepatoma cells to TRAIL-induced apoptosis. Oncol. Rep. 2006, 16, 389–392. [Google Scholar]
- Zhang, D.; Liu, J.; Wang, Y.; Chen, J.; Chen, T. shRNA-mediated silencing of Gli2 gene inhibits proliferation and sensitizes human hepatocellular carcinoma cells towards TRAIL-induced apoptosis. J. Cell Biochem. 2011, 112, 3140–3150. [Google Scholar]
- Chen, Q.; Lou, W.; Shen, J.; Ma, L.; Yang, Z.; Liu, L.; Luo, J.; Qian, C. Potent antitumor activity in experimental hepatocellular carcinoma by adenovirus-mediated coexpression of TRAIL and shrna against COX-2. Clin. Cancer Res. 2010, 16, 3696–3705. [Google Scholar]
- Kurita, S.; Higuchi, H.; Saito, Y.; Nakamoto, N.; Takaishi, H.; Tada, S.; Saito, H.; Gores, G.J.; Hibi, T. DNMT1 and DNMT3b silencing sensitizes human hepatoma cells to TRAIL-mediated apoptosis via up-regulation of TRAIL-R2/DR5 and caspase-8. Cancer Sci. 2010, 101, 1431–1439. [Google Scholar]
- Zhang, R.G.; Zhao, J.J.; Yang, L.Q.; Yang, S.M.; Wang, R.Q.; Chen, W.S.; Peng, G.Y.; Fang, D.C. RNA interference-mediated hTERT inhibition enhances TRAIL-induced apoptosis in resistant hepatocellular carcinoma cells. Oncol. Rep. 2010, 23, 1013–1019. [Google Scholar]
- Wang, C.; Qi, R.; Li, N.; Wang, Z.; An, H.; Zhang, Q.; Yu, Y.; Cao, X. Notch1 signaling sensitizes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53 degradation and up-regulating p53-dependent DR5 expression. J. Biol. Chem. 2009, 284, 16183–16190. [Google Scholar]
- Zhao, X.; Liu, Y.; Ma, Q.; Wang, X.; Jin, H.; Mehrpour, M.; Chen, Q. Caveolin-1 negatively regulates TRAIL-induced apoptosis in human hepatocarcinoma cells. Biochem. Biophys. Res. Commun. 2009, 378, 21–26. [Google Scholar]
- Pan, Q.; Liu, B.; Liu, J.; Cai, R.; Liu, X.; Qian, C. Synergistic antitumor activity of XIAP-shRNA and TRAIL expressed by oncolytic adenoviruses in experimental HCC. Acta Oncol. 2008, 47, 135–144. [Google Scholar]
- Schulze-Bergkamen, H.; Fleischer, B.; Schuchmann, M.; Weber, A.; Weinmann, A.; Krammer, P.H.; Galle, P.R. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer 2006, 6, 232. [Google Scholar]
- Thorburn, A.; Behbakht, K.; Ford, H. TRAIL receptor-targeted therapeutics: Resistance mechanisms and strategies to avoid them. Drug Resist. Updat. 2008, 11, 17–24. [Google Scholar]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–167. [Google Scholar]
- Bhatti, S.; Kozlov, S.; Farooqi, A.A.; Naqui, A.; Lavin, M.F.; Khanna, K.K. ATM protein kinase: The linchpin of cellular defenses to stress. Cell Mol. Life Sci. 2011, 68, 2977–3006. [Google Scholar]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar]
- Matsuoka, S.; Ballif, B.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.; Luo, J.L.; Bakalarski, C.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Rainery, M.D.; Charlton, M.E.; Sytanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar]
- Teoh, N.; Pyakurel, P.; Dan, Y.Y.; Swisshelm, K.; Hou, J.; Mitchell, C.; Fausto, N.; Gu, Y.; Farrell, G. Induction of p53 renders ATM-deficient mice refractory to hepatocarcinogenesis. Gastroenterology 2010, 138, 1155–1165. [Google Scholar]
- Lu, S.; Shen, K.C.; Wang, Y.; Brooks, S.C.; Wang, Y.A. Impaired hepatocyte survival and liver regeneration in ATM-deficient mice. Hum. Mol. Genet. 2005, 14, 3019–3025. [Google Scholar]
- Wu, G.S.; Burns, T.F.; McDonald, E.R.R.; Meng, R.D.; Kao, G.; Muschel, R.; Yen, T.; El-Deiry, W.S. Induction of the TRAIL receptor KILLER/DR5 in p53-dependent apoptosis but not growth arrest. Oncogene 1999, 18, 6411–6418. [Google Scholar]
- Stagni, V.; di Bari, M.G.; Condò, I.; Cursi, S.; Testi, R.; Larenthal, Y.; Cundari, E.; Barilà, D. ATM kinase activity modulates Fas sensitivity through the regulation of FLIP in lymphoid cells. Blood 2008, 111, 829–837. [Google Scholar]
- Solier, S.; Sordet, O.; Kohn, K.W.; Pommier, Y. Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol. Cell. Biol. 2009, 29, 68–82. [Google Scholar]
- Stagni, V.; Mingardi, M.; Santini, S.; Giaccari, D.; Barilà, D. ATM kinase activity modulates cFLIP protein levels: Potential interplay between DNA damage signalling and TRAIL-induced apoptosis. Carcinogenesis 2010, 31, 1956–1963. [Google Scholar]
- Dutton, A.; O’Neil, J.D.; Milner, A.E.; Reynolds, G.M.; Starczynski, J.; Crocker, J.; Young, L.S.; Murray, P.G. Expression of the cellular FLICE-inhibitory protein (c-FLIP) protects Hodgkin’s lymphoma cells from autonomous Fas-mediated death. Proc. Natl. Acad. Sci. USA 2004, 101, 6611–6616. [Google Scholar]
- Mathas, S.; Lietz, A.; Anagnostopoulos, I.; Hummel, F.; Wiesner, B.; Janz, M.; Jundt, F.; Hirsch, B.; Johrens-Leder, K.; Vornlocher, H.P.; et al. c-FLIP mediates resistance of hodgkin/reed-sternberg cells to death receptor-induced apoptosis. J. Exp. Med. 2004, 199, 1041–1052. [Google Scholar]
- Ganten, T.M.; Haas, T.L.; Sykora, J.; Stahl, H.; Sprick, M.R.; Fas, S.C.; Krueger, A.; Weigand, M.A.; Grosse-Wilde, A.; Stremmel, W.; et al. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ. 2004, 11, S86–S96. [Google Scholar]
- Ivanov, V.N.; Zhou, H.; Partridge, M.A.; Hei, T.K. Inhibition of ataxia telangiectasia mutated kinase activity enhances TRAIL-mediated apoptosis in human melanoma cells. Cancer Res. 2009, 69, 3510–3519. [Google Scholar]
- Lovric, M.M.; Hawkins, C.J. TRAIL treatment provokes mutations in surviving cells. Oncogene 2010, 29, 5048–5060. [Google Scholar]
- Yoon, J.H.; Ahn, S.G.; Lee, B.H.; Jung, S.H.; Oh, S.H. Role of autophagy in chemoresistance: Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem. Pharmacol. 2012, 83, 747–757. [Google Scholar]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Enhancement of tumor-TRAIL susceptibility by modulation of autophagy. Autophagy 2008, 4, 940–943. [Google Scholar]
- Herrero-Martín, G.; Høyer-Hansen, M.; García-García, C.; Fumarola, C.; Farkas, T.; López-Rivas, A.; Jäättelä, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar]
- Farooqi, A.A.; Waseem, S.; Ashraf, M.S.; Iqbal, M.J.; Bhatti, S. TRAIL and guardian angel of genome integrity: ATM boards TRAIL blazer. J. Cancer Res. Clin. Oncol. 2011, 137, 1283–1287. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stagni, V.; Santini, S.; Barilà, D. A New Player in the Development of TRAIL Based Therapies for Hepatocarcinoma Treatment: ATM Kinase. Cancers 2012, 4, 354-378. https://doi.org/10.3390/cancers4020354
Stagni V, Santini S, Barilà D. A New Player in the Development of TRAIL Based Therapies for Hepatocarcinoma Treatment: ATM Kinase. Cancers. 2012; 4(2):354-378. https://doi.org/10.3390/cancers4020354
Chicago/Turabian StyleStagni, Venturina, Simonetta Santini, and Daniela Barilà. 2012. "A New Player in the Development of TRAIL Based Therapies for Hepatocarcinoma Treatment: ATM Kinase" Cancers 4, no. 2: 354-378. https://doi.org/10.3390/cancers4020354
APA StyleStagni, V., Santini, S., & Barilà, D. (2012). A New Player in the Development of TRAIL Based Therapies for Hepatocarcinoma Treatment: ATM Kinase. Cancers, 4(2), 354-378. https://doi.org/10.3390/cancers4020354