Differentiation Therapy of Acute Myeloid Leukemia



{kind=link}
{kind=link}
Abstract
: Acute Myeloid Leukemia (AML) is a predominant acute leukemia among adults, characterized by accumulation of malignantly transformed immature myeloid precursors. A very attractive way to treat myeloid leukemia, which is now called ‘differentiation therapy’, was proposed as in vitro studies have shown that a variety of agents stimulate differentiation of the cell lines isolated from leukemic patients. One of the differentiation-inducing agents, all-trans retinoic acid (ATRA), which can induce granulocytic differentiation in myeloid leukemic cell lines, has been introduced into clinics to treat patients with acute promyelocytic leukemia (APL) in which a PML-RARA fusion protein is generated by a t(15;17)(q22;q12) chromosomal translocation. Because differentiation therapy using ATRA has significantly improved prognosis for patients with APL, many efforts have been made to find alternative differentiating agents. Since 1,25-dihydroxyvitamin D3 (1,25D) is capable of inducing in vitro monocyte/macrophage differentiation of myeloid leukemic cells, clinical trials have been performed to estimate its potential to treat patients with AML or myelodysplastic syndrome (MDS). Unfortunately therapeutic concentrations of 1,25D can induce potentially fatal systemic hypercalcemia, thus limiting clinical utility of that compound. Attempts to overcome this problem have focused on the synthesis of 1,25D analogs (VDAs) which retain differentiation inducing potential, but lack its hypercalcemic effects. This review aims to discuss current problems and potential solutions in differentiation therapy of AML.1. Introduction
Leukemia is a disease of the blood or bone marrow, which is characterized by increased numbers of abnormal white blood cells. The abnormality of leukemic cells lies in their inhibited differentiation and increased proliferation rate. Leukemia is divided into acute and chronic, and further subdivided into lymphocytic and myeloid [1]. Within these groups further divisions are often necessary. The most heterogeneous group being a group of acute myeloid leukemias (AML) [2]. In leukemias differentiation block occurs in early hematopoietic progenitors, and resulting malignant cells are named blast cells. Acute leukemias are diagnosed either on the basis of presence of over 20% of blasts in the blood or in bone marrow or on the basis of presence of specific cytogenetic or molecular abnormalities [1]. There are more than 200 known chromosome translocations and mutations in leukemic cells of patients diagnosed with AML [3]. Which of these 200 are initial mutations responsible for clonal expansion of abnormal cells, and which are accumulated during the progress of disease, is largely unknown. AML is rare in children, it constitutes 80–85% of acute leukemia in adults, and its incidence increases with age [4]. The progress in understanding molecular background of the disease has led to significant changes in the classification of AML subtypes. Before 2001, the French-American-British (FAB) classification was used, in which AML was divided into eight subtypes (M0—M7), based on the type of cell from which the leukemia developed and on its degree of maturity [5]. In 2001 WHO introduced a more accurate division that incorporates cytogenetic abnormalities and prognostic significance. WHO classification was further revised in 2008 and has classified AML into four main groups: AML with recurrent genetic abnormalities, AML with myelodysplasia-related changes, therapy-related myeloid neoplasms, and AML not otherwise specified [6]. It should be also remembered that the disease is not common. In our country, Poland, with a population of about 38 million people, there are about 1000 cases of AML diagnosed per year [7]. Taking together the numbers and the variety of genetic alterations, it becomes clear that finding of a specific treatment for AML is not an easy task.
2. A Paradigm of Targeted Cancer Therapies
The idea of targeted therapies is not new. In his speech at the ceremonial opening of the Georg-Speyer-Haus in September 1906, Paul Ehrlich proposed that drugs should work as “magic bullets” that kill pathogens, and leave normal tissue unaffected [8]. Ehrlich's “magic bullets” were directed towards microorganisms, but later on the idea was adopted for anticancer treatment. Of course, every drug has its target, and most of the currently used anticancer chemotherapeutic drugs target, in a more or less direct manner, synthesis of DNA and cell division. Since cancer cells are not the only ones that need to proliferate in human body, chemotherapeutic drugs are often toxic and cannot be considered as “magic bullets” or in other words, targeted drugs. Therefore targeted anticancer therapies should be based on compounds that interfere with cellular components that are altered or present only in cancer cells [9].
Paradigmatic targeted therapy is embodied by treatment of patients with chronic myeloid leukemia (CML) using Imatinib [10]. Over 90% of CML patients carry chromosomal abnormality called the Philadelphia (Ph) chromosome [11,12]. Therefore the drug that interferes selectively with the tyrosine kinase activity of the fusion protein Bcr-Abl, should be toxic only to CML malignant cells [13]. The introduction of Imatinib (and similar specific inhibitors of Bcr-Abl) has revolutionized treatment of CML patients and increased rates of complete hematological response to 97% and complete cytogenetic response to 85% [14].
The most important difficulty in finding appropriate regimens of targeted therapy for AML patients originates from the above mentioned heterogeneity of the disease. Among 200 known chromosome translocations and mutations in AML, some are more common than others. One of the most common mutations seen in AML is internal tandem duplication in fms-like tyrosine kinase 3 (FLT3-ITD). This mutation is apparent in about 25% of all AML patients and confers unfavorable prognosis [2,15]. There is also another mutation in FLT3 receptor, point mutation in the tyrosine kinase domain (FLT3-TDK), seen in approximately 7–8% of AML patients, with less defined prognostic significance [2,16]. FLT3 receptor tyrosine kinase is normally expressed in immature precursors of myeloid and B-lymphoid lineages [17,18]. FLT3 ligand (FLT3-L) together with other colony stimulating factors and interleukins can stimulate proliferation of hematopoietic cells [19,20]. The two above mentioned mutations result in a ligand-independent activation of the receptor and give survival advantage to blast cells over their normal counterparts [21]. Aberrant FLT3 receptor seemed to be an attractive therapeutic target in AML, so several small molecules FLT3 tyrosine kinase inhibitors (TKI) have been developed and examined in vitro and in vivo. Eight of them, after successful preclinical screening, have been introduced into clinical trials [22]. The data available at the moment show that TKIs, used as single agents did not fulfill expectations and that the quality of clinical response was unsatisfactory [22].
3. Differentiation Therapies for Leukemia
Leukemic cells are inhibited in their hematopoietic differentiation by either genetic abnormalities or by gene expression abnormalities [2]. These cells proliferate rapidly, but often do not express proteins important for function of their normal counterparts. Even though white blood cells counts are high in these patients, the immune functions are lacking. Therefore, finding a method of forced differentiation of leukemic cells always seemed attractive to researchers and clinicians. Differentiation therapy seems to be a particularly attractive solution for AML patients. These patients are often elderly and rapidly progressing disease causes poor tolerability of intensive cytotoxic protocols [1]. Forced differentiation of myloid precursors should, in principle, improve the immune status of patients without massive lysis of blast cells seen in some cytotoxic regimens [23].
It should be remembered that inhibited differentiation may result not only from presence of mutated proteins, but also from epigenetic changes like DNA hypermetylation or aberrant acetylation of histones [9]. On the contrary to the loss of gene function caused by mutations, epigenetic changes can be reversed via pharmacologic inhibition of DNA methyltransferases (DnmT) and histone deacetylases (HDAC). In normal cells, histone acetylation and DNA methylation are maintained in equilibrium, allowing temporal expression of the genes. In leukemic cells, this balance is disturbed, hypermethylation occurs, HDACs are overexpressed, what leads to the transcriptional repression [24]. Therefore, several HDAC and DnmT inhibitors have been under development for the treatment of patients with hematological malignancies, such as AML or MDS, either as monotherapies or in combination with other agents [25-28]. Treatment of leukemia cells with such inhibitors, results in chromatin remodeling that unblocks a set of genes whose transcriptional activation induces cellular differentiation, cell cycle arrest, apoptosis or autophagy [26,29,30]. miRs, small non-coding RNAs 19–25 nucleotides long, provide an additional level of control between proliferation and differentiation [31,32]. They regulate gene expression post-transcriptionally via degradation of target mRNAs or/and via inhibition of protein translation [33,34]. A single miR can control levels of hundreds different target genes. Many miRs have been linked to the specification of hematopoietic cell lineages, and have been found altered by chromosomal translocations associated with leukemia and therefore constitute potential therapeutic targets [35-38].
4. New Treatments for Acute Promyelocytic Leukemia as Examples of Differentiation Therapy
Acute Promyelocytic Leukemia (APL) is the first hematological malignancy in which therapeutic approach specifically targeting the underlying molecular lesion has been successfully introduced into clinical practice. APL is a subset of AML characterized by uncontrolled expansion of leukemic blast cells, blocked at promyelocytic stage of hematopoiesis, in the bone marrow [39]. Morphologically, it is classified as a subtype M3 of AML [5], cytogenetically is characterized by a reciprocal translocation between the long arms of chromosomes 15 and 17 [t(15;17)] [40-43]. These aberrations lead to the fusion between promyelocytic leukemia (PML) gene located on chromosome 15q21, and retinoic acid receptor α (RARA) gene from chromosome 17q21, and to the formation of the resultant chimeric oncoprotein PML-RARA [42,44]. The fusion transcript of PML-RARA is detectable in more than 95% of APL patients with t(15;17), and becomes a major player disturbing proper promyelocytic differentiation, as well as a molecular marker for this disease [41,45]. In the remaining minority of patients, alternative fusions of RARA may occur [46-49]. These findings enabled the introduction of all- trans-retinoic acid (ATRA) as a differentiation agent for APL treatment.
4.1. PML-RARA-Induced Transcriptional Repression in APL Cells
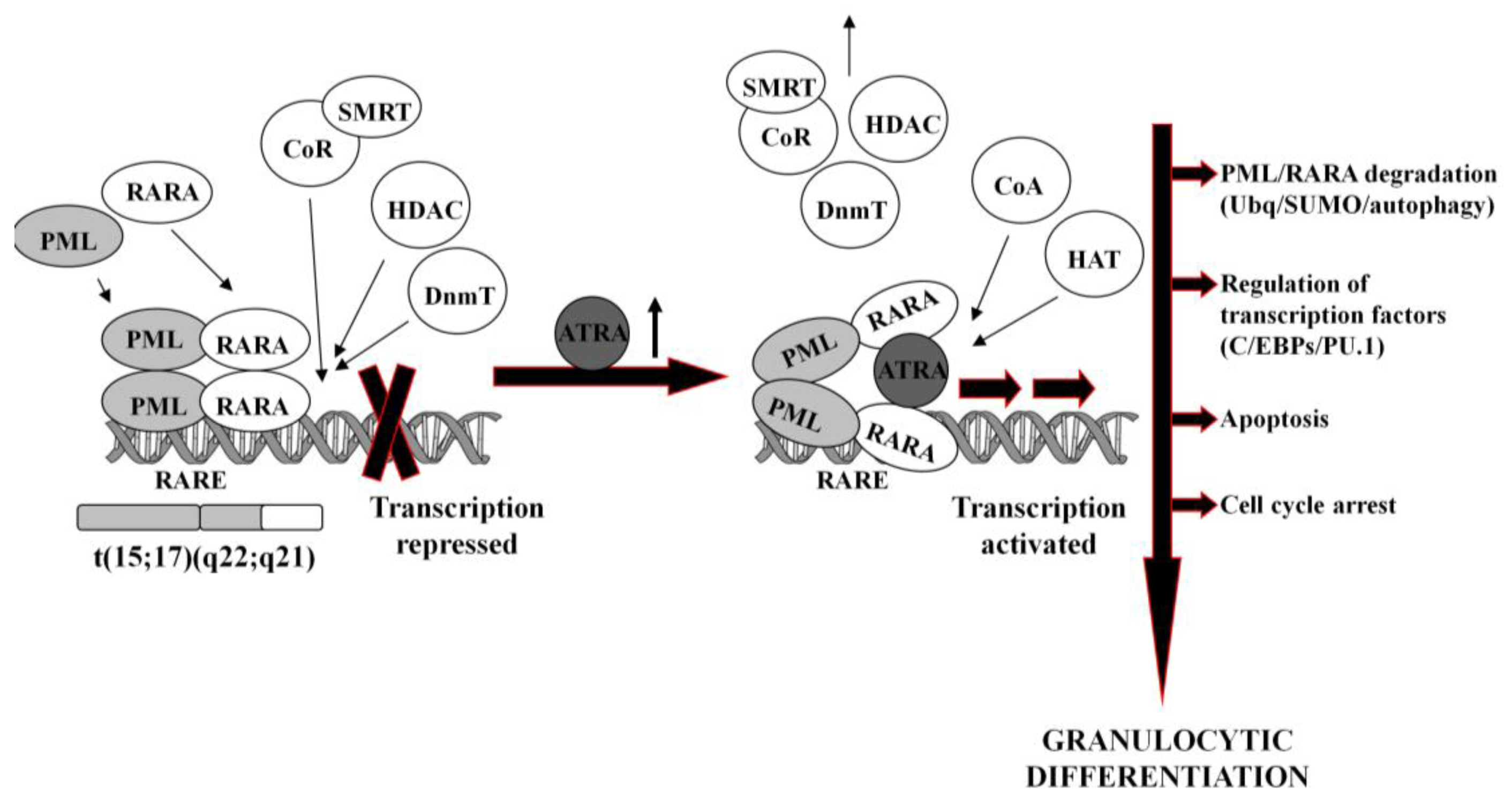
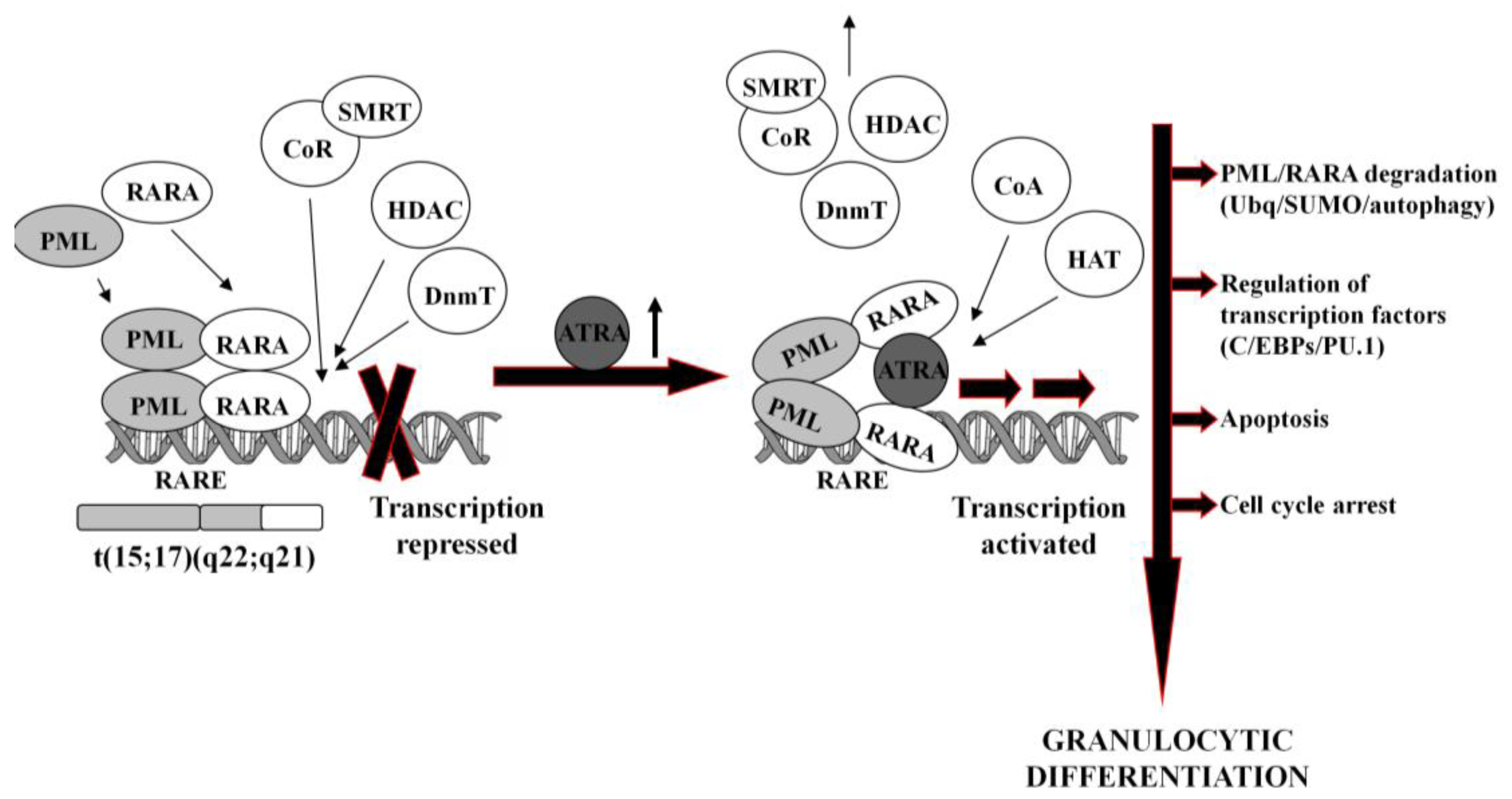
In normal hematopoietic cells, retinoids play multiple physiological roles in maturation and differentiation [50-52]. These compounds function through binding to their receptors (RAR and RXR), which belong to the superfamily of nuclear ligand-activated transcription factors [53,54]. In the absence of a ligand, RARA forms heterodimers with the RXR, binds to the retinoid acid response elements (RARE) in the promoter region of the target genes and recruits co-repressor (CoR) complex. CoR is composed of several proteins, including nuclear receptor co-repressor (NCoR), silencing mediator for retinoid and thyroid hormone receptors (SMRT) [55]. These proteins recruit DNA DnmT1 and DnmT3a and HDAC1 [56-58]. Deacetylated histones cause chromatin condensation and transcriptional repression [56,59]. At physiological concentrations (10−9–10−8M), ATRA binds to the RARA-RXR hetrodimer and induces dissociation of CoR, interchangeably with association of the co-activators (CoA) complex, containing histone acetyltransferase (HAT). Acetylated histones cause chromatin decondensation and activation of the transcription [56,59].
In APL, RARA heterodimerizes with promyelocytic leukemia (PML) nuclear protein, normally responsible for the formation of the nuclear bodies and regulation of the stem cells self-renewal [60,61]. PML-RARA fusion protein acts as an oncogene, causing enhanced proliferation and inhibited terminal differentiation of the hematopoietic cells. PML-RARA heterodimers act in a dominant negative manner over RARA, and have higher affinity to CoR and HDAC than RARA-RXR, resulting in enhanced hyper-methylation of the DNA [62,63]. Furthermore, PML-RARA oligomerizes in a different manner to RXR-RARA and may not only homodimerize, but also heterodimerize with wild-type PML and RXR [64]. This may cause sequesteration of those proteins and recruiting enzymes in a large complex, which augments transcriptional repression [62,65]. An example of genes repressed by PML-RARA is the one encoding protein p21Cip1, what may contribute to the final effect of enhanced proliferation of APL cells [64,66,67]. Heterodimerization with PML-RARA turns off pro-apoptotic and growth inhibiting properties of PML [68,69]. Interfering with the normal function of both, RARA and PML, the fusion protein reveals double dominant negative activity [68]. PML-RARA also binds promyelocytic leukemia zinc finger (PLZF) protein, and affects its functions as a growth suppressor [70]. Moreover, it appears that PML-RARA can enhance pro-proliferative and pro-survival functions of FLT3 [71,72]. It is important to note that PML-RARA often targets PU.1-regulated promoters through both protein-protein interactions, as well as DNA binding via RARE half sites [73]. Genes containing these PML-RARA-targeted promoters are transcriptionally suppressed in APL and most likely constitute a major mechanism of transcriptional repression occurring in APL [73]. Molecular disturbances mentioned above contribute to the blockage of granulocytic differentiation, and constitute the main reason for the inability of ATRA to unblock transcriptional repression at its physiological concentration.
4.2. ATRA-Induced Differentiation and Elimination of APL Cells
It is generally accepted that at pharmacological concentrations (10−7–10−6M) ATRA causes conformational changes of PML-RARA, which enable dissociation of the CoR and association of the CoA. As a result, chromatin structure becomes relaxed, transcriptional repression relieved, and APL cells undergo terminal differentiation into granulocytes (Figure 1) [74,75]. It has been reported that several miRs (i.e., miR-223, let-7a) play an important role of a regulatory circuit involving C/EBPα and NFI-A, master regulatory transcription factors that control granulocytic differentiation in ATRA-treated APL cells [36,76]. Moreover, it has been shown that several miRs (i.e., miR-210, miR23a/24-2) which are transcriptionally repressed by the APL-associated PML-RARA oncogene, become activated after treatment with ATRA [77-79].
ATRA-induced degradation of PML-RARA is the basic therapeutic mechanism in APL cells [42,80]. Recent results have revealed several ways leading to the destruction of the fusion oncogene, such as ubiquitination [81], sumoylation [82] or authophagy [83]. PML-RARA degradation is accompanied by the activation or inhibition of a number of various ATRA-response genes. These include transcription factors (for example C/EBPε or PU.1) [73,84], chromatin-regulating factors [63,85], cell cycle regulators [86,87], as well as protein synthesis inhibitors [88].
Effectiveness of ATRA in the treatment of APL is unquestionable. In more than 90% of the patients such a therapy leads to the complete remission [89,90]. Treatment regiments combining ATRA with arsenic trioxide (ASO) further improved curability of the patients, especially these with ATRA-resistance, usually occurring after long-term use of the drug [90,91]. The possible reason for the relapse is that ATRA used as a single agent, in spite of inducing complete remission, is unable to cause complete molecular remission. Therefore a fraction of leukemia initiating cells (LIC) may remain after initial successful ATRA treatment [80]. In contrast, ATRA/ASO combination therapy has been shown to produce final clearance of LICs [80,92]. In leukemic cells ASO supports ATRA-induced differentiation, but this is not enough for eradication of the disease. Even though ASO has been known as a therapeutic agent for ages, its mode of action remained obscure until recently [93]. At present we know that it affects cells in many different ways, one of the most important effects in APL cells is degradation of PML-RARA oncogene, a step necessary for eradication of LICs [94]. As mentioned above, similar effect of PML-RARA degradation may be obtained with ATRA alone, but only when its high intracellular levels are obtained. ATRA/ASO combination therapy synergizes molecular effects of both drugs [92]. At a molecular level ASO targets PML moiety in fusion PML-RARA protein where it triggers formation of arsenic-cysteine bonds that favor protein aggregation. PML-RARA in aggregates undergo ubiquitination, sumoylation and resulting degradation in proteasomes [93]. At a cellular level ASO induces apoptosis, mainly through the mitochondria-mediated intrinsic apoptotic pathway [95].
In its first description APL was considered to be the most malignant form of AML, accompanied by severe bleeding and short survival time, and now it is the most curable one [39,90]. Successful molecular-targeted therapy with ATRA/ASO, has opened a new area in cancer therapy, and raised the possibility that other diseases may be treated by different compounds in a similar way. Since 1,25-dihydroxyvitamin D3 (1,25D) is capable of inducing monocytic differentiation of AML cells in vitro [96,97], it is one of the best prospects for use in clinical applications [98].
4.3. ATRA in Non-APL Subtypes of AML
The possibility to induce differentiation of HL60 cells using ATRA has been known for over 30 years [99]. However, at the time of this discovery HL60 cells were believed to be APL, it only manifested later that they originate from subtype M2 of AML, according to FAB classification [100]. Further studies have shown that there are more non-APL AML cell lines that respond to ATRA. For example in THP-1 cells ATRA up-regulates expression of C/EBPα and β transcription factors [101], which are key regulators in myeloid cell differentiation and also important regulators of ATRA-induced differentiation of APL cells [102]. Such findings stimulated clinical attempts to combine ATRA with chemotherapy for non-APL AML patients. Some early clinical trials have been conducted, but resulting conclusions were confusing. Some trials did not show benefits of combination therapy for patients [103,104], while others did [105]. Therefore a search for more specific differentiation agents for non-APL AML is underway and will be discussed below.
5. 1,25D and Its Low-Calcemic Analogs for Differentiation Therapy
The major role of 1,25D in human body is maintenance of calcium/phosphate homeostasis, but many other so called non-classical actions of 1,25D are known. One of these non-classical actions is, the above mentioned, monocytic differentiation of AML cells [96,97]. In order to separate calcemic properties of 1,25D from other activities of the compound, many low-calcemic vitamin D analogs (VDAs) have been synthesized for various clinical purposes [106-108]. Some VDAs have shown very promising anti-leukemic activities in vitro and in vivo [109-113]. Encouraged by early observations of 1,25D-induced differentiation of AML cells, few clinical trials were conducted to test the ability of 1,25D to treat myelodysplastic syndrome (MDS) and AML [114,115]. Since hypercalcemia is a limiting factor in clinical use of 1,25D in cancer patients, a low-calcemic analog, paricalcitol was also used to treat MDS in a small clinical trial [116]. Results of these trials were disappointing, therefore at present the use of VDAs in combination therapy is postulated. Several different combinations of drugs for use together with VDAs have been proposed and in most of them the goal is to potentiate differentiating effects of 1,25D or VDAs. This way calcemic effects could be avoided by lowering doses of 1,25D or VDAs necessary to obtain differentiation of leukemic cells. A series of papers has shown that antioxidants, such as carnosic acid, silibinin and curcumin effectively potentiate 1,25D-induced cell differentiation in vitro [117-119] and extend the life span of mice inoculated with murine leukemia [110,120]. Similar effect of enhanced differentiation could be obtained using everolismus, immunosuppressant used in transplantation medicine and in oncology, which inhibits mammalian target of rapamycin (mTOR) [121]. Interestingly inhibitors of p38 kinases α and β [122], as well as inhibitors of phospholipase A2 [123] and non-specific inhibitors of cyclooxygenase (COX) [124] do the same. All compounds mentioned above share the ability to interfere with 1,25D-induced cell signaling in leukemic cells and, what is important, most of them are accepted drugs. The second strategy is to combine 1,25D or VDAs with agents that trigger cell death, for example with nutlin-3, which enhanced pro-apoptotic and downregulated anti-apoptotic proteins [125]. Many studies have shown that 1,25D potentiates the anti-tumor activities of chemotherapeutics agents [126], predominantly in solid cancers. However, there are also examples of clinical usefulness of 1,25D and cytarabine combination, which prolonged remission in elderly patients with acute AML and MDS [127,128].
Another approach is to select from a great variety of AMLs, only these which are susceptible to 1,25D. A good analogy exists in ATRA treatment, which is very effective in APL patients, but not in other subtypes of AML. It has been shown that individual responses of ex vivo cultured blast cells from patients with AML to differentiation-inducing effect of 1,25D are variable [118]. The analysis performed in order to look for correlations between mutations that are often diagnosed in blast cells of AML patients, with the susceptibility of the blasts towards VDA-induced differentiation, has shown that the most susceptible cells are these that carry monosomy 7 or partial loss of 7q [129]. Monosomy 7 or losses in the long arm of this chromosome occur in about 18% of the AML cases [130] and are often connected with prior MDS or with earlier therapy with alkylating agents [131]. Adult patients with this mutation have a very aggressive disease, great susceptibility to infections and poor prognosis [131]. If the correlation found in ex vivo cultured blasts could be translated into therapeutic use of VDAs in patients with monosomy 7 or partial loss of 7q, the differentiation-inducing activity of analogs together with their immunostimulating potential [132] might have beneficial effects in this group of patients.
5.1. 1,25D Signaling Pathways of Cell Differentiation in Brief
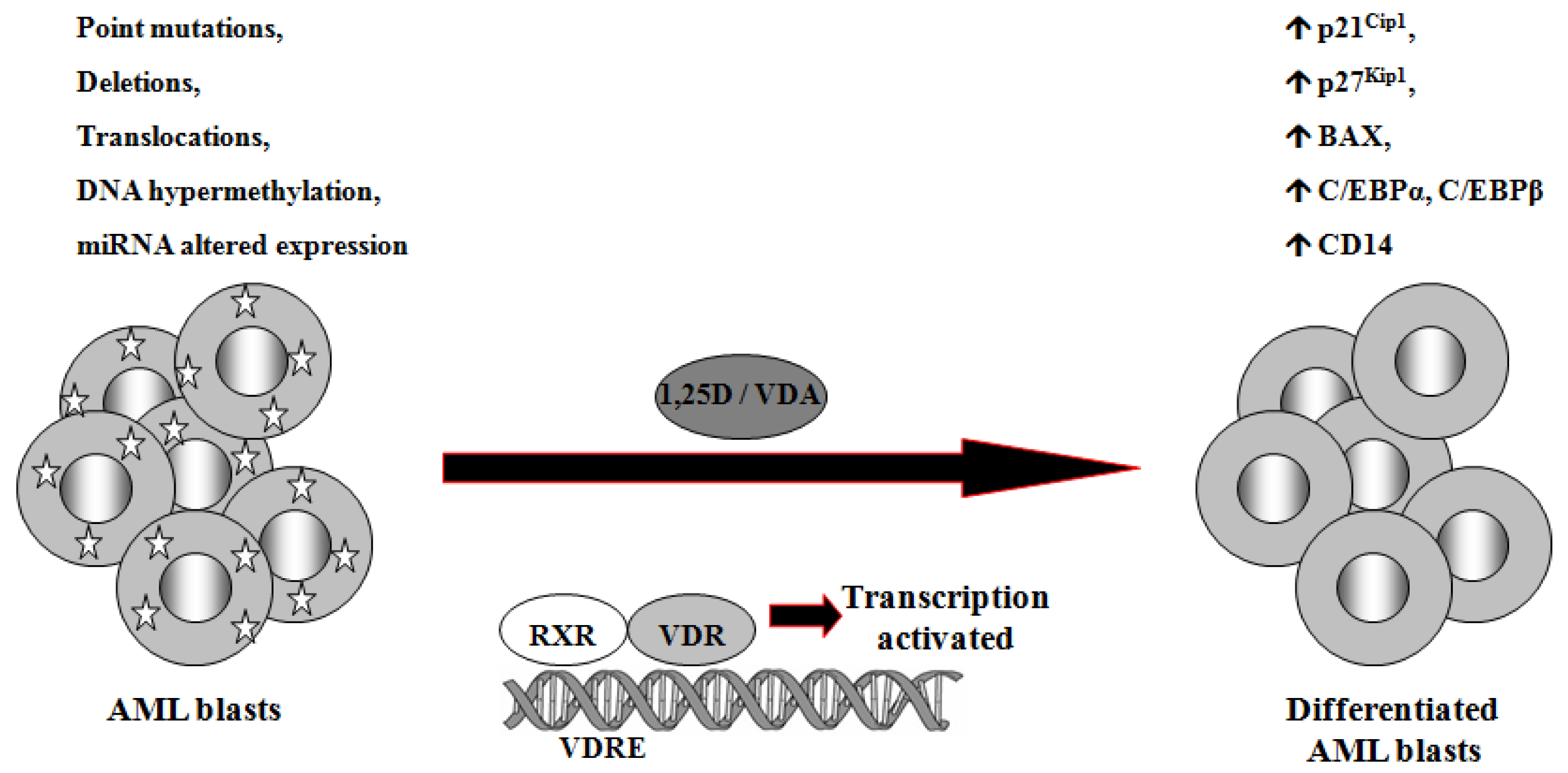
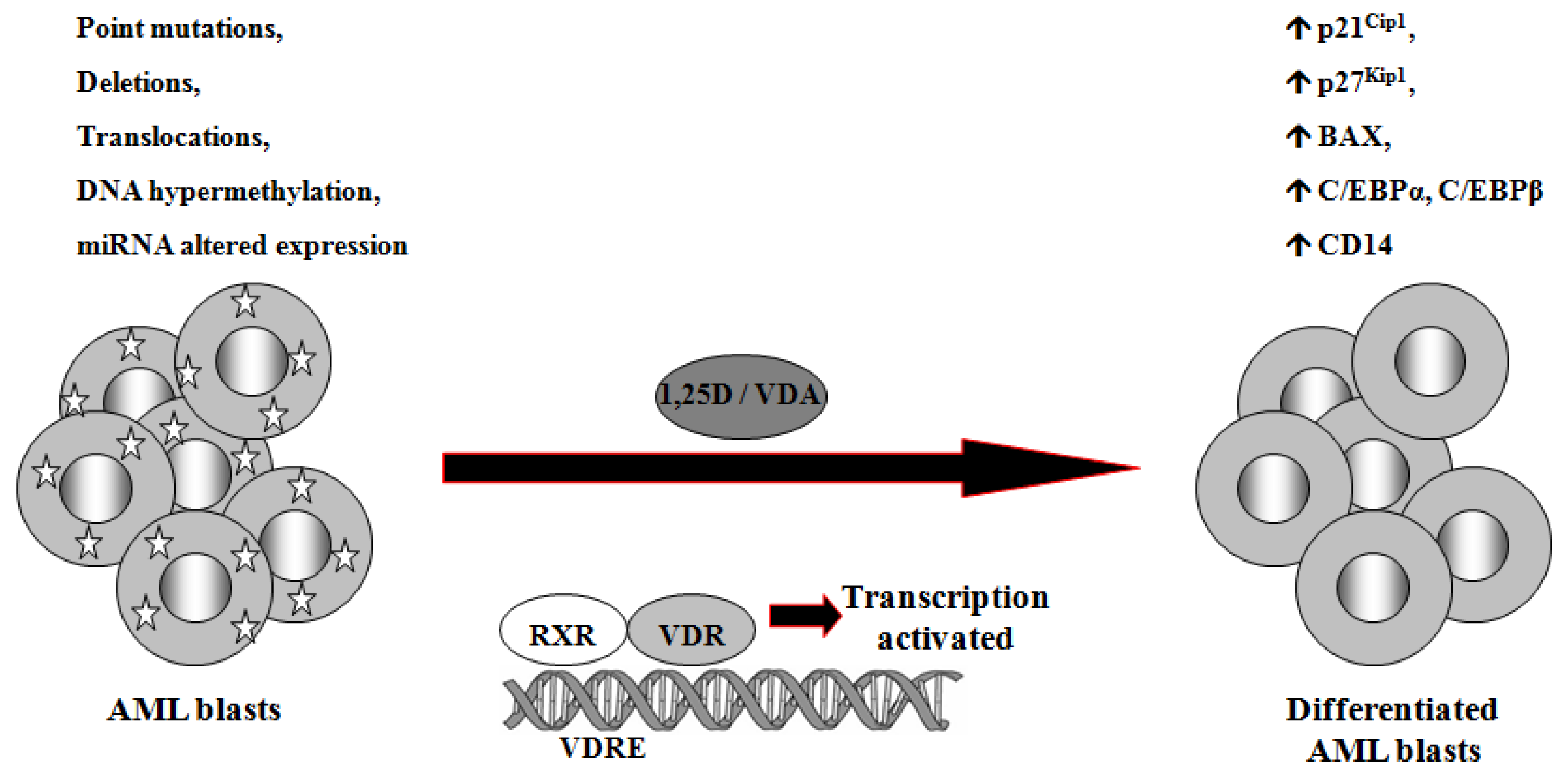
1,25D is one of the steroid hormones that exert biological activity through interaction with their specific nuclear receptors. Vitamin D receptor (VDR) belongs to the same superfamily of nuclear receptors as RAR and RXR. VDR is a ligand-induced transcription factor and a major regulator of 1,25D effects [133]. After ligation with 1,25D or VDAs, VDR becomes protected from degradation [134] and translocates from cell cytosol to the nucleus [135,136]. VDAs with high pro-differentiating potential efficiently stabilize VDR protein and cause its accumulation in the nuclei of the target cells [111]. VDR target genes are connected with the calcium/phosphate homeostasis, but also with anti-proliferative, pro-apoptotic and pro-differentiating actions of 1,25D in non-calcemic tissues. Among such genes are inhibitors of cell cycle, such as p21Cip1 and p27Kip1 [137], pro-apoptotic Bax [138] and transcription factors of monocytic lineage differentiation, such as C/EBPα and β [139,140]. C/EBPα and β regulate transcription of many downstream genes that encode proteins important for proper macrophage function, such as CD14 cell surface molecule, a component of the innate immune system, acting as a co-receptor of bacterial lipopolysaccharide [139,141]. Regulation of target genes by 1,25D may be obtained not only through transcription. It has been documented that 1,25D-induced increase in p27Kip1 at both mRNA and protein levels results from decreased expression of in p27Kip1 inhibitors miR181a/b. Forced expression of pre-miR181a in leukemic cells not only abrogated 1,25D-induced increase in p27Kip1, but also blunted differentiation effect and reduced cell cycle arrest in 1,25D-treated cells [142]. However, VDR seems not to be crucial in normal hematopoiesis, since VDR null mice do not show major defects in blood cells development [143]. Therefore it is likely that 1,25D and VDAs through upregulation of C/EBPα and β transcription factors, their downstream target genes, and by other 1,25D-dependent mechanisms might bypass normal pathways of myeloid differentiation [144], which are blocked in some leukemic blasts (Figure 2).
6. Conclusions
Significant clinical improvement of patients with APL treated with ATRA raised reasonable hope for other agents, such as 1,25D and VDAs, to be effective differentiation-inducing drugs. Further improvements of APL therapy with ASO and detailed understanding of molecular mechanisms of both drugs have shown that therapies may be tailored for specific abnormalities present in neoplastic cells. Novel insights into the etiology of leukemia are of major importance for clinical utility in future drugs [144-146] and may help to select susceptible targets for differentiation therapy using 1,25D and VDAs. It is obvious that the new therapeutic approach should be directed towards leukemic cells, without general cytotoxicity to the organism.
Acknowledgments
This work was supported by the Polish Ministry of Science and Higher Education (Grant Number 2132/B/P01/2008/34) and by the Foundation for Polish Science to E.G.
References
- Hoffbrand, A.; Moss, P.; Pettit, J. Essential Haematolog, 5th ed.; Blackwell Publishing: Oxford, United Kingdom, UK, 2006. [Google Scholar]
- Lowenberg, B. Acute myeloid leukemia: The challenge of capturing disease variety. Hematology 2008, 1–11. [Google Scholar]
- Lowenberg, B.; Griffin, J.; Tallman, M. Acute myeloid leukemia and acute promyelocytic leukemia. Hematology 2003, 82–101. [Google Scholar]
- Sandler, D.; Ross, J. Epidemiology of acute leukemia in children and adults. Semin. Oncol. 1997, 24, 3–16. [Google Scholar]
- Bennett, J.; Catovsky, D.; Daniel, M.; Flandrin, G.; Galton, D.; Gralnick, H.; Sultan, C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar]
- Swerdlow, S.; Campo, E.; Harris, N.; Jaffe, E.; Pileri, S.; Stein, H.; Theile, J.; Vardiman, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC Press: Lyon, France, 2008; pp. 109–138. [Google Scholar]
- Hematologia Molekularna, Patogeneza, Patomechanizmy i Metody Badawcze; Witt, M., Szczepanski, T., Dawidowska, M., Eds.; Ośrodek Wydawnictw Naukowych: Poznań, Poland, 2009.
- Biography of Ehrlich, in Whonamedit? A dictionary of medical eponyms Home Page. Available online: http://www.whonamedit.com/doctor.cfm/83.html/ (accessed 10 December 2010).
- Zelent, A.; Petrie, K.; Chen, Z.; Lotan, R.; Lubbert, M.; Tallman, M.; Ohno, R.; Degos, L.; Waxman, S. Molecular target-based treatment of human cancer: Summary of the 10th international conference on differentiation therapy. Cancer Res. 2005, 65, 1117–1123. [Google Scholar]
- An, X.; Tiwari, A.; Sun, Y.; Ding, P.; Ashby, C.J.; Chen, Z. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: A review. Leuk. Res. 2010, 34, 1255–1268. [Google Scholar]
- Nowell, P.; Hungerford, D. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497. [Google Scholar]
- Erikson, J.; Griffin, C.; ar-Rushdi, A.; Valtieri, M.; Hoxie, J.; Finan, J.; Emanuel, B.; Rovera, G.; Nowell, P.; Croce, C. Heterogeneity of chromosome 22 breakpoint in Philadelphia-positive (Ph+) acute lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 1986, 83, 1807–1811. [Google Scholar]
- Druker, B.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.; Fanning, S.; Zimmermann, J.; Lydon, N. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar]
- Cilloni, D.; Saglio, G. CML: A model for targeted therapy. Best Pract. Res. Clin. Haematol. 2009, 22, 285–294. [Google Scholar]
- Stirewalt, D.; Radich, J. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar]
- Rosnet, O.; Birnbaum, D. Hematopoietic receptors of class III receptor-type tyrosine kinases. Crit. Rev. Oncog. 1993, 4, 595–613. [Google Scholar]
- Rosnet, O.; Schiff, C.; Pébusque, M.; Marchetto, S.; Tonnelle, C.; Toiron, Y.; Birg, F.; Birnbaum, D. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood 1993, 82, 1110–1119. [Google Scholar]
- Lyman, S. Biology of flt3 ligand and receptor. Int. J. Hematol. 1995, 62, 63–73. [Google Scholar]
- McKenna, H.; Stocking, K.; Miller, R.; Brasel, K.; De Smedt, T.; Maraskovsky, E.; Maliszewski, C.; Lynch, D.; Smith, J.; Pulendran, B.; et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000, 95, 3489–3497. [Google Scholar]
- Tse, K.; Mukherjee, G.; Small, D. Constitutive activation of FLT3 stimulates multiple intracellular signal transducers and results in transformation. Leukemia 2000, 14, 1766–1776. [Google Scholar]
- Kindler, T.; Lipka, D.; Fischer, T. FLT3 as a therapeutic target in AML: Still challenging after all these years. Blood 2010, 116, 5089–5102. [Google Scholar]
- Mughal, T.; Ejaz, A.; Foringer, J.; Coiffier, B. An integrated clinical approach for the identification, prevention, and treatment of tumor lysis syndrome. Cancer Treat. Rev. 2010, 36, 164–176. [Google Scholar]
- Ferguson, L.; Tatham, A.; Lin, Z.; Denny, W. Epigenetic regulation of gene expression as an anticancer drug target. Curr. Cancer Drug Targets 2011, 11, 199–212. [Google Scholar]
- Blum, W.; Marcucci, G. Targeting epigenetic changes in acute myeloid leukemia. Clin. Adv. Hematol. Oncol. 2005, 3, 855–865. [Google Scholar]
- Wei, Y.; Kadia, T.; Tong, W.; Zhang, M.; Jia, Y.; Yang, H.; Hu, Y.; Viallet, J.; O'Brien, S.; Garcia-Manero, G. The combination of a histone deacetylase inhibitor with the BH3-mimetic GX15-070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Autophagy 2010, 6, 976–978. [Google Scholar]
- de Lima, M.; Giralt, S.; Thall, P.; de Padua Silva, L.; Jones, R.; Komanduri, K.; Braun, T.; Nguyen, H.; Champlin, R.; Garcia-Manero, G. Maintenance therapy with low-dose azacitidine after allogeneic hematopoietic stem cell transplantation for recurrent acute myelogenous leukemia or myelodysplastic syndrome: A dose and schedule finding study. Cancer 2010, 116, 5420–5431. [Google Scholar]
- Quintás-Cardama, A.; Santos, F.; Garcia-Manero, G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25, 226–235. [Google Scholar]
- Maeda, T.; Towatari, M.; Kosugi, H.; Saito, H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood 2000, 96, 3847–3856. [Google Scholar]
- Venugopal, B.; Evans, T. Developing histone deacetylase inhibitors as anti-cancer therapeutics. Curr. Med. Chem. 2011, 18, 1658–1671. [Google Scholar]
- Bartel, D. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Schickel, R.; Boyerinas, B.; Park, S.; Peter, M. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959–5974. [Google Scholar]
- Engels, B.; Hutvagner, G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene 2006, 25, 6163–6169. [Google Scholar]
- Zeng, Y. Principles of micro-RNA production and maturation. Oncogene 2006, 25, 6156–6162. [Google Scholar]
- Chen, C.; Li, L.; Lodish, H.; Bartel, D. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar]
- Garzon, R.; Pichiorri, F.; Palumbo, T.; Visentini, M.; Aqeilan, R.; Cimmino, A.; Wang, H.; Sun, H.; Volinia, S.; Alder, H.; et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene 2007, 26, 4148–4157. [Google Scholar]
- Fabbri, M.; Garzon, R.; Andreeff, M.; Kantarjian, H.; Garcia-Manero, G.; Calin, G. MicroRNAs and noncoding RNAs in hematological malignancies: Molecular, clinical and therapeutic implications. Leukemia 2008, 22, 1095–1105. [Google Scholar]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar]
- Hillestad, L. Acute promyelocytic leukemia. Acta Med. Scand. 1957, 159, 189–194. [Google Scholar]
- Rowley, J.; Golomb, H.; Dougherty, C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977, 1, 549–550. [Google Scholar]
- Kakizuka, A.; Miller, W.J.; Umesono, K.; Warrell, R.J.; Frankel, S.; Murty, V.; Dmitrovsky, E.; Evans, R. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663–674. [Google Scholar]
- de Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar]
- Borrow, J.; Goddard, A.; Sheer, D.; Solomon, E. Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science 1990, 249, 1577–1580. [Google Scholar]
- Dong, S.; Geng, J.; Tong, J.; Wu, Y.; Cai, J.; Sun, G.; Chen, S.; Wang, Z.; Larsen, C.; Berger, R.; Chen, S.; Chen, Z. Breakpoint clusters of the PML gene in acute promyelocytic leukemia: Primary structure of the reciprocal products of the PML-RARA gene in a patient with t(15;17). Gene. Chromosome.Canc. 1993, 6, 133–139. [Google Scholar]
- Akagi, T.; Shih, L.; Kato, M.; Kawamata, N.; Yamamoto, G.; Sanada, M.; Okamoto, R.; Miller, C.; Liang, D.; Ogawa, S.; Koeffler, H. Hidden abnormalities and novel classification of t(15;17) acute promyelocytic leukemia (APL) based on genomic alterations. Blood 2009, 113, 1741–1748. [Google Scholar]
- Rego, E.; Ruggero, D.; Tribioli, C.; Cattoretti, G.; Kogan, S.; Redner, R.; Pandolfi, P. Leukemia with distinct phenotypes in transgenic mice expressing PML/RAR alpha, PLZF/RAR alpha or NPM/RAR alpha. Oncogene 2006, 25, 1974–1979. [Google Scholar]
- McConnell, M.; Licht, J. The PLZF gene of t (11;17)-associated APL. Curr. Top. Microbiol. Immunol. 2007, 313, 31–48. [Google Scholar]
- Falini, B.; Nicoletti, I.; Bolli, N.; Martelli, M.; Liso, A.; Gorello, P.; Mandelli, F.; Mecucci, C.; Martelli, M. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica 2007, 92, 519–532. [Google Scholar]
- Redner, R. Variations on a theme: The alternate translocations in APL. Leukemia 2002, 16, 1927–1932. [Google Scholar]
- Collins, S. Retinoic acid receptors, hematopoiesis and leukemogenesis. Curr. Opin. Hematol. 2008, 15, 346–351. [Google Scholar]
- Tsai, S.; Collins, S. A dominant negative retinoic acid receptor blocks neutrophil differentiation at the promyelocyte stage. Proc. Natl. Acad. Sci. USA 1993, 90, 7153–7157. [Google Scholar]
- Drumea, K.; Yang, Z.; Rosmarin, A. Retinoic acid signaling in myelopoiesis. Curr. Opin. Hematol. 2008, 15, 37–41. [Google Scholar]
- Zhang, J.; Wang, J.; Chen, S.; Chen, Z. Mechanisms of all-trans retinoic acid-induced differentiation of acute promyelocytic leukemia cells. J. Biosci. 2000, 25, 275–284. [Google Scholar]
- Freedman, L. Increasing the complexity of coactivation in nuclear receptor signaling. Cell 1999, 97, 5–8. [Google Scholar]
- Hou, Z.; Peng, H.; White, D.; Negorev, D.; Maul, G.; Feng, Y.; Longmore, G.; Waxman, S.; Zelent, A.; Rauscher, F.r. LIM protein Ajuba functions as a nuclear receptor corepressor and negatively regulates retinoic acid signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 2938–2943. [Google Scholar]
- Chen, J.; Evans, R. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [Google Scholar]
- Nagy, L.; Kao, H.; Chakravarti, D.; Lin, R.; Hassig, C.; Ayer, D.; Schreiber, S.; Evans, R. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 1997, 89, 373–380. [Google Scholar]
- Lo-Coco, F.; Ammatuna, E. The biology of acute promyelocytic leukemia and its impact on diagnosis and treatment. Hematology 2006, 514, 156–161. [Google Scholar]
- Nowak, D.; Stewart, D.; Koeffler, H. Differentiation therapy of leukemia: 3 decades of development. Blood 2009, 113, 3655–3665. [Google Scholar]
- Dyck, J.; Maul, G.; Miller, W.J.; Chen, J.; Kakizuka, A.; Evans, R. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 1994, 76, 333–343. [Google Scholar]
- Bernardi, R.; Pandolfi, P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell. Bio.l 2007, 8, 1006–1016. [Google Scholar]
- Melnick, A.; Licht, J. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 1999, 93, 3167–3215. [Google Scholar]
- Di Croce, L.; Raker, V.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; Minucci, S.; Pelicci, P. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002, 295, 1079–1082. [Google Scholar]
- Kamashev, D.; Vitoux, D.; De Thé, H. PML-RARA-RXR oligomers mediate retinoid and rexinoid/cAMP cross-talk in acute promyelocytic leukemia cell differentiation. J. Exp. Med. 2004, 199, 1163–1174. [Google Scholar]
- Lin, R.; Evans, R. Acquisition of oncogenic potential by RAR chimeras in acute promyelocytic leukemia through formation of homodimers. Mol. Cell. 2000, 5, 821–830. [Google Scholar]
- Hoemme, C.; Peerzada, A.; Behre, G.; Wang, Y.; McClelland, M.; Nieselt, K.; Zschunke, M.; Disselhoff, C.; Agrawal, S.; Isken, F.; et al. Chromatin modifications induced by PML-RARalpha repress critical targets in leukemogenesis as analyzed by ChIP-Chip. Blood 2008, 111, 2887–2895. [Google Scholar]
- Viale, A.; De Franco, F.; Orleth, A.; Cambiaghi, V.; Giuliani, V.; Bossi, D.; Ronchini, C.; Ronzoni, S.; Muradore, I.; Monestiroli, S.; et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 2009, 457, 51–56. [Google Scholar]
- Salomoni, P.; Pandolfi, P. The role of PML in tumor suppression. Cell 2002, 108, 165–170. [Google Scholar]
- Jensen, K.; Shiels, C.; Freemont, P. PML protein isoforms and the RBCC/TRIM motif. Oncogene 2001, 20, 7223–7233. [Google Scholar]
- Scaglioni, P.; Pandolfi, P. The theory of APL revisited. Curr. Top. Microbiol. Immunol. 2007, 313, 85–100. [Google Scholar]
- Shih, L.; Kuo, M.; Liang, D.; Huang, C.; Lin, T.; Wu, J.; Wang, P.; Dunn, P.; Lai, C. Internal tandem duplication and Asp835 mutations of the FMS-like tyrosine kinase 3 (FLT3) gene in acute promyelocytic leukemia. Cancer 2003, 98, 1206–1216. [Google Scholar]
- Sritana, N.; Auewarakul, C. KIT and FLT3 receptor tyrosine kinase mutations in acute myeloid leukemia with favorable cytogenetics: Two novel mutations and selective occurrence in leukemia subtypes and age groups. Exp. Mol. Pathol. 2008, 85, 227–231. [Google Scholar]
- Wang, K.; Wang, P.; Shi, J.; Zhu, X.; He, M.; Jia, X.; Yang, X.; Qiu, F.; Jin, W.; Qian, M.; et al. PML/RARalpha targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell 2010, 17, 186–197. [Google Scholar]
- Glass, C.; Rosenfeld, M. The coregulator exchange in transcriptional functions of nuclear receptors. Gene. Dev. 2000, 14, 121–141. [Google Scholar]
- Minucci, S.; Maccarana, M.; Cioce, M.; De Luca, P.; Gelmetti, V.; Segalla, S.; Di Croce, L.; Giavara, S.; Matteucci, C.; Gobbi, A.; et al. Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol. Cell. 2000, 5, 811–820. [Google Scholar]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar]
- Saumet, A.; Vetter, G.; Bouttier, M; Portales-Casamar, E.; Wasserman, W.; Maurin, T.; Mari, B.; Barbry, P.; Vallar, L.; Friederich, E.; et al. Transcriptional repression of microRNA genes by PML-RARA increases expression of key cancer proteins in acute promyelocytic leukemia. Blood 2009, 113, 412–421. [Google Scholar]
- De Marchis, M.; Ballarino, M.; Salvatori, B.; Puzzolo, M.; Bozzoni, I.; Fatica, A. A new molecular network comprising PU.1, interferon regulatory factor proteins and miR-342 stimulates ATRA-mediated granulocytic differentiation of acute promyelocytic leukemia cells. Leukemia 2009, 23, 856–862. [Google Scholar]
- Careccia, S.; Mainardi, S.; Pelosi, A.; Gurtner, A.; Diverio, D.; Riccioni, R.; Testa, U.; Pelosi, E.; Piaggio, G.; Sacchi, A.; et al. A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene 2009, 28, 4034–4040. [Google Scholar]
- Nasr, R.; Guillemin, M.; Ferhi, O.; Soilihi, H.; Peres, L.; Berthier, C.; Rousselot, P.; Robledo-Sarmiento, M.; Lallemand-Breitenbach, V.; Gourmel, B.; et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 2008, 14, 1333–1342. [Google Scholar]
- Kitareewan, S.; Pitha-Rowe, I.; Sekula, D.; Lowrey, C.; Nemeth, M.; Golub, T.; Freemantle, S.; Dmitrovsky, E. UBE1L is a retinoid target that triggers PML/RARalpha degradation and apoptosis in acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 3806–3811. [Google Scholar]
- Huang, Y.; Qiu, J.; Chen, G.; Dong, S. Coiled-coil domain of PML is essential for the aberrant dynamics of PML-RARalpha, resulting in sequestration and decreased mobility of SMRT. Biochem. Biophys. Res. Commun. 2008, 365, 258–265. [Google Scholar]
- Isakson, P.; Bjørås, M.; Bøe, S.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar]
- Park, D.; Chumakov, A.; Vuong, P.; Chih, D.; Gombart, A.; Miller, W.J.; Koeffler, H. CCAAT/enhancer binding protein epsilon is a potential retinoid target gene in acute promyelocytic leukemia treatment. J. Clin. Invest. 1999, 103, 1399–1408. [Google Scholar]
- Carbone, R.; Botrugno, O.; Ronzoni, S.; Insinga, A.; Di Croce, L.; Pelicci, P.; Minucci, S. Recruitment of the histone methyltransferase SUV39H1 and its role in the oncogenic properties of the leukemia-associated PML-retinoic acid receptor fusion protein. Mol. Cell. Biol. 2006, 26, 1288–1296. [Google Scholar]
- Liu, M.; Iavarone, A.; Freedman, L. Transcriptional activation of the human p21(WAF1/CIP1) gene by retinoic acid receptor. Correlation with retinoid induction of U937 cell differentiation. J. Biol. Chem. 1996, 271, 31723–31728. [Google Scholar]
- Bocchia, M.; Xu, Q.; Wesley, U.; Xu, Y.; Korontsvit, T.; Loganzo, F.; Albino, A.; Scheinberg, D. Modulation of p53, WAF1/p21 and BCL-2 expression during retinoic acid-induced differentiation of NB4 promyelocytic cells. Leuk. Res. 1997, 21, 439–447. [Google Scholar]
- Harris, M.; Ozpolat, B.; Abdi, F.; Gu, S.; Legler, A.; Mawuenyega, K.; Tirado-Gomez, M.; Lopez-Berestein, G.; Chen, X. Comparative proteomic analysis of all-trans-retinoic acid treatment reveals systematic posttranscriptional control mechanisms in acute promyelocytic leukemia. Blood 2004, 104, 1314–1323. [Google Scholar]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Breccia, M.; Gallo, E.; Rambaldi, A.; Paoloni, F.; Fioritoni, G.; Ferrara, F.; Specchia, G.; et al. Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood 2010, 116, 3171–3179. [Google Scholar]
- Wang, Z.; Chen, Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar]
- Shen, Y.; Shen, Z.; Yan, H.; Chen, J.; Zeng, X.; Li, J.; Li, X.; Wu, W.; Xiong, S.; Zhao, W.; et al. Studies on the clinical efficacy and pharmacokinetics of low-dose arsenic trioxide in the treatment of relapsed acute promyelocytic leukemia: A comparison with conventional dosage. Leukemia 2001, 15, 735–741. [Google Scholar]
- Nasr, R.; de Thé, H. Eradication of acute promyelocytic leukemia-initiating cells by PML/RARA-targeting. Int. J. Hematol. 2010, 91, 742–747. [Google Scholar]
- Chen, S.; Zhou, G.; Zhang, X.; Mao, J.; de The, H.; Chen, Z. From an old remedy to a magic bullet: Molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood 2011. Epub ahead of print. [Google Scholar]
- Ablain, J.; de The, H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 2011. Epub ahead of print. [Google Scholar]
- Chen, G.; Shi, X.; Tang, W.; Xiong, S.; Zhu, J.; Cai, X.; Han, Z.; Ni, J.; Shi, G.; Jia, P.; et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 1997, 89, 3345–3353. [Google Scholar]
- Koeffler, H. Induction of differentiation of human acute myelogenous leukemia cells: Therapeutic implications. Blood 1983, 62, 709–721. [Google Scholar]
- Miyaura, C.; Abe, E.; Kuribayashi, T.; Tanaka, H.; Konno, K.; Nishii, Y.; Suda, T. 1alpha,25-Dihydroxyvitamin D3 induces differentiation of human myeloid leukemia cells. Biochem. Biophys. Res. Commun. 1981, 102, 937–943. [Google Scholar]
- Krishnan, A.; Trump, D.; Johnson, C.; Feldman, D. The role of vitamin D in cancer prevention and treatment. Endocrinol. Metab. Clin. North Am. 2010, 39, 401–418. [Google Scholar]
- Breitman, T.; Selonick, S.; Collins, S. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar]
- Morosetti, R.; Park, D.; Chumakov, A.; Grillier, I.; Shiohara, M.; Gombart, A.; Nakamaki, T.; Weinberg, K.; Koeffler, H. A novel, myeloid transcription factor, C/EBPepsilon, is upregulated during granulocytic, but not monocytic, differentiation. Blood 1997, 90, 2591–2600. [Google Scholar]
- Lee, I.; Lee, J.; Lee, M.; Lee, S.; Kim, I. Involvement of CCAAT/enhancer-binding protein alpha in haptoglobin gene expression by all-trans-retinoic acid. Biochem. Biophys. Res. Commun. 2002, 294, 956–961. [Google Scholar]
- Duprez, E.; Wagner, K.; Koch, H.; Tenen, D. C/EBPbeta: A major PML-RARA-responsive gene in retinoic acid-induced differentiation of APL cells. EMBO J. 2003, 22, 5806–5816. [Google Scholar]
- Seiter, K.; Feldman, E.; Halicka, D.; Deptala, A.; Traganos, F.; Burke, H.; Hoang, A.; Goff, H.; Pozzuoli, M.; Kancherla, R.; Darzynkiewicz, Z.; Ahmed, T. Clinical and laboratory evaluation of all-trans retinoic acid modulation of chemotherapy in patients with acute myelogenous leukaemia. Br. J. Haematol. 2000, 108, 40–47. [Google Scholar]
- Bassan, R.; Chiodini, B.; Lerede, T.; Giussani, U.; Oldani, E.; Buelli, M.; Rossi, A.; Viero, P.; Rambaldi, A.; Barbui, T. Prolonged administration of all-trans retinoic acid in combination with intensive chemotherapy and G-CSF for adult acute myelogenous leukemia: Single-centre pilot study in different risk groups. Hematol. J. 2002, 3, 193–200. [Google Scholar]
- Ustün, C.; Beksac, M.; Dalva, K.; Koc, H.; Konuk, N.; Ilhan, O.; Ozcan, M.; Topcuoglu, P.; Sertkaya, D.; Hayran, M. In vivo use of all-trans retinoic acid prior to induction chemotherapy improves complete remission rate and increases rhodamine 123 uptake in patients with de novo acute myeloid leukemia. Med. Oncol. 2002, 19, 59–67. [Google Scholar]
- Bouillion, R.; Okamura, W.; Norman, A. Structure-function relationships in the vitamin D endocrine system. Endocr. Rev. 1995, 16, 200–216. [Google Scholar]
- Jones, G.; Strugnell, S.; DeLuca, H. Current understanding of the molecular actions of vitamin D. Physiol. Rev. 1998, 78, 1193–1231. [Google Scholar]
- Jones, G. Vitamin D analogs. Endocrinol. Metab. Clin. North Am. 2010, 39, 447–472. [Google Scholar]
- Takahashi, E.; Nakagawa, K.; Suhara, Y.; Kittaka, A.; Nihei, K.; Konno, K.; Takayama, H.; Ozono, K.; Okano, T. Biological activities of 2alpha-substituted analogues of 1alpha,25-dihydroxyvitamin D3 in transcriptional regulation and human promyelocytic leukemia (HL-60) cell proliferation and differentiation. Biol. Pharm. Bull. 2006, 29, 2246–2250. [Google Scholar]
- Sharabani, H.; Izumchenko, E.; Wang, Q.; Kreinin, R.; Steiner, M.; Barvish, Z.; Kafka, M.; Sharoni, Y.; Levy, J.; Uskokovic, M.; et al. Cooperative antitumor effects of vitamin D3 derivatives and rosemary preparations in a mouse model of myeloid leukemia. Int. J. Cancer 2006, 118, 3012–3021. [Google Scholar]
- Gocek, E.; Kielbinski, M.; Wylob, P.; Kutner, A.; Marcinkowska, E. Side-chain modified vitamin D analogs induce rapid accumulation of VDR in the cell nuclei proportionately to their differentiation-inducing potential. Steroids 2008, 73, 1359–1366. [Google Scholar]
- Maehr, H.; Lee, H.; Perry, B.; Suh, N.; Uskokovic, M. Calcitriol derivatives with two different side chains at C-20. V. Potent inhibitors of mammary carcinogenesis and inducers of leukemia differentiation. J. Med. Chem. 2009, 52, 5505–5519. [Google Scholar]
- Slominski, A.; Janjetovic, Z.; Fuller, B.; Zmijewski, M.; Tuckey, R.; Nguyen, M.; Sweatman, T.; Li, W.; Zjawiony, J.; Miller, D.; Chen, T.; Lozanski, G.; Holick, M. Products of vitamin D3 or 7-dehydrocholesterol metabolism by cytochrome P450scc show anti-leukemia effects, having low or absent calcemic activity. PLoS One 2010, 5. [Google Scholar]
- Koeffler, H.; Hirji, K.; Itri, L. 1,25-Dihydroxyvitamin D3: In vivo and in vitro effects on human preleukemic and leukemic cells. Cancer Treat. Rep. 1985, 69, 1399–1407. [Google Scholar]
- Takahashi, T.; Ichiba, S.; Okuno, Y.; Sugiyama, H.; Sakai, Y.; Imura, H.; Iho, S.; Hoshino, T.; Suzuki, A.; Okada, T. Therapeutic effectiveness of vitamin D3 in patients with myelodysplastic syndromes, leukemias and myeloproliferative disorders. Rinsho Ketsueki 1989, 30, 1–10. [Google Scholar]
- Okamoto, R.; Akagi, T.; Koeffler, H. Vitamin D compounds and myelodysplastic syndrome. Leuk. Lymphoma 2008, 49, 12–13. [Google Scholar]
- Danilenko, M.; Wang, X.; Studzinski, G. Carnosic acid and promotion of monocytic differentiation of HL60-G cells initiated by other agents. J. Natl. Cancer Inst. 2001, 93, 1224–1233. [Google Scholar]
- Wang, Q.; Harrison, J.; Uskokovic, M.; Kutner, A.; Studzinski, G. Translational study of vitamin D differentiation therapy of myeloid leukemia: Effects of the combination with a p38 MAPK inhibitor and an antioxidant. Leukemia 2005, 19, 1812–1817. [Google Scholar]
- Danilenko, M.; Wang, Q.; Wang, X.; Levy, J.; Sharoni, Y.; Studzinski, G. Carnosic acid potentiates the antioxidant and prodifferentiation effects of 1α,25-dihydroxyvitamin D3 in leukemia cells, but does not promote elevation of basal levels of intracellular calcium. Cancer Res. 2003, 63, 1325–1332. [Google Scholar]
- Shabtay, A.; Sharabani, H.; Barvish, Z.; Kafka, M.; Amichay, D.; Levy, J.; Sharoni, Y.; Uskokovic, M.; Studzinski, G.; Danilenko, M. Synergistic antileukemic activity of carnosic acid-rich rosemary extract and the 19-nor Gemini vitamin D analogue in a mouse model of systemic acute myeloid leukemia. Oncology 2008, 75, 203–214. [Google Scholar]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Ni, L.; Koeffler, H.; Yokoyama, A. Inhibition of mTORC1 by RAD001 (everolimus) potentiates the effects of 1,25-dihydroxyvitamin D3 to induce growth arrest and differentiation of AML cells in vitro and in vivo. Exp. Hematol. 2010, 38, 666–676. [Google Scholar]
- Zhang, J.; Harrison, J.; Studzinski, G. Isoforms of p38MAPK gamma and delta contribute to differentiation of human AML cells induced by 1,25-dihydroxyvitamin D3. Exp. Cell. Res. 2011, 317, 117–130. [Google Scholar]
- Marcinkowska, E.; Kutner, A. Side-chain modified vitamin D analogs require activation of both PI 3-K and erk1,2 signal transduction pathways to induce differentiation of human promyelocytic leukemia cells. Acta Biochim. Pol. 2002, 49, 393–406. [Google Scholar]
- Jamshidi, F.; Zhang, J.; Harrison, J.; Wang, X.; Studzinski, G. Induction of differentiation of human leukemia cells by combinations of COX inhibitors and 1,25-dihydroxyvitamin D3 involves Raf1 but not Erk 1/2 signaling. Cell Cycle 2008, 917–924. [Google Scholar]
- Thompson, T.; Andreeff, M.; Studzinski, G.; Vassilev, L. 1,25-dihydroxyvitamin D3 enhances the apoptotic activity of MDM2 antagonist nutlin-3a in acute myeloid leukemia cells expressing wild-type p53. Mol. Cancer Ther. 2010, 9, 1158–1168. [Google Scholar]
- Ma, Y.; Trump, D.; Johnson, C. Vitamin D in combination cancer treatment. J. Cancer 2010, 1, 101–107. [Google Scholar]
- Slapak, C.; Desforges, J.; Fogaren, T.; Miller, K. Treatment of acute myeloid leukemia in the elderly with low-dose cytarabine, hydroxyurea, and calcitriol. Am. J. Hematol. 1992, 41, 178–183. [Google Scholar]
- Ferrero, D.; Campa, E.; Dellacasa, C.; Campana, S.; Foli, C.; Boccadoro, M. Differentiating agents + low-dose chemotherapy in the management of old/poor prognosis patients with acute myeloid leukemia or myelodysplastic syndrome. Haematologica 2004, 89, 619–620. [Google Scholar]
- Gocek, E.; Kielbinski, M.; Baurska, H.; Haus, O.; Kutner, A.; Marcinkowska, E. Different susceptibilities to 1,25-dihydroxyvitamin D3-induced differentiation of AML cells carrying various mutations. Leuk. Res. 2010, 34, 649–657. [Google Scholar]
- Brozek, I.; Babinska, M.; Kardas, I.; Wozniak, A.; Balcerska, A.; Hellmann, A.; Limon, J. Cytogenetic analysis and clinical significance of chromosome 7 aberrations in acute leukaemia. J. Appl. Genet. 2003, 44, 401–412. [Google Scholar]
- Galili, N.; Cerny, J.; Raza, A. Current treatment options: Impact of cytogenetics on the course of myelodysplasia. Curr. Treat Opt. Oncol. 2007, 8, 117–128. [Google Scholar]
- Wang, T.; Nestel, F.; Bourdeau, V.; Nagai, Y.; Wang, Q.; Liao, J.; Tavera-Mendoza, L.; Lin, R.; Hanrahan, J.; Mader, S.; White, J. Cutting edge: 1,25-Dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 2004, 173, 2909–2912. [Google Scholar]
- Dusso, A.; Brown, A.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Renal. Physiol. 2005, 289, F8–F28. [Google Scholar]
- Arbour, N.; Prahl, J.; DeLuca, H. Stabilization of the vitamin D receptor in rat osteosarcoma cells through the action of 1,25-dihydroxyvitamin D3. Mol. Endocrinol. 1993, 7, 1307–1312. [Google Scholar]
- Racz, A.; Barsony, J. Hormone-dependent translocation of vitamin D receptors is linked to transactivation. J. Biol. Chem. 1999, 274, 19352–19360. [Google Scholar]
- Gocek, E.; Kielbinski, M.; Marcinkowska, E. Activation of intracellular signaling pathways is necessary for an increase in VDR expression and its nuclear translocation. FEBS Lett. 2007, 581, 1751–1757. [Google Scholar]
- Verlinden, L.; Verstuyf, A.; Convents, R.; Marcelis, S.; Van Camp, M.; Bouillon, R. Action of 1,25(OH)2D3 on the cell cycle genes, cyclin D1, p21 and p27 in MCF-7 cells. Mol. Cell. Endocrinol. 1998, 142, 57–65. [Google Scholar]
- Kizildag, S.; Ates, H.; Kizildag, S. Treatment of K562 cells with 1,25-dihydroxyvitamin D3 induces distinct alterations in the expression of apoptosis-related genes BCL2, BAX, BCL(XL), and p21. Ann. Hematol. 2009, 89, 1–7. [Google Scholar]
- Pan, Z.; Hetherington, C.; Zhang, D. CCAAT/enhancer-binding protein activates the CD14 promoter and mediates transforming growth factor beta signaling in monocyte development. J. Biol. Chem. 1999, 274, 23242–23248. [Google Scholar]
- Marcinkowska, E; Garay, E; Gocek, E; Chrobak, A; Wang, X; Studzinski, GP. Regulation of C/EBPbeta isoforms by MAPK pathways in HL60 cells induced to differentiate by 1,25-dihydroxyvitamin D3. Exp. Cell. Res. 2006, 312, 2054–2065. [Google Scholar]
- Triantafilou, M.; Triantafilou, K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. 2002, 23, 301–304. [Google Scholar]
- Wang, X.; Gocek, E.; Liu, C.; Studzinski, G. MicroRNAs181 regulate the expression of p27Kip1 in human myeloid leukemia cells induced to differentiate by 1,25-dihydroxyvitamin D3. Cell Cycle 2009, 18, 736–741. [Google Scholar]
- Yoshizawa, T.; Handa, Y.; Uematsu, Y.; Takeda, S.; Sekine, K.; Yoshihara, Y.; Kawakami, T.; Arioka, K.; Sato, H.; Uchiyama, Y.; et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat. Genet. 1997, 16, 391–396. [Google Scholar]
- Hughes, P.; Marcinkowska, E.; Gocek, E.; Studzinski, G.; Brown, G. Vitamin D3-driven signals for myeloid cell differentiation—Implications for differentiation therapy. Leuk. Res. 2010, 34, 553–565. [Google Scholar]
- Lübbert, M.; Müller-Tidow, C.; Hofmann, W.; Koeffler, H. Advances in the treatment of acute myeloid leukemia: From chromosomal aberrations to biologically targeted therapy. J. Cell. Biochem. 2008, 104, 2059–2070. [Google Scholar]
- Petrie, K.; Zelent, A.; Waxman, S. Differentiation therapy of acute myeloid leukemia: Past, present and future. Curr. Opin. Hematol. 2009, 16, 84–91. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gocek, E.; Marcinkowska, E. Differentiation Therapy of Acute Myeloid Leukemia. Cancers 2011, 3, 2402-2420. https://doi.org/10.3390/cancers3022402
Gocek E, Marcinkowska E. Differentiation Therapy of Acute Myeloid Leukemia. Cancers. 2011; 3(2):2402-2420. https://doi.org/10.3390/cancers3022402
Chicago/Turabian StyleGocek, Elzbieta, and Ewa Marcinkowska. 2011. "Differentiation Therapy of Acute Myeloid Leukemia" Cancers 3, no. 2: 2402-2420. https://doi.org/10.3390/cancers3022402
APA StyleGocek, E., & Marcinkowska, E. (2011). Differentiation Therapy of Acute Myeloid Leukemia. Cancers, 3(2), 2402-2420. https://doi.org/10.3390/cancers3022402