The Enigmatic Roles of Caspases in Tumor Development



Abstract
:1. Structure and Activation of Caspases
2. Is loss of Caspase Activity Involved in Cancer?
2.1. Somatic Mutations of Caspase Genes in Human Cancer
2.1.1. Executioner caspases

{kind=link}
| Caspase | Cancer type | mutated | silenced | LOH | Function | Reference |
|---|---|---|---|---|---|---|
| Executioner caspases | ||||||
| Caspase-3 | Colon | 2.1% (98) | 10 | |||
| NHL | 0.8% (129) | 10 | ||||
| Lung | 1.7% (181) | 10 | ||||
| Gastric | 5% (60) | 24 | ||||
| Caspase-6 | Colorectal | 2% (100) | 21 | |||
| Gastric | 48% (120) | 25 | ||||
| Caspase-7 | Esophagal | 1% (50) | impaired | 19 | ||
| Gastric | 67% (120) | 25 | ||||
| Colon | 85% (26) | 23 | ||||
| Initiator caspases | ||||||
| Caspase-2 | Gastric | 65% (120) | 25 | |||
| Caspase-8 | Gastric | 8% (162) | 1 LOH, 6 het, 6 NI | impaired | 44 | |
| Liver | 13% (69) | impaired | 42 | |||
| Colon | 5.1% (98) | impaired | 43 | |||
| Neuroblastoma | 75% (140) | 53 | ||||
| Medulloblastoma | 52% (27) | 47 | ||||
| SCLC | 79% (34) | 45 | ||||
| Caspase-9 | Colon | 46% (26) | 23 | |||
| Gastric | 10% (60) | 24 | ||||
| Caspase-10 | Colon | 2.1% (47) | 37 | |||
| Gastric | 3% (99) | 2 LOH | impaired | 41 | ||
| NHL | 14.5% (117) | impaired | 38 | |||
| Gastric | 5% (60) | 24 | ||||
2.1.2. Initiator caspases
2.2. Evidence from Transgenic Mice
2.3. In Vitro Transformation of Human Cells
3. Why Caspase Genes Possibly Need Not be Altered in Tumors?
3.1. Scenario 1: Dominant-Negative Splice Variants of Caspases
3.2. Scenario 2: Endogeneous Inhibitors of Apoptosis Block Caspases in Tumor Cells
3.3. Scenario 3: Pathways Upstream of Caspase Activation are Disrupted
3.4. Scenario 4: CICD Circumvents the Need for Caspase Dysfunction
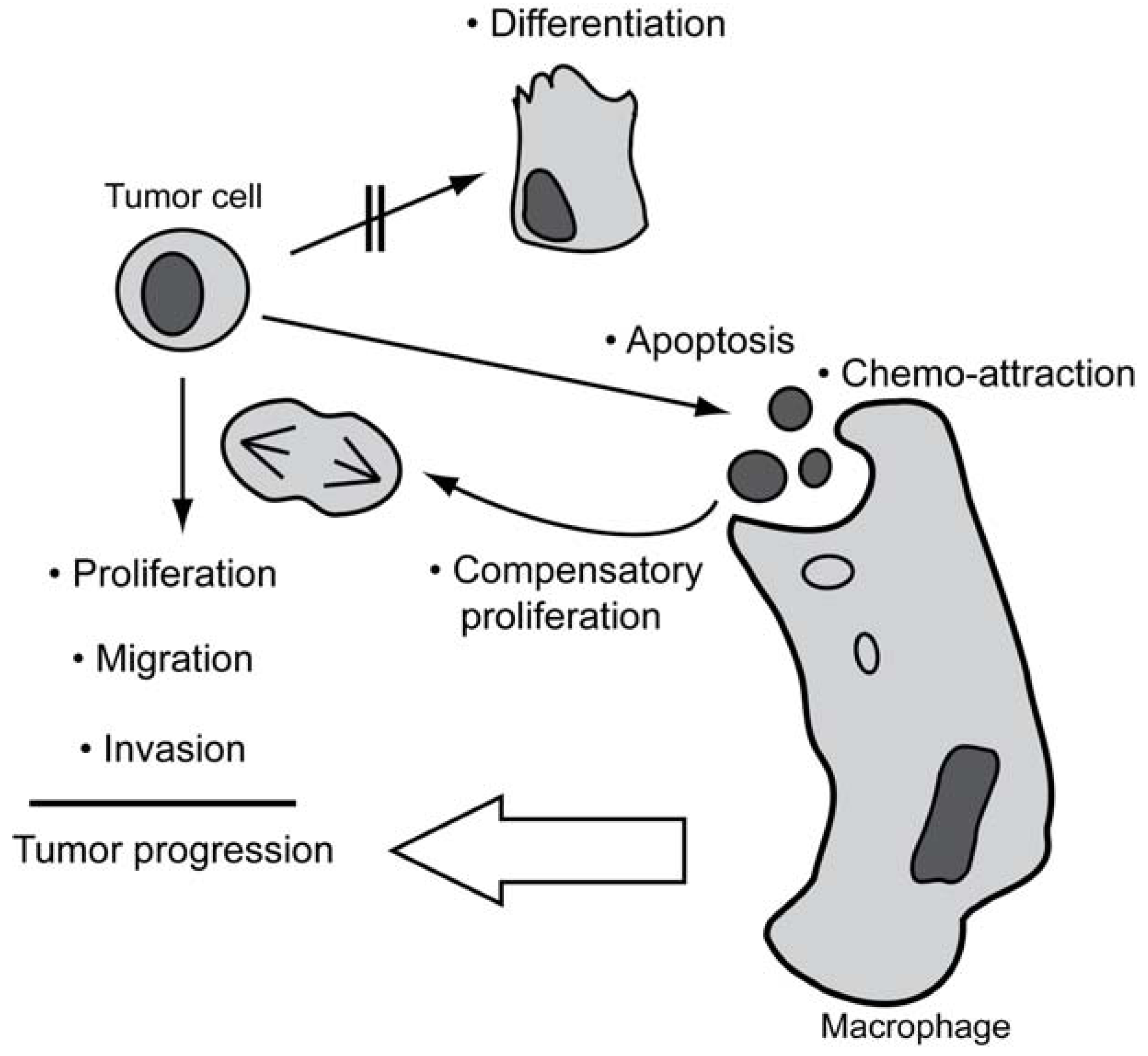
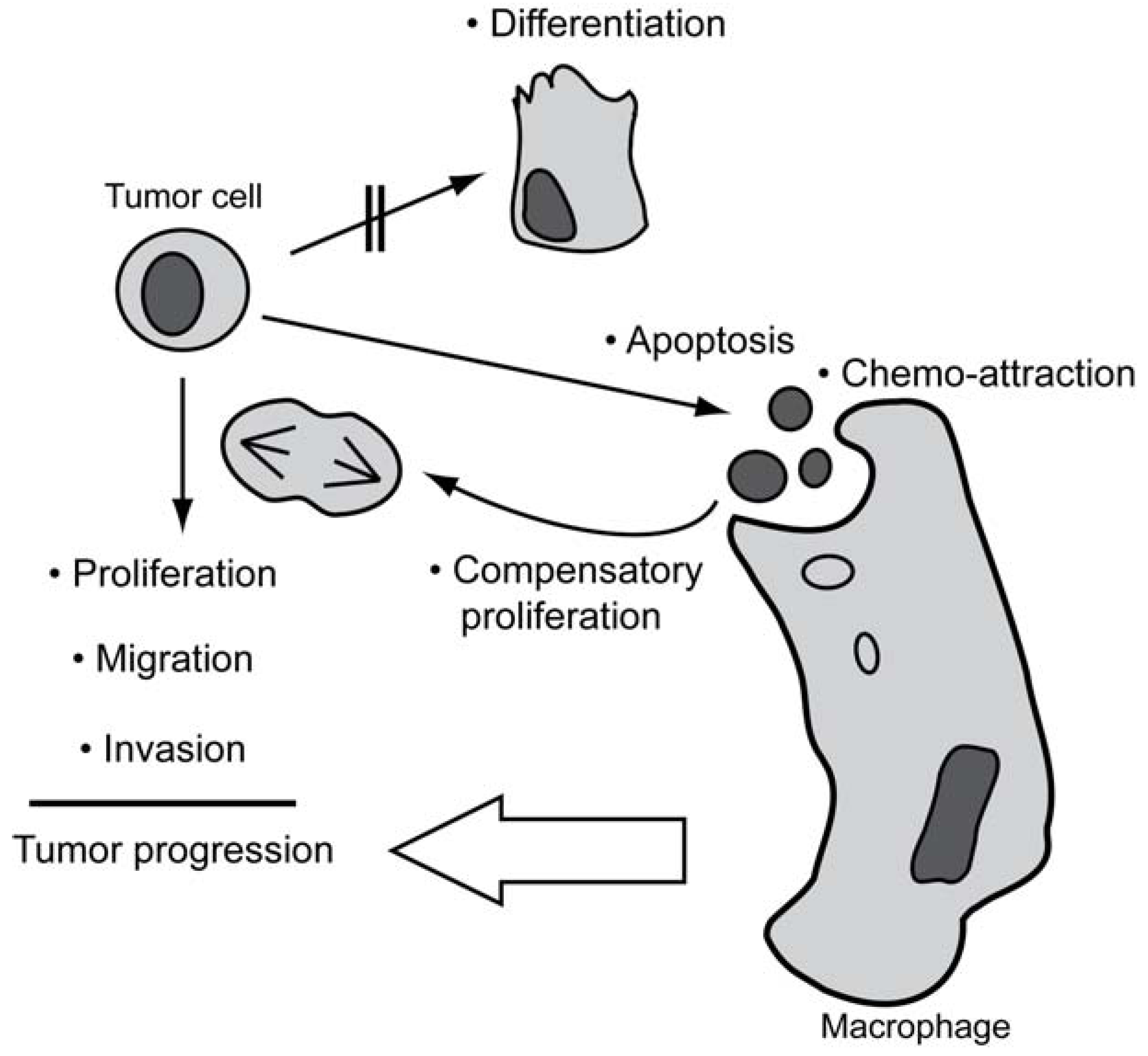
4. The Occurrence of Spontaneous Apoptosis in Tumors
5. Non-Apoptotic Functions of Caspases Possibly Involved in Tumorigenesis
5.1. Caspases in Proliferation
5.2. Caspases in Migration and Invasion
| Function | Caspase | Experimental system | Catalytic activity required? | Ref. |
|---|---|---|---|---|
| Proliferation | Caspase-3 | • mitotic check-point control of HeLa cells | yes | [169] |
| • c-myc-induced hyperproliferation of pancreatic beta-cells in vivo | n.d. | [64] | ||
| Caspase-7 | • caspase-7 knockdown leads to mitotic arrest in HepG2 cells | n.d. | [170] | |
| Caspase-6 | • reentry into cell cycle of quiescent B-cells | no | [171] | |
| Caspase-8 | • reentry into cell cycle of quiescent T-cells | n.d. | [58] | |
| • cytokine-induced proliferation of hematopoietic progenitors | yes | [172] | ||
| Migration | Caspase-3 | • laminin-induced migration of ovarian carcinoma cells | yes | [177] |
| Caspase-8 | • Caspase-8-deficient embryonic fibroblasts display reduced motility | n.d. | [173] | |
| • EGFR-mediated migration of neuroblastoma cells | no | [176] | ||
| • interaction between Caspase-8 and Calpain 2 involved in migration of neuroblastoma cells | no | [175] | ||
| Invasion | Caspase-3 | • invasive behavior of rat hepatoma cells | yes | [178] |
| Caspase-8 | • Caspase-8 cleaves ROCK in TRAIL- stimulated colon cancer cells | yes | [91] |
6. Concluding Remarks
Acknowledgements
References
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell. Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Medema, J.P.; Scaffidi, C.; Kischkel, F.C.; Shevchenko, A.; Mann, M.; Krammer, P.H.; Peter, M.E. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). Embo. J. 1997, 16, 2794–2804. [Google Scholar] [CrossRef]
- Sprick, M.R.; Rieser, E.; Stahl, H.; Grosse-Wilde, A.; Weigand, M.A.; Walczak, H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002, 21, 4520–4530. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Tinel, A.; Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef]
- Denecker, G.; Ovaere, P.; Vandenabeele, P.; Declercq, W. Caspase-14 reveals its secrets. J. Cell. Biol. 2008, 180, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; Los, M. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Park, W.S.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Somatic mutations of CASP3 gene in human cancers. Hum. Genet. 2004, 115, 112–115. [Google Scholar]
- Kim, M.S.; Park, S.W.; Kim, Y.R.; Lee, J.Y.; Lim, H.W.; Song, S.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of caspase genes in prostate carcinomas. APMIS 118, 308–312.
- Anwar, S.; Ambros, R.A.; Jennings, T.A.; Ross, J.S.; Beza, A.; Mian, B.; Nazeer, T. Expression of cysteine protease protein 32 in prostatic adenocarcinoma correlates with tumor grade. Arch. Pathol. Lab. Med. 2004, 128, 649–652. [Google Scholar]
- O'Donovan, N.; Crown, J.; Stunell, H.; Hill, A.D.; McDermott, E.; O'Higgins, N.; Duffy, M.J. Caspase 3 in breast cancer. Clin. Cancer Res. 2003, 9, 738–742. [Google Scholar]
- O'Neill, A.J.; Boran, S.A.; O'Keane, C.; Coffey, R.N.; Hegarty, N.J.; Hegarty, P.; Gaffney, E.F.; Fitzpatrick, J.M.; Watson, R.W. Caspase 3 expression in benign prostatic hyperplasia and prostate carcinoma. Prostate 2001, 47, 183–188. [Google Scholar] [CrossRef]
- Sohn, J.H.; Kim, D.H.; Choi, N.G.; Park, Y.E.; Ro, J.Y. Caspase-3/CPP32 immunoreactivity and its correlation with frequency of apoptotic bodies in human prostatic carcinomas and benign nodular hyperplasias. Histopathology 2000, 37, 555–560. [Google Scholar] [CrossRef]
- Vakkala, M.; Paakko, P.; Soini, Y. Expression of caspases 3, 6 and 8 is increased in parallel with apoptosis and histological aggressiveness of the breast lesion. Br. J. Cancer 1999, 81, 592–599. [Google Scholar] [CrossRef]
- Winter, R.N.; Kramer, A.; Borkowski, A.; Kyprianou, N. Loss of caspase-1 and caspase-3 protein expression in human prostate cancer. Cancer Res. 2001, 61, 1227–1232. [Google Scholar]
- Krajewski, S.; Krajewska, M.; Turner, B.C.; Pratt, C.; Howard, B.; Zapata, J.M.; Frenkel, V.; Robertson, S.; Ionov, Y.; Yamamoto, H.; Perucho, M.; Takayama, S.; Reed, J.C. Prognostic significance of apoptosis regulators in breast cancer. Endocr. Relat. Cancer 1999, 6, 29–40. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, H.S.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Park, J.Y.; Cho, Y.G.; Kim, C.J.; Park, Y.G.; Nam, S.W.; Jeong, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Inactivating mutations of CASPASE-7 gene in human cancers. Oncogene 2003, 22, 8048–8052. [Google Scholar] [CrossRef]
- Yoo, N.J.; Soung, Y.H.; Lee, S.H.; Kim, K.M.; Lee, S.H. Absence of CASP7 and CASP8 mutation in gastrointestinal lymphomas. Eur. J. Haematol. 2007, 79, 86–87. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, M.R.; Soung, Y.H.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of the CASP6 gene in colorectal and gastric carcinomas. Apmis 2006, 114, 646–650. [Google Scholar] [CrossRef]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Park, W.S.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Somatic mutation of pro-apoptosis caspase-6 gene is rare in breast and lung carcinomas. Pathology 2006, 38, 358–359. [Google Scholar] [CrossRef]
- Palmerini, F.; Devilard, E.; Jarry, A.; Birg, F.; Xerri, L. Caspase 7 downregulation as an immunohistochemical marker of colonic carcinoma. Hum. Pathol. 2001, 32, 461–467. [Google Scholar] [CrossRef]
- Yoo, N.J.; Kim, H.S.; Kim, S.Y.; Park, W.S.; Kim, S.H.; Lee, J.Y.; Lee, S.H. Stomach cancer highly expresses both initiator and effector caspases; an immunohistochemical study. Apmis 2002, 110, 825–832. [Google Scholar] [CrossRef]
- Yoo, N.J.; Lee, J.W.; Kim, Y.J.; Soung, Y.H.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Lee, J.Y.; Lee, S.H. Loss of caspase-2, -6 and -7 expression in gastric cancers. Apmis 2004, 112, 330–335. [Google Scholar]
- Estrov, Z.; Thall, P.F.; Talpaz, M.; Estey, E.H.; Kantarjian, H.M.; Andreeff, M.; Harris, D.; Van, Q.; Walterscheid, M.; Kornblau, S.M. Caspase 2 and caspase 3 protein levels as predictors of survival in acute myelogenous leukemia. Blood 1998, 92, 3090–3097. [Google Scholar]
- Devarajan, E.; Sahin, A.A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.H.; Tora, A.D.; Mehta, K. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Park, W.S.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of proapoptotic caspase-9 gene in common human carcinomas. Apmis 2006, 114, 292–297. [Google Scholar] [CrossRef]
- Abel, F.; Sjoberg, R.M.; Ejeskar, K.; Krona, C.; Martinsson, T. Analyses of apoptotic regulators CASP9 and DFFA at 1P36.2, reveal rare allele variants in human neuroblastoma tumours. Br. J. Cancer 2002, 86, 596–604. [Google Scholar] [CrossRef]
- Catchpoole, D.R.; Lock, R.B. The potential tumour suppressor role for caspase-9 (CASP9) in the childhood malignancy, neuroblastoma. Eur. J. Cancer 2001, 37, 2217–2221. [Google Scholar] [CrossRef]
- Yoo, N.J.; Lee, S.H.; Jeong, E.G.; Lee, S.H. Expression of phosphorylated caspase-9 in gastric carcinomas. Apmis 2007, 115, 354–359. [Google Scholar]
- Allan, L.A.; Clarke, P.R. Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. Febs. J. 2009, 276, 6063–6073. [Google Scholar] [CrossRef]
- Kumar, S.; White, D.L.; Takai, S.; Turczynowicz, S.; Juttner, C.A.; Hughes, T.P. Apoptosis regulatory gene NEDD2 maps to human chromosome segment 7q34-35, a region frequently affected in haematological neoplasms. Hum. Genet. 1995, 95, 641–644. [Google Scholar]
- Svingen, P.A.; Karp, J.E.; Krajewski, S.; Mesner, P.W., Jr.; Gore, S.D.; Burke, P.J.; Reed, J.C.; Lazebnik, Y.A.; Kaufmann, S.H. Evaluation of Apaf-1 and procaspases-2, -3, -7, -8, and -9 as potential prognostic markers in acute leukemia. Blood 2000, 96, 3922–3931. [Google Scholar]
- Kim, Y.R.; Kim, K.M.; Yoo, N.J.; Lee, S.H. Mutational analysis of CASP1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and 14 genes in gastrointestinal stromal tumors. Hum. Pathol. 2009, 40, 868–871. [Google Scholar] [CrossRef]
- Grenet, J.; Teitz, T.; Wei, T.; Valentine, V.; Kidd, V.J. Structure and chromosome localization of the human CASP8 gene. Gene 1999, 226, 225–232. [Google Scholar] [CrossRef]
- Oh, J.E.; Kim, M.S.; Ahn, C.H.; Kim, S.S.; Han, J.Y.; Lee, S.H.; Yoo, N.J. Mutational analysis of CASP10 gene in colon, breast, lung and hepatocellular carcinomas. Pathology 42, 73–76.
- Shin, M.S.; Kim, H.S.; Kang, C.S.; Park, W.S.; Kim, S.Y.; Lee, S.N.; Lee, J.H.; Park, J.Y.; Jang, J.J.; Kim, C.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Inactivating mutations of CASP10 gene in non-Hodgkin lymphomas. Blood 2002, 99, 4094–4099. [Google Scholar] [CrossRef]
- Soini, Y.; Paakko, P. Apoptosis and expression of caspases 3, 6 and 8 in malignant non-Hodgkin's lymphomas. Apmis 1999, 107, 1043–1050. [Google Scholar] [CrossRef]
- Kim, M.S.; Oh, J.E.; Min, C.K.; Lee, S.; Chung, N.G.; Yoo, N.J.; Lee, S.H. Mutational analysis of CASP10 gene in acute leukaemias and multiple myelomas. Pathology 2009, 41, 484–487. [Google Scholar] [CrossRef]
- Park, W.S.; Lee, J.H.; Shin, M.S.; Park, J.Y.; Kim, H.S.; Lee, J.H.; Kim, Y.S.; Lee, S.N.; Xiao, W.; Park, C.H.; Lee, S.H.; Yoo, N.J.; Lee, J.Y. Inactivating mutations of the caspase-10 gene in gastric cancer. Oncogene 2002, 21, 2919–2925. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Sung, Y.J.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Caspase-8 gene is frequently inactivated by the frameshift somatic mutation 1225_1226delTG in hepatocellular carcinomas. Oncogene 2005, 24, 141–147. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, J.W.; Soung, Y.H.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Park, J.Y.; Cho, Y.G.; Kim, C.J.; Jeong, S.W.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology 2003, 125, 708–715. [Google Scholar] [CrossRef]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Jang, J.; Park, Y.G.; Park, W.S.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 2005, 65, 815–821. [Google Scholar]
- Shivapurkar, N.; Toyooka, S.; Eby, M.T.; Huang, C.X.; Sathyanarayana, U.G.; Cunningham, H.T.; Reddy, J.L.; Brambilla, E.; Takahashi, T.; Minna, J.D.; Chaudhary, P.M.; Gazdar, A.F. Differential inactivation of caspase-8 in lung cancers. Cancer Biol. Ther. 2002, 1, 65–69. [Google Scholar] [CrossRef]
- Takita, J.; Yang, H.W.; Bessho, F.; Hanada, R.; Yamamoto, K.; Kidd, V.; Teitz, T.; Wei, T.; Hayashi, Y. Absent or reduced expression of the caspase 8 gene occurs frequently in neuroblastoma, but not commonly in Ewing sarcoma or rhabdomyosarcoma. Med. Pediatr. Oncol. 2000, 35, 541–543. [Google Scholar] [CrossRef]
- Zuzak, T.J.; Steinhoff, D.F.; Sutton, L.N.; Phillips, P.C.; Eggert, A.; Grotzer, M.A. Loss of caspase-8 mRNA expression is common in childhood primitive neuroectodermal brain tumour/medulloblastoma. Eur. J. Cancer 2002, 38, 83–91. [Google Scholar] [CrossRef]
- Mazumder, S.; Almasan, A. Is caspase-8 a neuroendocrine lung tumor suppressor? Cancer Biol. Ther. 2002, 1, 70–71. [Google Scholar]
- ç, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [CrossRef]
- Hopkins-Donaldson, S.; Ziegler, A.; Kurtz, S.; Bigosch, C.; Kandioler, D.; Ludwig, C.; Zangemeister-Wittke, U.; Stahel, R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003, 10, 356–364. [Google Scholar] [CrossRef]
- Harada, K.; Toyooka, S.; Shivapurkar, N.; Maitra, A.; Reddy, J.L.; Matta, H.; Miyajima, K.; Timmons, C.F.; Tomlinson, G.E.; Mastrangelo, D.; Hay, R.J.; Chaudhary, P.M.; Gazdar, A.F. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002, 62, 5897–5901. [Google Scholar]
- Pingoud-Meier, C.; Lang, D.; Janss, A.J.; Rorke, L.B.; Phillips, P.C.; Shalaby, T.; Grotzer, M.A. Loss of caspase-8 protein expression correlates with unfavorable survival outcome in childhood medulloblastoma. Clin. Cancer Res. 2003, 9, 6401–6409. [Google Scholar]
- Fulda, S.; Poremba, C.; Berwanger, B.; Hacker, S.; Eilers, M.; Christiansen, H.; Hero, B.; Debatin, K.M. Loss of caspase-8 expression does not correlate with MYCN amplification, aggressive disease, or prognosis in neuroblastoma. Cancer Res. 2006, 66, 10016–10023. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pedini, F.; Mollinari, C.; Condorelli, G.; Bonci, D.; Bez, A.; Colombo, A.; Parati, E.; Peschle, C.; De Maria, R. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J. Exp. Med. 2004, 200, 1257–1266. [Google Scholar] [CrossRef]
- Stupack, D.G.; Teitz, T.; Potter, M.D.; Mikolon, D.; Houghton, P.J.; Kidd, V.J.; Lahti, J.M.; Cheresh, D.A. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature 2006, 439, 95–99. [Google Scholar] [CrossRef]
- Zheng, T.S.; Flavell, R.A. Divinations and surprises: Genetic analysis of caspase function in mice. Exp. Cell. Res. 2000, 256, 67–73. [Google Scholar] [CrossRef]
- Kang, T.B.; Ben-Moshe, T.; Varfolomeev, E.E.; Pewzner-Jung, Y.; Yogev, N.; Jurewicz, A.; Waisman, A.; Brenner, O.; Haffner, R.; Gustafsson, E.; Ramakrishnan, P.; Lapidot, T.; Wallach, D. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 2004, 173, 2976–2984. [Google Scholar]
- Salmena, L.; Lemmers, B.; Hakem, A.; Matysiak-Zablocki, E.; Murakami, K.; Au, P.Y.; Berry, D.M.; Tamblyn, L.; Shehabeldin, A.; Migon, E.; Wakeham, A.; Bouchard, D.; Yeh, W.C.; McGlade, J.C.; Ohashi, P.S.; Hakem, R. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003, 17, 883–895. [Google Scholar] [CrossRef]
- Scott, C.L.; Schuler, M.; Marsden, V.S.; Egle, A.; Pellegrini, M.; Nesic, D.; Gerondakis, S.; Nutt, S.L.; Green, D.R.; Strasser, A. Apaf-1 and caspase-9 do not act as tumor suppressors in myc-induced lymphomagenesis or mouse embryo fibroblast transformation. J. Cel.l Biol. 2004, 164, 89–96. [Google Scholar] [CrossRef]
- Soengas, M.S.; Alarcon, R.M.; Yoshida, H.; Giaccia, A.J.; Hakem, R.; Mak, T.W.; Lowe, S.W. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 1999, 284, 156–159. [Google Scholar] [CrossRef]
- Krelin, Y.; Zhang, L.; Kang, T.B.; Appel, E.; Kovalenko, A.; Wallach, D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008, 15, 1350–1355. [Google Scholar] [CrossRef]
- Ho, L.H.; Taylor, R.; Dorstyn, L.; Cakouros, D.; Bouillet, P.; Kumar, S. A tumor suppressor function for caspase-2. Proc. Natl. Acad. Sci. USA 2009, 106, 5336–5341. [Google Scholar] [CrossRef]
- Reddy, J.P.; Peddibhotla, S.; Bu, W.; Zhao, J.; Haricharan, S.; Du, Y.C.; Podsypanina, K.; Rosen, J.M.; Donehower, L.A.; Li, Y. Defining the ATM-mediated barrier to tumorigenesis in somatic mammary cells following ErbB2 activation. Proc. Natl. Acad. Sci. USA 107, 3728–3733.
- Radziszewska, A.; Schroer, S.A.; Choi, D.; Tajmir, P.; Radulovich, N.; Ho, J.C.; Wang, L.; Liadis, N.; Hakem, R.; Tsao, M.S.; Penn, L.Z.; Evan, G.I.; Woo, M. Absence of caspase-3 protects pancreatic {beta}-cells from c-Myc-induced apoptosis without leading to tumor formation. J. Biol. Chem. 2009, 284, 10947–10956. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef]
- Hahn, W.C.; Weinberg, R.A. Rules for making human tumor cells. N. Engl. J. Med. 2002, 347, 1593–1603. [Google Scholar] [CrossRef]
- Rangarajan, A.; Hong, S.J.; Gifford, A.; Weinberg, R.A. Species- and cell type-specific requirements for cellular transformation. Cancer Cell 2004, 6, 171–183. [Google Scholar] [CrossRef]
- Evan, G.; Littlewood, T. A matter of life and cell death. Science 1998, 281, 1317–1322. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Kroemer, G. Apoptosis and genomic instability. Nat. Rev. Mol. Cell. Biol. 2004, 5, 752–762. [Google Scholar] [CrossRef]
- Clarke, A.R.; Purdie, C.A.; Harrison, D.J.; Morris, R.G.; Bird, C.C.; Hooper, M.L.; Wyllie, A.H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 1993, 362, 849–852. [Google Scholar] [CrossRef]
- Pajares, M.J.; Ezponda, T.; Catena, R.; Calvo, A.; Pio, R.; Montuenga, L.M. Alternative splicing: An emerging topic in molecular and clinical oncology. Lancet Oncol. 2007, 8, 349–357. [Google Scholar] [CrossRef]
- Droin, N.; Beauchemin, M.; Solary, E.; Bertrand, R. Identification of a caspase-2 isoform that behaves as an endogenous inhibitor of the caspase cascade. Cancer Res. 2000, 60, 7039–7047. [Google Scholar]
- Seol, D.W.; Billiar, T.R. A caspase-9 variant missing the catalytic site is an endogenous inhibitor of apoptosis. J. Biol. Chem. 1999, 274, 2072–2076. [Google Scholar] [CrossRef]
- Wang, H.; Wang, P.; Sun, X.; Luo, Y.; Wang, X.; Ma, D.; Wu, J. Cloning and characterization of a novel caspase-10 isoform that activates NF-kappa B activity. Biochim. Biophys. Acta. 2007, 1770, 1528–1537. [Google Scholar]
- Wang, L.; Miura, M.; Bergeron, L.; Zhu, H.; Yuan, J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 1994, 78, 739–750. [Google Scholar] [CrossRef]
- Huang, Y.; Shin, N.H.; Sun, Y.; Wang, K.K. Molecular cloning and characterization of a novel caspase-3 variant that attenuates apoptosis induced by proteasome inhibition. Biochem. Biophys. Res. Commun. 2001, 283, 762–769. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R.; Oudin, C.; Riedinger, J.M.; Bonnetain, F.; Lizard-Nacol, S. Overexpression of caspase-3s splice variant in locally advanced breast carcinoma is associated with poor response to neoadjuvant chemotherapy. Clin. Cancer Res. 2006, 12, 5794–5800. [Google Scholar] [CrossRef]
- Horiuchi, T.; Himeji, D.; Tsukamoto, H.; Harashima, S.; Hashimura, C.; Hayashi, K. Dominant expression of a novel splice variant of caspase-8 in human peripheral blood lymphocytes. Biochem. Biophys. Res. Commun. 2000, 272, 877–881. [Google Scholar] [CrossRef]
- Himeji, D.; Horiuchi, T.; Tsukamoto, H.; Hayashi, K.; Watanabe, T.; Harada, M. Characterization of caspase-8L: A novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood 2002, 99, 4070–4078. [Google Scholar] [CrossRef]
- Mohr, A.; Zwacka, R.M.; Jarmy, G.; Buneker, C.; Schrezenmeier, H.; Dohner, K.; Beltinger, C.; Wiesneth, M.; Debatin, K.M.; Stahnke, K. Caspase-8L expression protects CD34+ hematopoietic progenitor cells and leukemic cells from CD95-mediated apoptosis. Oncogene 2005, 24, 2421–2429. [Google Scholar] [CrossRef]
- Miller, M.A.; Karacay, B.; Zhu, X.; O'Dorisio, M.S.; Sandler, A.D. Caspase 8L, a novel inhibitory isoform of caspase 8, is associated with undifferentiated neuroblastoma. Apoptosis 2006, 11, 15–24. [Google Scholar] [CrossRef]
- Yu, J.W.; Shi, Y. FLIP and the death effector domain family. Oncogene 2008, 27, 6216–6227. [Google Scholar] [CrossRef]
- Du, X.; Bao, G.; He, X.; Zhao, H.; Yu, F.; Qiao, Q.; Lu, J.; Ma, Q. Expression and biological significance of c-FLIP in human hepatocellular carcinomas. J. Exp. Clin. Cancer Res. 2009, 28, 24. [Google Scholar] [CrossRef]
- Korkolopoulou, P.; Saetta, A.A.; Levidou, G.; Gigelou, F.; Lazaris, A.; Thymara, I.; Scliri, M.; Bousboukea, K.; Michalopoulos, N.V.; Apostolikas, N.; Konstantinidou, A.; Tzivras, M.; Patsouris, E. c-FLIP expression in colorectal carcinomas: Association with Fas/FasL expression and prognostic implications. Histopathology 2007, 51, 150–156. [Google Scholar] [CrossRef]
- Ryang, D.Y.; Joo, Y.E.; Chung, K.M.; Lim, S.R.; Jeong, H.K.; Kim, H.I.; Lee, W.S.; Park, C.H.; Kim, H.S.; Choi, S.K.; Rew, J.S.; Lee, J.H.; Park, C.S. Expression of c-FLIP in gastric cancer and its relation to tumor cell proliferation and apoptosis. Korean J. Intern. Med. 2007, 22, 263–269. [Google Scholar] [CrossRef]
- van Houdt, I.S.; Muris, J.J.; Hesselink, A.T.; Kramer, D.; Cillessen, S.A.; Moesbergen, L.M.; Vos, W.; Hooijberg, E.; Meijer, C.J.; Kummer, J.A.; Oudejans, J.J. Expression of c-FLIP is primarily detected in diffuse large B-cell lymphoma and Hodgkin's lymphoma and correlates with lack of caspase 8 activation. Histopathology 2007, 51, 778–784. [Google Scholar] [CrossRef]
- Kim, S.Y.; Song, S.Y.; Kim, M.S.; Lee, J.Y.; Lee, H.M.; Choi, H.Y.; Yoo, N.J.; Lee, S.H. Immunohistochemical analysis of Fas and FLIP in prostate cancers. Apmis 2009, 117, 28–33. [Google Scholar] [CrossRef]
- Coffey, F.; Manser, T. Expression of cellular FLIP by B cells is required for their participation in an immune response. J. Immunol. 184, 4871–4879. [CrossRef]
- Zhang, H.; Rosenberg, S.; Coffey, F.J.; He, Y.W.; Manser, T.; Hardy, R.R.; Zhang, J. A role for cFLIP in B cell proliferation and stress MAPK regulation. J. Immunol. 2009, 182, 207–215. [Google Scholar]
- Ehrenschwender, M.; Siegmund, D.; Wicovsky, A.; Kracht, M.; Dittrich-Breiholz, O.; Spindler, V.; Waschke, J.; Kalthoff, H.; Trauzold, A.; Wajant, H. Mutant PIK3CA licenses TRAIL and CD95L to induce non-apoptotic caspase-8-mediated ROCK activation. Cell Death Differ. 2010, 17, 1435–1447. [Google Scholar] [CrossRef]
- El-Gazzar, A.; Wittinger, M.; Perco, P.; Anees, M.; Horvat, R.; Mikulits, W.; Grunt, T.W.; Mayer, B.; Krainer, M. The role of c-FLIP(L) in ovarian cancer: Chaperoning tumor cells from immunosurveillance and increasing their invasive potential. Gynecol. Oncol. 117, 451–459.
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef]
- Nachmias, B.; Ashhab, Y.; Ben-Yehuda, D. The inhibitor of apoptosis protein family (IAPs): An emerging therapeutic target in cancer. Semin. Cancer Biol. 2004, 14, 231–243. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Leo, E.; Stennicke, H.R.; Welsh, K.; Salvesen, G.S.; Reed, J.C. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. Embo. J. 1999, 18, 5242–5251. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nakabayashi, Y.; Takahashi, R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 8662–8667. [Google Scholar] [CrossRef]
- Burstein, D.E.; Idrees, M.T.; Li, G.; Wu, M.; Kalir, T. Immunohistochemical detection of the X-linked inhibitor of apoptosis protein (XIAP) in cervical squamous intraepithelial neoplasia and squamous carcinoma. Ann. Diagn. Pathol. 2008, 12, 85–89. [Google Scholar] [CrossRef]
- De Oliveira Lima, F.; De Oliveira Costa, H.; Barrezueta, L.F.; Fujiyama Oshima, C.T.; Silva, J.A., Jr.; Gomes, T.S.; Pinheiro, N., Jr.; Neto, R.A.; Franco, M. Immunoexpression of inhibitors of apoptosis proteins and their antagonist SMAC/DIABLO in colorectal carcinoma: Correlation with apoptotic index, cellular proliferation and prognosis. Oncol. Rep. 2009, 22, 295–303. [Google Scholar]
- Emanuel, P.O.; Phelps, R.G.; Mudgil, A.; Shafir, M.; Burstein, D.E. Immunohistochemical detection of XIAP in melanoma. J. Cutan. Pathol. 2008, 35, 292–297. [Google Scholar] [CrossRef]
- Ferreira, C.G.; van der Valk, P.; Span, S.W.; Ludwig, I.; Smit, E.F.; Kruyt, F.A.; Pinedo, H.M.; van Tinteren, H.; Giaccone, G. Expression of X-linked inhibitor of apoptosis as a novel prognostic marker in radically resected non-small cell lung cancer patients. Clin. Cancer Res. 2001, 7, 2468–2474. [Google Scholar]
- Jaffer, S.; Orta, L.; Sunkara, S.; Sabo, E.; Burstein, D.E. Immunohistochemical detection of antiapoptotic protein X-linked inhibitor of apoptosis in mammary carcinoma. Hum. Pathol. 2007, 38, 864–870. [Google Scholar] [CrossRef]
- Krajewska, M.; Krajewski, S.; Banares, S.; Huang, X.; Turner, B.; Bubendorf, L.; Kallioniemi, O.P.; Shabaik, A.; Vitiello, A.; Peehl, D.; Gao, G.J.; Reed, J.C. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin. Cancer Res. 2003, 9, 4914–4925. [Google Scholar]
- Nagi, C.; Xiao, G.Q.; Li, G.; Genden, E.; Burstein, D.E. Immunohistochemical detection of X-linked inhibitor of apoptosis in head and neck squamous cell carcinoma. Ann. Diagn. Pathol. 2007, 11, 402–406. [Google Scholar] [CrossRef]
- Xiao, G.Q.; Unger, P.D.; Burstein, D.E. Immunohistochemical detection of X-linked inhibitor of apoptosis (XIAP) in neoplastic and other thyroid disorders. Ann. Diagn. Pathol. 2007, 11, 235–240. [Google Scholar] [CrossRef]
- Akyurek, N.; Ren, Y.; Rassidakis, G.Z.; Schlette, E.J.; Medeiros, L.J. Expression of inhibitor of apoptosis proteins in B-cell non-Hodgkin and Hodgkin lymphomas. Cancer 2006, 107, 1844–1851. [Google Scholar] [CrossRef]
- Parton, M.; Krajewski, S.; Smith, I.; Krajewska, M.; Archer, C.; Naito, M.; Ahern, R.; Reed, J.; Dowsett, M. Coordinate expression of apoptosis-associated proteins in human breast cancer before and during chemotherapy. Clin. Cancer Res. 2002, 8, 2100–2108. [Google Scholar]
- Liston, P.; Fong, W.G.; Kelly, N.L.; Toji, S.; Miyazaki, T.; Conte, D.; Tamai, K.; Craig, C.G.; McBurney, M.W.; Korneluk, R.G. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat. Cell. Biol. 2001, 3, 128–133. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Yoo, N.J.; Kim, H.S.; Kim, S.Y.; Park, W.S.; Park, C.H.; Jeon, H.M.; Jung, E.S.; Lee, J.Y.; Lee, S.H. Immunohistochemical analysis of Smac/DIABLO expression in human carcinomas and sarcomas. APMIS 2003, 111, 382–388. [Google Scholar] [CrossRef]
- Byun, D.S.; Cho, K.; Ryu, B.K.; Lee, M.G.; Kang, M.J.; Kim, H.R.; Chi, S.G. Hypermethylation of XIAP-associated factor 1, a putative tumor suppressor gene from the 17p13.2 locus, in human gastric adenocarcinomas. Cancer Res. 2003, 63, 7068–7075. [Google Scholar]
- Huang, J.; Yao, W.Y.; Zhu, Q.; Tu, S.P.; Yuan, F.; Wang, H.F.; Zhang, Y.P.; Yuan, Y.Z. XAF1 as a prognostic biomarker and therapeutic target in pancreatic cancer. Cancer Sci. 101, 559–567.
- Kempkensteffen, C.; Fritzsche, F.R.; Johannsen, M.; Weikert, S.; Hinz, S.; Dietel, M.; Riener, M.O.; Moch, H.; Jung, K.; Krause, H.; Miller, K.; Kristiansen, G. Down-regulation of the pro-apoptotic XIAP associated factor-1 (XAF1) during progression of clear-cell renal cancer. BMC Cancer 2009, 9, 276. [Google Scholar] [CrossRef]
- Lee, M.G.; Huh, J.S.; Chung, S.K.; Lee, J.H.; Byun, D.S.; Ryu, B.K.; Kang, M.J.; Chae, K.S.; Lee, S.J.; Lee, C.H.; Kim, J.I.; Chang, S.G.; Chi, S.G. Promoter CpG hypermethylation and downregulation of XAF1 expression in human urogenital malignancies: Implication for attenuated p53 response to apoptotic stresses. Oncogene 2006, 25, 5807–5822. [Google Scholar] [CrossRef]
- Ma, T.L.; Ni, P.H.; Zhong, J.; Tan, J.H.; Qiao, M.M.; Jiang, S.H. Low expression of XIAP-associated factor 1 in human colorectal cancers. Chin. J. Dig. Dis. 2005, 6, 10–14. [Google Scholar] [CrossRef]
- Sakemi, R.; Yano, H.; Ogasawara, S.; Akiba, J.; Nakashima, O.; Fukahori, S.; Sata, M.; Kojiro, M. X-linked inhibitor of apoptosis (XIAP) and XIAP-associated factor-1 expressions and their relationship to apoptosis in human hepatocellular carcinoma and non-cancerous liver tissues. Oncol. Rep. 2007, 18, 65–70. [Google Scholar]
- Shibata, T.; Mahotka, C.; Wethkamp, N.; Heikaus, S.; Gabbert, H.E.; Ramp, U. Disturbed expression of the apoptosis regulators XIAP, XAF1, and Smac/DIABLO in gastric adenocarcinomas. Diagn. Mol. Pathol. 2007, 16, 1–8. [Google Scholar] [CrossRef]
- Galban, S.; Duckett, C.S. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 17, 54–60. [CrossRef]
- Van Themsche, C.; Leblanc, V.; Parent, S.; Asselin, E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J. Biol. Chem. 2009, 284, 20462–20466. [Google Scholar] [CrossRef]
- Schile, A.J.; Garcia-Fernandez, M.; Steller, H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008, 22, 2256–2266. [Google Scholar] [CrossRef]
- Hwang, C.; Oetjen, K.A.; Kosoff, D.; Wojno, K.J.; Albertelli, M.A.; Dunn, R.L.; Robins, D.M.; Cooney, K.A.; Duckett, C.S. X-linked inhibitor of apoptosis deficiency in the TRAMP mouse prostate cancer model. Cell Death Differ. 2008, 15, 831–840. [Google Scholar] [CrossRef]
- Mehrotra, S.; Languino, L.R.; Raskett, C.M.; Mercurio, A.M.; Dohi, T.; Altieri, D.C. IAP regulation of metastasis. Cancer Cell. 2010, 17, 53–64. [Google Scholar] [CrossRef]
- Ozoren, N.; El-Deiry, W.S. Cell surface Death Receptor signaling in normal and cancer cells. Semin. Cancer Biol. 2003, 13, 135–147. [Google Scholar] [CrossRef]
- Buneker, C.; Mohr, A.; Zwacka, R.M. The TRAIL-receptor-1: TRAIL-receptor-3 and -4 ratio is a predictor for TRAIL sensitivity of cancer cells. Oncol. Rep. 2009, 21, 1289–1295. [Google Scholar]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.; Eder, A.; Mao, M.; Bast, R.C., Jr.; Boyd, D.; Mills, G.B. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 1999, 18, 6635–6640. [Google Scholar] [CrossRef]
- Vousden, K.H. p53: Death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; Orntoft, T.; Lukas, J.; Bartek, J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Nuciforo, P.G.; Luise, C.; Capra, M.; Pelosi, G.; d'Adda di Fagagna, F. Complex engagement of DNA damage response pathways in human cancer and in lung tumor progression. Carcinogenesis 2007, 28, 2082–2088. [Google Scholar] [CrossRef]
- Jager, R.; Herzer, U.; Schenkel, J.; Weiher, H. Overexpression of Bcl-2 inhibits alveolar cell apoptosis during involution and accelerates c-myc-induced tumorigenesis of the mammary gland in transgenic mice. Oncogene 1997, 15, 1787–1795. [Google Scholar]
- Naik, P.; Karrim, J.; Hanahan, D. The rise and fall of apoptosis during multistage tumorigenesis: Down-modulation contributes to tumor progression from angiogenic progenitors. Genes Dev. 1996, 10, 2105–2116. [Google Scholar] [CrossRef]
- Broker, L.E.; Kruyt, F.A.; Giaccone, G. Cell death independent of caspases: A review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef]
- Carter, B.Z.; Kornblau, S.M.; Tsao, T.; Wang, R.Y.; Schober, W.D.; Milella, M.; Sung, H.G.; Reed, J.C.; Andreeff, M. Caspase-independent cell death in AML: Caspase inhibition in vitro with pan-caspase inhibitors or in vivo by XIAP or Survivin does not affect cell survival or prognosis. Blood 2003, 102, 4179–4186. [Google Scholar] [CrossRef]
- Colell, A.; Ricci, J.E.; Tait, S.; Milasta, S.; Maurer, U.; Bouchier-Hayes, L.; Fitzgerald, P.; Guio-Carrion, A.; Waterhouse, N.J.; Li, C.W.; Mari, B.; Barbry, P.; Newmeyer, D.D.; Beere, H.M.; Green, D.R. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 2007, 129, 983–997. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Caspase-independent cell death: Leaving the set without the final cut. Oncogene 2008, 27, 6452–6461. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kroemer, G. Necroptosis: A specialized pathway of programmed necrosis. Cell 2008, 135, 1161–1163. [Google Scholar] [CrossRef]
- Liao, D.J.; Dickson, R.B. Cell death in MMTV-c-myc transgenic mouse mammary tumors may not be typical apoptosis. Lab. Invest. 2003, 83, 1437–1449. [Google Scholar] [CrossRef]
- Bai, M.; Agnantis, N.J.; Kamina, S.; Demou, A.; Zagorianakou, P.; Katsaraki, A.; Kanavaros, P. In vivo cell kinetics in breast carcinogenesis. Breast Cancer Res. 2001, 3, 276–283. [Google Scholar] [CrossRef]
- Gandhi, A.; Holland, P.A.; Knox, W.F.; Potten, C.S.; Bundred, N.J. Evidence of significant apoptosis in poorly differentiated ductal carcinoma in situ of the breast. Br. J. Cancer 1998, 78, 788–794. [Google Scholar] [CrossRef]
- Ioffe, O.B.; Papadimitriou, J.C.; Drachenberg, C.B. Correlation of proliferation indices, apoptosis, and related oncogene expression (bcl-2 and c-erbB-2) and p53 in proliferative, hyperplastic, and malignant endometrium. Hum. Pathol. 1998, 29, 1150–1159. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Dinjens, W.N.; Bosman, F.T. Proliferation and apoptosis in proliferative lesions of the colon and rectum. Virchows Arch. 1997, 431, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Lipponen, P. Apoptosis in breast cancer: Relationship with other pathological parameters. Endocr. Relat. Cancer 1999, 6, 13–16. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Roddey, G.; McDonnell, T.J.; Shen, Y.; Cleary, K.R.; Stephens, L.C. Increased apoptosis accompanies neoplastic development in the human colorectum. Clin. Cancer Res. 1996, 2, 1999–2006. [Google Scholar]
- Sitorus, R.S.; Gumay, S.; van der Valk, P. The apoptotic paradox in retinoblastoma. Ann. NY Acad. Sci. 2009, 1171, 77–86. [Google Scholar]
- Staunton, M.J.; Gaffney, E.F. Tumor type is a determinant of susceptibility to apoptosis. Am. J. Clin. Pathol. 1995, 103, 300–307. [Google Scholar]
- Targa, A.C.; Cesar, A.C.; Cury, P.M.; Silva, A.E. Apoptosis in different gastric lesions and gastric cancer: Relationship with Helicobacter pylori, overexpression of p53 and aneuploidy. Genet. Mol. Res. 2007, 6, 554–565. [Google Scholar]
- Tormanen, U.; Eerola, A.K.; Rainio, P.; Vahakangas, K.; Soini, Y.; Sormunen, R.; Bloigu, R.; Lehto, V.P.; Paakko, P. Enhanced apoptosis predicts shortened survival in non-small cell lung carcinoma. Cancer Res. 1995, 55, 5595–5602. [Google Scholar]
- Watanabe, S.; Miyata, Y.; Kanda, S.; Iwata, T.; Hayashi, T.; Kanetake, H.; Sakai, H. Expression of X-linked inhibitor of apoptosis protein in human prostate cancer specimens with and without neo-adjuvant hormonal therapy. J. Cancer Res. Clin. Oncol. 136, 787–793.
- Soini, Y.; Paakko, P.; Lehto, V.P. Histopathological evaluation of apoptosis in cancer. Am. J. Pathol. 1998, 153, 1041–1053. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Potten, C.S. What is an apoptotic index measuring? A commentary. Br. J. Cancer 1996, 74, 1743–1748. [Google Scholar] [CrossRef]
- Rubio, C.A. Apoptosis in human tumours. Br. J. Cancer 2002, 86, 1661. [Google Scholar] [CrossRef]
- Grekou, A.N.; Toliou, T.; Stravoravdi, P.; Patakiouta, F.; Tsoukalas, T.; Pinakidis, M.; Keramidas, G. Correlation of apoptosis with the distribution and composition of lymphocytic infiltrate in human breast carcinomas. Anticancer Res. 1996, 16, 3991–3995. [Google Scholar]
- Erwig, L.P.; Henson, P.M. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008, 15, 243–250. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Wodarz, D.; Komarova, N. Can loss of apoptosis protect against cancer? Trends Genet. 2007, 23, 232–237. [Google Scholar] [CrossRef]
- Enderling, H.; Anderson, A.R.; Chaplain, M.A.; Beheshti, A.; Hlatky, L.; Hahnfeldt, P. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res. 2009, 69, 8814–8821. [Google Scholar] [CrossRef]
- Gurova, K.V.; Gudkov, A.V. Paradoxical role of apoptosis in tumor progression. J. Cell. Biochem. 2003, 88, 128–137. [Google Scholar] [CrossRef]
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; Xu, Y.; Autenrieth, I.B.; Schulze-Osthoff, K.; Belka, C.; Stuhler, G.; Wesselborg, S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef]
- Fan, Y.; Bergmann, A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev. Cell 2008, 14, 399–410. [Google Scholar] [CrossRef]
- Chera, S.; Ghila, L.; Dobretz, K.; Wenger, Y.; Bauer, C.; Buzgariu, W.; Martinou, J.C.; Galliot, B. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev. Cell 2009, 17, 279–289. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef]
- Fujita, J.; Crane, A.M.; Souza, M.K.; Dejosez, M.; Kyba, M.; Flavell, R.A.; Thomson, J.A.; Zwaka, T.P. Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2008, 2, 595–601. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Jacobson, M.D.; Raff, M.C. A role for caspases in lens fiber differentiation. J. Cell. Biol. 1998, 140, 153–158. [Google Scholar] [CrossRef]
- Murray, T.V.; McMahon, J.M.; Howley, B.A.; Stanley, A.; Ritter, T.; Mohr, A.; Zwacka, R.; Fearnhead, H.O. A non-apoptotic role for caspase-9 in muscle differentiation. J. Cell. Sci. 2008, 121, 3786–3793. [Google Scholar] [CrossRef]
- Oomman, S.; Strahlendorf, H.; Dertien, J.; Strahlendorf, J. Bergmann glia utilize active caspase-3 for differentiation. Brain Res. 2006, 1078, 19–34. [Google Scholar] [CrossRef]
- Sordet, O.; Rebe, C.; Plenchette, S.; Zermati, Y.; Hermine, O.; Vainchenker, W.; Garrido, C.; Solary, E.; Dubrez-Daloz, L. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 2002, 100, 4446–4453. [Google Scholar] [CrossRef]
- Hsu, S.L.; Yu, C.T.; Yin, S.C.; Tang, M.J.; Tien, A.C.; Wu, Y.M.; Huang, C.Y. Caspase 3, periodically expressed and activated at G2/M transition, is required for nocodazole-induced mitotic checkpoint. Apoptosis 2006, 11, 765–771. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yamauchi, L.; Hunter, T.; Kikkawa, U.; Kamada, S. Possible involvement of caspase-7 in cell cycle progression at mitosis. Genes Cells 2008, 13, 609–621. [Google Scholar] [CrossRef]
- Olson, N.E.; Graves, J.D.; Shu, G.L.; Ryan, E.J.; Clark, E.A. Caspase activity is required for stimulated B lymphocytes to enter the cell cycle. J. Immunol. 2003, 170, 6065–6072. [Google Scholar]
- Pellegrini, M.; Bath, S.; Marsden, V.S.; Huang, D.C.; Metcalf, D.; Harris, A.W.; Strasser, A. FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells. Blood 2005, 106, 1581–1589. [Google Scholar] [CrossRef]
- Helfer, B.; Boswell, B.C.; Finlay, D.; Cipres, A.; Vuori, K.; Bong Kang, T.; Wallach, D.; Dorfleutner, A.; Lahti, J.M.; Flynn, D.C.; Frisch, S.M. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 2006, 66, 4273–4278. [Google Scholar] [CrossRef]
- Barbero, S.; Barila, D.; Mielgo, A.; Stagni, V.; Clair, K.; Stupack, D. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. J Biol Chem 2008, 283, 13031–13034. [Google Scholar] [CrossRef]
- Barbero, S.; Mielgo, A.; Torres, V.; Teitz, T.; Shields, D.J.; Mikolon, D.; Bogyo, M.; Barila, D.; Lahti, J.M.; Schlaepfer, D.; Stupack, D.G. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009, 69, 3755–3763. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.; Howes, A.; Vuori, K. Critical role for caspase-8 in epidermal growth factor signaling. Cancer Res. 2009, 69, 5023–5029. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, D.; Zhao, Z.; Xiao, Y.; Sengupta, S.; Xiao, Y.; Zhang, R.; Lauber, K.; Wesselborg, S.; Feng, L.; Rose, T.M.; Shen, Y.; Zhang, J.; Prestwich, G.; Xu, Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J. Biol. Chem. 2006, 281, 29357–29368. [Google Scholar] [CrossRef]
- Mukai, M.; Kusama, T.; Hamanaka, Y.; Koga, T.; Endo, H.; Tatsuta, M.; Inoue, M. Cross talk between apoptosis and invasion signaling in cancer cells through caspase-3 activation. Cancer Res. 2005, 65, 9121–9125. [Google Scholar] [CrossRef]
- Labi, V.; Erlacher, M.; Krumschnabel, G.; Manzl, C.; Tzankov, A.; Pinon, J.; Egle, A.; Villunger, A. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 2010, 24, 1602–1607. [Google Scholar] [CrossRef]
- Michalak, E.M.; Vandenberg, C.J.; Delbridge, A.R.; Wu, L.; Scott, C.L.; Adams, J.M.; Strasser, A. Apoptosis-promoted tumorigenesis: Gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 2010, 24, 1608–1613. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jäger, R.; Zwacka, R.M. The Enigmatic Roles of Caspases in Tumor Development. Cancers 2010, 2, 1952-1979. https://doi.org/10.3390/cancers2041952
Jäger R, Zwacka RM. The Enigmatic Roles of Caspases in Tumor Development. Cancers. 2010; 2(4):1952-1979. https://doi.org/10.3390/cancers2041952
Chicago/Turabian StyleJäger, Richard, and Ralf M. Zwacka. 2010. "The Enigmatic Roles of Caspases in Tumor Development" Cancers 2, no. 4: 1952-1979. https://doi.org/10.3390/cancers2041952