Modulation of Radiation-Induced Genetic Damage by HCMV in Peripheral Blood Lymphocytes from a Brain Tumor Case-Control Study

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Culture Preparation for Genomic Instability Measurements
2.3. Cultures Infected with HCMV
2.4. Cultures Treated with Radiation
2.5. Cultures Infected with HCMV and Treated with Radiation
2.6. CBMN-Cyt Assay Evaluation
2.7. Statistical Analysis
3. Results and Discussion
3.1. Subject Characteristics
3.2. Baseline Chromosome Damage Frequencies
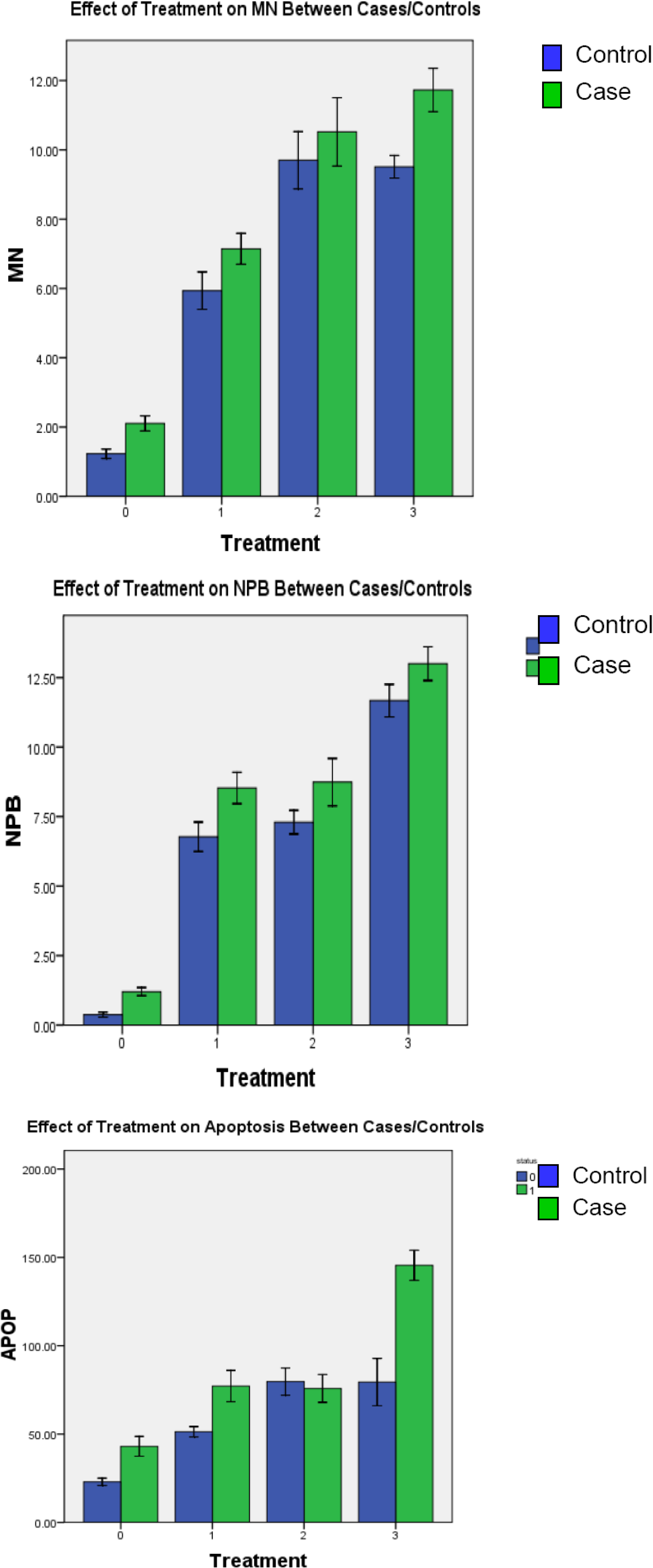
3.3. Induced Chromosome Damage Frequencies
3.3.1. Radiation-Induced Chromosome Damage
3.3.2. HCMV-induced Chromosome Damage
3.3.3. HCMV + Radiation–induced Chromosome Damage

{kind=link}
| Controls Mean ± SEM | Cases Mean ± SEM | |
| Micronucleus | ||
| Baselineb | 1.23 ± 0.07 | 2.10 ± 0.11 |
| Radiation | 5.94 ± 0.27 | 7.14 ± 0.22 |
| Virus | 9.70 ± 0.41 | 10.52 ± 0.49 |
| Virus + Radiation | 9.51 ± 0.16 | 11.73 ± 0.31 |
| Paired T-test | p-valuea | p-valuea |
| Radiation vs. Virus | <0.01 | <0.01 |
| Radiation vs. Virus + Radiation | <0.01 | <0.01 |
| Virus vs. Virus + Radiation | 0.631 | <0.01 |
| Nucleoplasmic Bridges | ||
| Baselineb | 0.37 ± 0.04 | 1.20 ± 0.07 |
| Radiation | 6.78 ± 0.26 | 8.53 ± 0.28 |
| Virus | 7.30 ± 0.21 | 8.74 ± 0.43 |
| Virus + Radiation | 11.67 ± 0.29 | 13.00 ± 0.30 |
| Paired T-test | p-valuea | p-valuea |
| Radiation vs. Virus | 0.036 | 0.647 |
| Radiation vs. Virus + Radiation | <0.01 | <0.01 |
| Virus vs. Virus + Radiation | <0.01 | <0.01 |
| Apoptosis | ||
| Baselineb | 23.00 ± 1.03 | 43.04 ± 2.80 |
| Radiation | 51.32 ± 1.47 | 77.17 ± 4.42 |
| Virus | 79.71 ± 3.84 | 75.83 ± 3.92 |
| Virus + Radiation | 89.02 ± 5.86 | 143.95 ± 3.46 |
| Paired T-test | p-valuea | p-valuea |
| Radiation vs. Virus | <0.01 | 0.736 |
| Radiation vs. Virus + Radiation | <0.01 | <0.01 |
| Virus vs. Virus + Radiation | 0.012 | <0.01 |
3.4. Baseline and Induced Cell Death
3.5. Discussion
4. Conclusions
Conflict of Interest Statement
References
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W. J. Human Cytomegalovirus Infection and Expression in Human Malignant Glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar]
- Schwartzbaum, J.A.; Fisher, J.L.; Aldape, K.D.; Wrensch, M. Epidemiology and Molecular Pathology of Glioma. Nat. Clin. Pract. Neurol. 2006, 2, 494–503, quiz 1 p following 516. [Google Scholar]
- American Cancer Society. Estimated New Cancer Cases and Deaths by Sex, US (2009). Available online: http://www.cancer.org/downloads/STT/500809web.pdf (Accessed on 22 March 2010).
- Wrensch, M.; Fisher, J.L.; Schwartzbaum, J.A.; Bondy, M.; Berger, M.; Aldape, K.D. The Molecular Epidemiology of Gliomas in Adults. Neurosurg. Focus 2005, 19, E5. [Google Scholar]
- Fisher, J.L.; Schwartzbaum, J.A.; Wrensch, M.; Wiemels, J.L. Epidemiology of Brain Tumors. Neurol. Clin. 2007, 25, 867–90. [Google Scholar] [CrossRef]
- Wang, L.E.; Sturgis, E. M.; Eicher, S.A.; Spitz, M.R.; Hong, W.K.; Wei, Q. Mutagen Sensitivity to Benzo(a)Pyrene Diol Epoxide and the Risk of Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 1998, 4, 1773–1778. [Google Scholar]
- Gurney, J.G.; Kadan-Lottick, N. Brain and Other Central Nervous System Tumors: Rates, Trends, and Epidemiology. Curr. Opin. Oncol. 2001, 13, 160–166. [Google Scholar] [CrossRef]
- Deng, C.Z.; AbuBakar, S.; Fons, M.P.; Boldogh, I.; Albrecht, T. Modulation of the Frequency of Human Cytomegalovirus-Induced Chromosome Aberrations by Camptothecin. Virology 1992, 189, 397–401. [Google Scholar] [CrossRef]
- Deng, C.Z.; AbuBakar, S.; Fons, M.P.; Boldogh, I.; Hokanson, J.; Au, W.W.; Albrecht, T. Cytomegalovirus-Enhanced Induction of Chromosome Aberrations in Human Peripheral Blood Lymphocytes Treated with Potent Genotoxic Agents. Environ. Mol. Mutagen. 1992, 19, 304–310. [Google Scholar]
- Bonassi, S.; Fenech, M.; Lando, C.; Lin, Y.P.; Ceppi, M.; Chang, W.P.; Holland, N.; Kirsch-Volders, M.; Zeiger, E.; Ban, S.; Barale, R.; Bigatti, M.P.; Bolognesi, C.; Jia, C.; Di Giorgio, M.; Ferguson, L.R.; Fucic, A.; Lima, O.G.; Hrelia, P.; Krishnaja, A.P.; Lee, T. K.; Migliore, L.; Mikhalevich, L.; Mirkova, E.; Mosesso, P.; Muller, W.U.; Odagiri, Y.; Scarffi, M.R.; Szabova, E.; Vorobtsova, I.; Vral, A.; Zijno, A. HUman MicroNucleus Project: International Database Comparison for Results with the Cytokinesis-Block Micronucleus Assay in Human Lymphocytes: I. Effect of Laboratory Protocol, Scoring Criteria, and Host Factors on the Frequency of Micronuclei. Environ. Mol. Mutagen. 2001, 37, 31–45. [Google Scholar]
- Bondy, M.L.; Kyritsis, A.P.; Gu, J.; de Andrade, M.; Cunningham, J.; Levin, V.A.; Bruner, J.M.; Wei, Q. Mutagen Sensitivity and Risk of Gliomas: A Case-Control Analysis. Cancer Res. 1996, 56, 1484–1486. [Google Scholar]
- Bondy, M. L.; Wang, L.E.; El-Zein, R.; de Andrade, M.; Selvan, M.S.; Bruner, J.M.; Levin, V.A.; Alfred Yung, W.K.; Adatto, P.; Wei, Q. Gamma-Radiation Sensitivity and Risk of Glioma. J. Natl. Cancer Inst. 2001, 93, 1553–1557. [Google Scholar] [CrossRef]
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il'yasova, D.; Kruchko, C.; McCarthy, B. J.; Rajaraman, P.; Schwartzbaum, J.A.; Sadetzki, S.; Schlehofer, B.; Tihan, T.; Wiemels, J.L.; Wrensch, M.; Buffler, P.A. Brain Tumor Epidemiology Consortium Brain Tumor Epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar]
- Liu, Y.; Scheurer, M.E.; El-Zein, R.; Cao, Y.; Do, K.A.; Gilbert, M.; Aldape, K.D.; Wei, Q.; Etzel, C.; Bondy, M.L. Association and Interactions between DNA Repair Gene Polymorphisms and Adult Glioma. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 204–214. [Google Scholar] [CrossRef]
- Hodges, L.C.; Smith, J.L.; Garrett, A.; Tate, S. Prevalence of Glioblastoma Multiforme in Subjects with Prior Therapeutic Radiation. J. Neurosci. Nurs. 1992, 24, 79–83. [Google Scholar]
- Edick, M. J.; Cheng, C.; Yang, W.; Cheok, M.; Wilkinson, M.R.; Pei, D.; Evans, W.E.; Kun, L.E.; Pui, C.H.; Relling, M.V. Lymphoid Gene Expression as a Predictor of Risk of Secondary Brain Tumors. Genes Chromosomes Cancer 2005, 42, 107–116. [Google Scholar]
- Relling, M. V.; Rubnitz, J. E.; Rivera, G. K.; Boyett, J. M.; Hancock, M. L.; Felix, C. A.; Kun, L. E.; Walter, A. W.; Evans, W. E.; Pui, C. H. High Incidence of Secondary Brain Tumours After Radiotherapy and Antimetabolites. Lancet 1999, 354, 34–39. [Google Scholar]
- Shiraishi, J.; Tsugata, M.; Masuda, R.; Mori, Y.; Suzuki, K.; Takemura, T. Type AB Thymoma Accompanied by Pure Red Cell Aplasia and Good Syndrome with CMV Infection of Tumor Cells. Pathol. Int. 2008, 58, 489–493. [Google Scholar] [CrossRef]
- Ho, W.Z.; Harouse, J.M.; Rando, R.F.; Gonczol, E.; Srinivasan, A.; Plotkin, S.A. Reciprocal Enhancement of Gene Expression and Viral Replication between Human Cytomegalovirus and Human Immunodeficiency Virus Type 1. J. Gen. Virol. 1990, 71, 97–103. [Google Scholar] [CrossRef]
- Albrecht, T.; Deng, C.Z.; Abdel-Rahman, S.Z.; Fons, M.; Cinciripini, P.; El-Zein, R.A. Differential Mutagen Sensitivity of Peripheral Blood Lymphocytes from Smokers and Nonsmokers: Effect of Human Cytomegalovirus Infection. Environ. Mol. Mutagen. 2004, 43, 169–178. [Google Scholar]
- Boldogh, I.; Baskar, J.F.; Mar, E.C.; Huang, E. S. Human Cytomegalovirus and Herpes Simplex Type 2 Virus in Normal and Adenocarcinomatous Prostate Glands. J. Natl. Cancer Inst. 1983, 70, 819–826. [Google Scholar]
- Boldogh, I.; AbuBakar, S.; Fons, M.P.; Deng, C.Z.; Albrecht, T. Activation of Cellular Oncogenes by Clinical Isolates and Laboratory Strains of Human Cytomegalovirus. J. Med. Virol. 1991, 34, 241–247. [Google Scholar] [CrossRef]
- Scheurer, M.E.; Bondy, M.L.; Aldape, K.D.; Albrecht, T.; El-Zein, R. Detection of Human Cytomegalovirus in Different Histological Types of Gliomas. Acta Neuropathol. 2008, 116, 79–86. [Google Scholar] [CrossRef]
- Albrecht, T.; Boldogh, I.; Fons, M.; Lee, C.H.; AbuBakar, S.; Russell, J.M.; Au, W.W. Cell-Activation Responses to Cytomegalovirus Infection Relationship to the Phasing of CMV Replication and to the Induction of Cellular Damage. Subcell. Biochem. 1989, 15, 157–202. [Google Scholar]
- Boldogh, I.; AbuBakar, S.; Albrecht, T. Activation of Proto-Oncogenes: An Immediate Early Event in Human Cytomegalovirus Infection. Science 1990, 247, 561–564. [Google Scholar]
- Ohagen, A.; Gibaja, V.; Horrigan, J.; Lunderville, D.; Jayarama, V.; Marcello, J.; Chapman, J.; Lazo, A. Induction of Latent Human Cytomegalovirus by Conventional Gamma Radiation and Prevention by Treatment with INACTINE PEN110. Vox Sang. 2004, 87, 1–9. [Google Scholar]
- Egami, T.; Ohuchida, K.; Mizumoto, K.; Onimaru, M.; Toma, H.; Nishio, S.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation Enhances Adenoviral Gene Therapy in Pancreatic Cancer Via Activation of Cytomegalovirus Promoter and Increased Adenovirus Uptake. Clin. Cancer Res. 2008, 14, 1859–1867. [Google Scholar] [CrossRef]
- Bresnahan, W.A.; Hultman, G.E.; Shenk, T. Replication of Wild-Type and Mutant Human Cytomegalovirus in Life-Extended Human Diploid Fibroblasts. J. Virol. 2000, 74, 10816–10818. [Google Scholar] [CrossRef]
- Schroer, J.; Shenk, T. Inhibition of Cyclooxygenase Activity Blocks Cell-to-Cell Spread of Human Cytomegalovirus. Proc. Natl. Acad. Sci. USA 2008, 105, 19468–19473. [Google Scholar] [CrossRef]
- Shanley, J.D. Human Cytomegalovirus Replicates in Gamma-Radiated Fibroblasts. J. Med. Virol. 1986, 20, 347–355. [Google Scholar]
- AbuBakar, S.; Boldogh, I.; Albrecht, T. Human Cytomegalovirus. Stimulation of 3H. Release from 3H.-Arachidonic Acid Prelabelled Cells. Arch. Virol. 1990, 113, 255–266. [Google Scholar]
- Solomon, E.; Borrow, J.; Goddard, A.D. Chromosome Aberrations and Cancer. Science 1991, 254, 1153–1160. [Google Scholar]
- Fenech, M.; Chang, W.P.; Kirsch-Volders, M.; Holland, N.; Bonassi, S.; Zeiger, E. HUman MicronNucleus project HUMN Project: Detailed Description of the Scoring Criteria for the Cytokinesis–Block Micronucleus Assay using Isolated Human Lymphocyte Cultures. Mutat. Res. 2003, 534, 65–75. [Google Scholar]
- Fenech, M. Cytokinesis-Block Micronucleus Cytome Assay. Nat. Protoc. 2007, 2, 1084–1104. [Google Scholar] [CrossRef]
- Wu, X.; Gu, J.; Spitz, M. R. Mutagen Sensitivity: A Genetic Predisposition Factor for Cancer. Cancer Res. 2007, 67, 3493–3495. [Google Scholar]
- El-Zein, R.A.; Schabath, M.B.; Etzel, C.J.; Lopez, M.S.; Franklin, J. D.; Spitz, M.R. Cytokinesis-Blocked Micronucleus Assay as a Novel Biomarker for Lung Cancer Risk. Cancer Res. 2006, 66, 6449–6456. [Google Scholar]
- El-Zein, R.A.; Fenech, M.; Lopez, M.S.; Spitz, M.R.; Etzel, C.J. Cytokinesis-Blocked Micronucleus Cytome Assay Biomarkers Identify Lung Cancer Cases Amongst Smokers. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 1111–1119. [Google Scholar]
- Cheng, T.J.; Christiani, D.C.; Xu, X.; Wain, J.C.; Wiencke, J.K.; Kelsey, K.T. Increased Micronucleus Frequency in Lymphocytes from Smokers with Lung Cancer. Mutat. Res. 1996, 349, 43–50. [Google Scholar] [CrossRef]
- Mozdarani, H.; Mansouri, Z.; Haeri, S.A. Cytogenetic Radiosensitivity of g0-Lymphocytes of Breast and Esophageal Cancer Patients as Determined by Micronucleus Assay. J. Radiat. Res. (Tokyo) 2005, 46, 111–116. [Google Scholar]
- Evans, H.J.; O'Riordan, M.L. Human Peripheral Blood Lymphocytes for the Analysis of Chromosome Aberrations in Mutagen Tests. Mutat. Res. 1975, 31, 135–148. [Google Scholar]
- AbuBakar, S.; Au, W.W.; Legator, M. S.; Albrecht, T. Induction of Chromosome Aberrations and Mitotic Arrest by Cytomegalovirus in Human Cells. Environ. Mol. Mutagen. 1988, 12, 409–420. [Google Scholar] [CrossRef]
- Murgia, E.; Ballardin, M.; Bonassi, S.; Rossi, A.M.; Barale, R. Validation of Micronuclei Frequency in Peripheral Blood Lymphocytes as Early Cancer Risk Biomarker in a Nested Case-Control Study. Mutat. Res. 2008, 639, 27–34. [Google Scholar]
- Marekova, M.; Vavrova, J.; Vokurkova, D.; Psutka, J. Modulation of Ionizing Radiation-Induced Apoptosis and Cell Cycle Arrest by all-Trans Retinoic Acid in Promyelocytic Leukemia Cells (HL-60). Physiol. Res. 2003, 52, 599–606. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Fruehauf, J. P.; Meyskens, F.L., Jr. Reactive Oxygen Species: A Breath of Life Or Death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar]
- Lee, J.H.; Kim, S.Y.; Kil, I.S.; Park, J.W. Regulation of Ionizing Radiation-Induced Apoptosis by Mitochondrial NADP+-Dependent Isocitrate Dehydrogenase. J. Biol. Chem. 2007, 282, 13385–13394. [Google Scholar] [CrossRef]
- Mi, J.; Bolesta, E.; Brautigan, D.L.; Larner, J.M. PP2A Regulates Ionizing Radiation-Induced Apoptosis through Ser46 Phosphorylation of p53. Mol. Cancer. Ther. 2009, 8, 135–140. [Google Scholar] [CrossRef]
- Yount, G.L.; Afshar, G.; Ries, S.; Korn, M.; Shalev, N.; Basila, D.; McCormick, F.; Haas-Kogan, D. A. Transcriptional Activation of TRADD Mediates p53-Independent Radiation-Induced Apoptosis of Glioma Cells. Oncogene 2001, 20, 2826–2835. [Google Scholar]
- Dion, M.; Hamelin, C. Relationship between Enhanced Reactivation and Mutagenesis of u.v.-Radiated Human Cytomegalovirus in Normal Human Cells. EMBO J. 1987, 6, 397–399. [Google Scholar]
- Albrecht, T.; Fons, M.P.; Deng, C.Z.; Boldogh, I. Increased Frequency of Specific Locus Mutation Following Human Cytomegalovirus Infection. Virology 1997, 230, 48–61. [Google Scholar] [CrossRef]
- Zhu, H.; Shen, Y.; Shenk, T. Human Cytomegalovirus IE1 and IE2 Proteins Block Apoptosis. J. Virol. 1995, 69, 7960–7970. [Google Scholar]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. The Story of Human Cytomegalovirus and Cancer: Increasing Evidence and Open Questions. Neoplasia 2009, 11, 1–9. [Google Scholar]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A Cytomegalovirus-Encoded Inhibitor of Apoptosis that Suppresses Caspase-8 Activation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 7829–7834. [Google Scholar]
- Gaspar, M.; Shenk, T. Human Cytomegalovirus Inhibits a DNA Damage Response by Mislocalizing Checkpoint Proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 2821–2826. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rourke, E.A.; Lopez, M.S.; Monroy, C.M.; Scheurer, M.E.; Etzel, C.J.; Albrecht, T.; Bondy, M.L.; El-Zein, R.A. Modulation of Radiation-Induced Genetic Damage by HCMV in Peripheral Blood Lymphocytes from a Brain Tumor Case-Control Study. Cancers 2010, 2, 420-435. https://doi.org/10.3390/cancers2020420
Rourke EA, Lopez MS, Monroy CM, Scheurer ME, Etzel CJ, Albrecht T, Bondy ML, El-Zein RA. Modulation of Radiation-Induced Genetic Damage by HCMV in Peripheral Blood Lymphocytes from a Brain Tumor Case-Control Study. Cancers. 2010; 2(2):420-435. https://doi.org/10.3390/cancers2020420
Chicago/Turabian StyleRourke, Elizabeth A., Mirtha S. Lopez, Claudia M. Monroy, Michael E. Scheurer, Carol J. Etzel, Thomas Albrecht, Melissa L. Bondy, and Randa A. El-Zein. 2010. "Modulation of Radiation-Induced Genetic Damage by HCMV in Peripheral Blood Lymphocytes from a Brain Tumor Case-Control Study" Cancers 2, no. 2: 420-435. https://doi.org/10.3390/cancers2020420
APA StyleRourke, E. A., Lopez, M. S., Monroy, C. M., Scheurer, M. E., Etzel, C. J., Albrecht, T., Bondy, M. L., & El-Zein, R. A. (2010). Modulation of Radiation-Induced Genetic Damage by HCMV in Peripheral Blood Lymphocytes from a Brain Tumor Case-Control Study. Cancers, 2(2), 420-435. https://doi.org/10.3390/cancers2020420