
HuR (ELAVL1) Stabilizes SOX9 mRNA and Promotes Migration and Invasion in Breast Cancer Cells
 , ,
, ,
Abstract
:Simple Summary
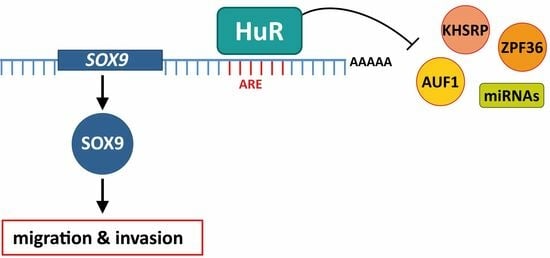
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Plasmids
2.2. RNA-Seq Analysis
2.3. RNA Extraction, cDNA Synthesis and Quantitative PCR (qPCR)
2.4. mRNA Stability
2.5. Ribonucleoprotein Immunoprecipitation Assay (RIP)
2.6. RNA Pull Down of SOX9 ARE
2.7. 3′ UTR Luciferase Reporter Assays
2.8. Western Blot Analysis
2.9. Migration and Invasion Assays
2.10. Statistical Analysis
3. Results
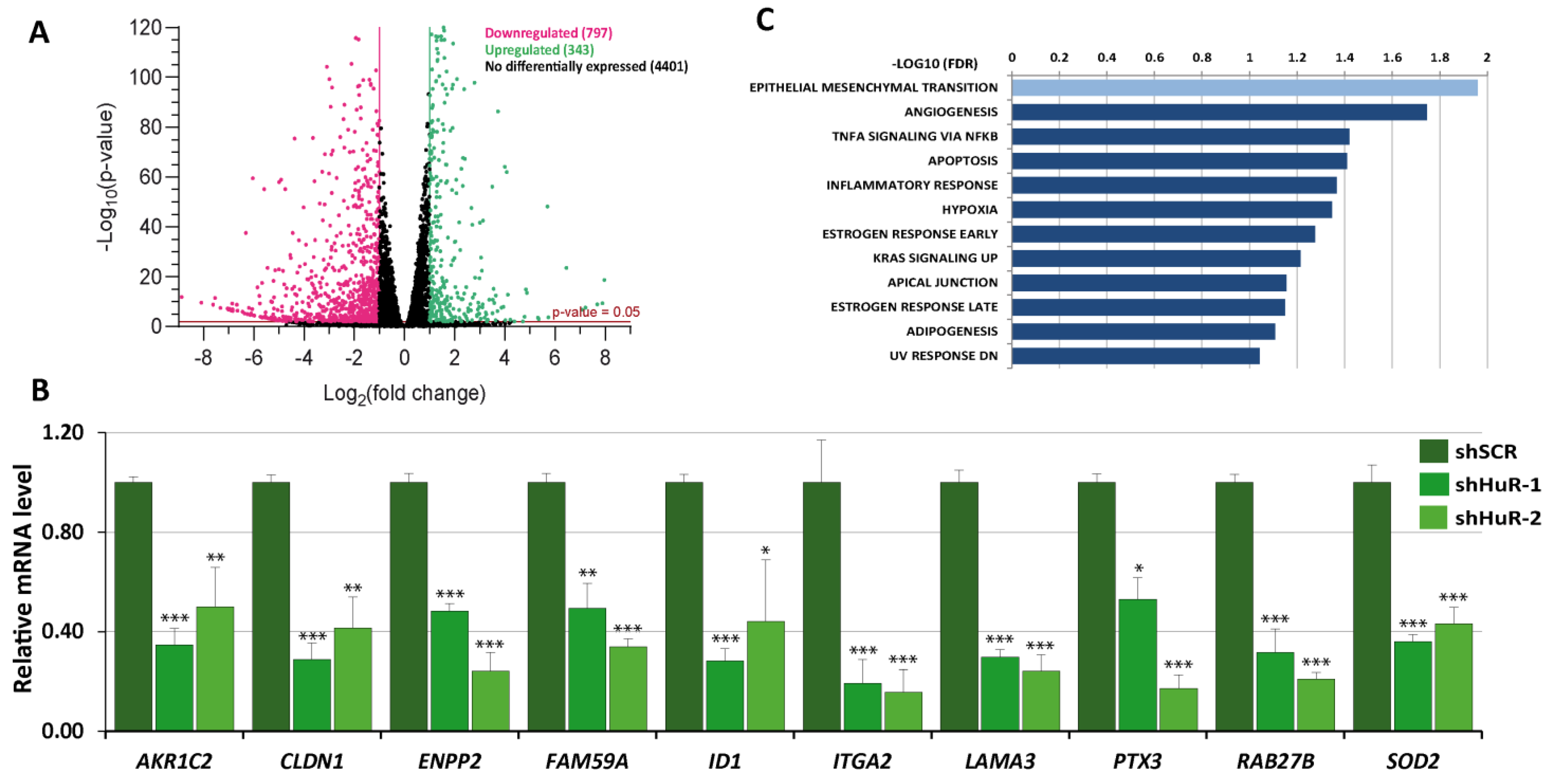
3.1. Gene Expression Profile of HuR-Silenced Cells
3.2. Selection of Putative HuR Targets
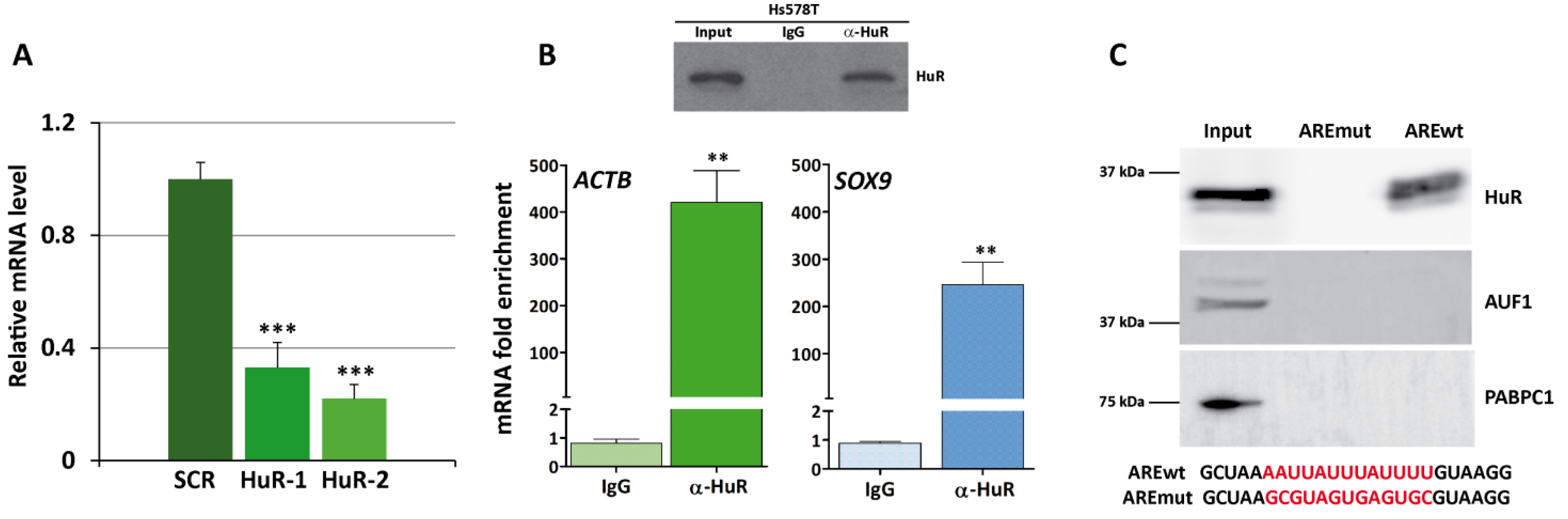
3.3. HuR Binds to SOX9 mRNA
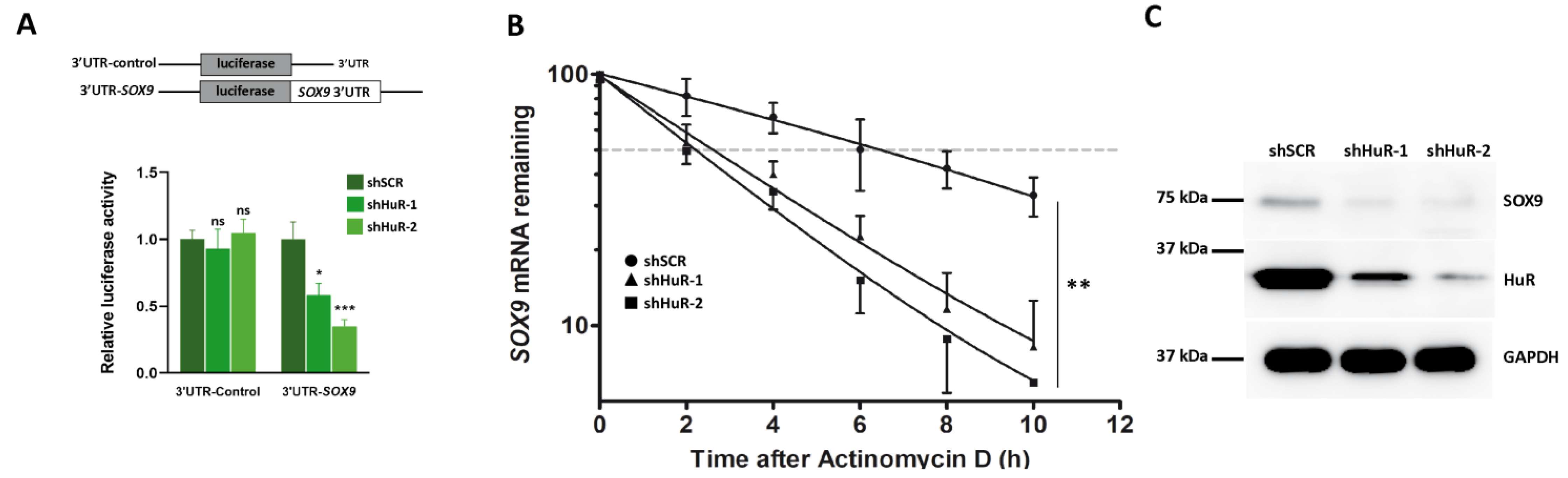
3.4. HuR Stabilizes SOX9 mRNA
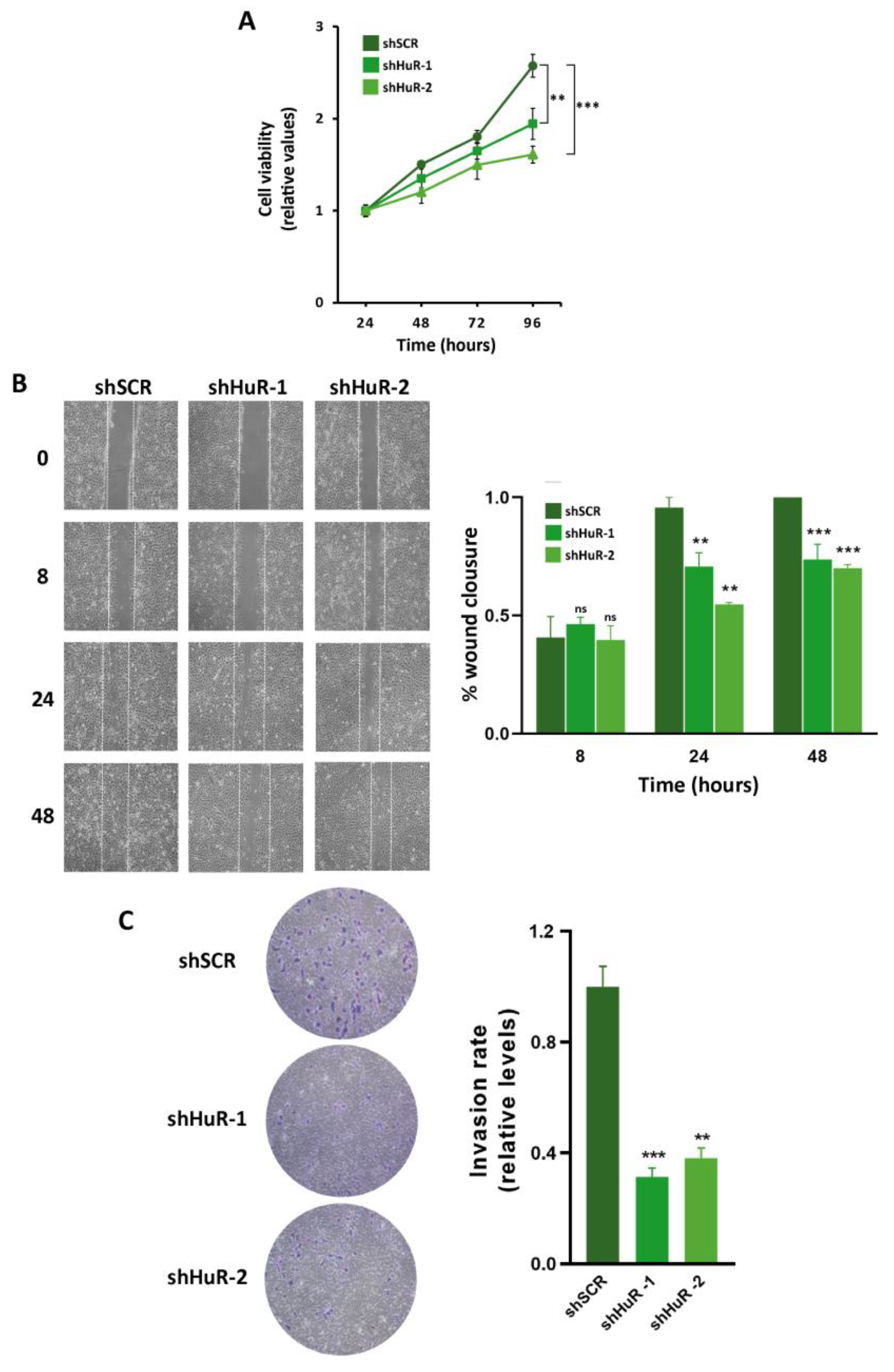
3.5. Proliferation, Migration and Invasion Are Decreased in HuR-Silenced Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerstberger, S.; Hafner, M.; Ascano, M.; Tuschl, T. Evolutionary Conservation and Expression of Human RNA-Binding Proteins and Their Role in Human Genetic Disease. In Systems Biology of RNA Binding Proteins; Springer: New York, NY, USA, 2014; pp. 1–55. [Google Scholar]
- Lukong, K.E.; Chang, K.; Khandjian, E.W.; Richard, S. RNA-Binding Proteins in Human Genetic Disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-Binding Proteins in Human Genetic Disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Lee, Y.; Lee, J.-S. RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives. Cancers 2020, 12, 2699. [Google Scholar] [CrossRef]
- Qin, H.; Ni, H.; Liu, Y.; Yuan, Y.; Xi, T.; Li, X.; Zheng, L. RNA-Binding Proteins in Tumor Progression. J. Hematol. Oncol. 2020, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, X.; Chen, J. RNA-Binding Proteins in Regulating MRNA Stability and Translation: Roles and Mechanisms in Cancer. Semin. Cancer Biol. 2022, 86, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, Z.; Lei, Z.; Zhang, H.-T. RNA-Binding Proteins and Cancer Metastasis. Semin. Cancer Biol. 2022, 86, 748–768. [Google Scholar] [CrossRef]
- Schultz, C.W.; Preet, R.; Dhir, T.; Dixon, D.A.; Brody, J.R. Understanding and Targeting the Disease-related RNA Binding Protein Human Antigen R (HuR). WIREs RNA 2020, 11, e1581. [Google Scholar] [CrossRef]
- Khabar, K.S.A. Hallmarks of Cancer and AU-rich Elements. WIREs RNA 2017, 8, e1368. [Google Scholar] [CrossRef]
- Srikantan, S. HuR Function in Disease. Front. Biosci. 2012, 17, 189. [Google Scholar] [CrossRef]
- Kotta-Loizou, I.; Vasilopoulos, S.N.; Coutts, R.H.A.; Theocharis, S. Current Evidence and Future Perspectives on HuR and Breast Cancer Development, Prognosis, and Treatment. Neoplasia 2016, 18, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple Functions of the RNA-Binding Protein HuR in Cancer Progression, Treatment Responses and Prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and BHLH Factors in Tumour Progression: An Alliance against the Epithelial Phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory Networks Defining EMT during Cancer Initiation and Progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT Transcription Factors in Cancer Development Re-Evaluated: Beyond EMT and MET. Biochim. Biophys. Acta (BBA) Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef]
- Peinado, H.; Portillo, F.; Cano, A. Transcriptional Regulation of Cadherins during Development and Carcinogenesis. Int. J. Dev. Biol. 2004, 48, 365–375. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional Regulation of Cell Polarity in EMT and Cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef]
- Xu, W.; Chen, L.; Liu, J.; Zhang, Z.; Wang, R.; Zhang, Q.; Li, H.; Xiang, J.; Fang, L.; Xu, P.; et al. LINC00152 induced by TGF-β promotes metastasis via HuR in lung adenocarcinoma. Cell Death Dis. 2022, 13, 772–784. [Google Scholar] [CrossRef]
- Dong, R.; Chen, P.; Polireddy, K.; Wu, X.; Wang, T.; Ramesh, R.; Dixon, D.A.; Xu, L.; Aubé, J.; Chen, Q. An RNA-Binding Protein, Hu-Antigen R, in Pancreatic Cancer Epithelial to Mesenchymal Transition, Metastasis, and Cancer Stem Cells. Mol. Cancer Ther. 2020, 19, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.Z.; McCarthy, G.A.; Carroll, J.R.; Di Niro, R.; Pelz, C.; Jain, A.; Sutton, T.L.; Holly, H.D.; Nevler, A.; Schultz, C.W.; et al. The RNA-Binding Protein HuR Posttranscriptionally Regulates the Protumorigenic Activator YAP1 in Pancreatic Ductal Adenocarcinoma. Mol. Cell. Biol. 2022, 42, e00018-22. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Carelli, S.; Raimondi, I.; D’Agostino, V.; Castiglioni, I.; Zucal, C.; Moro, G.; Luciani, A.; Ghilardi, G.; Monti, E.; et al. The Ribonucleic Complex HuR-MALAT1 Represses CD133 Expression and Suppresses Epithelial-Mesenchymal Transition in Breast Cancer. Cancer Res. 2016, 76, 2626–2636. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.; Denduluri, S.; Zhang, B.; Wang, Z.; Yin, L.; Yan, Z.; Kang, R.; Shi, L.L.; Mok, J.; Lee, M.J.; et al. The Versatile Functions of Sox9 in Development, Stem Cells, and Human Diseases. Genes Dis. 2014, 1, 149–161. [Google Scholar] [CrossRef]
- Grimm, D.; Bauer, J.; Wise, P.; Krüger, M.; Simonsen, U.; Wehland, M.; Infanger, M.; Corydon, T.J. The Role of SOX Family Members in Solid Tumours and Metastasis. Semin. Cancer Biol. 2020, 67, 122–153. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Madhu Krishna, B.; Singhal, J.; Horne, D.; Awasthi, S.; Salgia, R.; Singhal, S.S. SOX9: The Master Regulator of Cell Fate in Breast Cancer. Biochem. Pharmacol. 2020, 174, 113789. [Google Scholar] [CrossRef]
- Panda, M.; Tripathi, S.K.; Biswal, B.K. SOX9: An Emerging Driving Factor from Cancer Progression to Drug Resistance. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1875, 188517. [Google Scholar] [CrossRef]
- Ma, Y.; Shepherd, J.; Zhao, D.; Bollu, L.R.; Tahaney, W.M.; Hill, J.; Zhang, Y.; Mazumdar, A.; Brown, P.H. SOX9 Is Essential for Triple-Negative Breast Cancer Cell Survival and Metastasis. Mol. Cancer Res. 2020, 18, 1825–1838. [Google Scholar] [CrossRef]
- Capaccione, K.M.; Hong, X.; Morgan, K.M.; Liu, W.; Bishop, M.J.; Liu, L.; Markert, E.; Deen, M.; Minerowicz, C.; Bertino, J.R.; et al. Sox9 Mediates Notch1-Induced Mesenchymal Features in Lung Adenocarcinoma. Oncotarget 2014, 5, 3636–3650. [Google Scholar] [CrossRef]
- Zhou, H.; Li, G.; Huang, S.; Feng, Y.; Zhou, A. SOX9 Promotes Epithelial-mesenchymal Transition via the Hippo-YAP Signaling Pathway in Gastric Carcinoma Cells. Oncol. Lett. 2019, 18, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.; Hitti, E.; Khabar, K.S.A. ARED-Plus: An Updated and Expanded Database of AU-Rich Element-Containing MRNAs and Pre-MRNAs. Nucleic Acids Res. 2018, 46, D218–D220. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. StarBase v2.0: Decoding MiRNA-CeRNA, MiRNA-NcRNA and Protein–RNA Interaction Networks from Large-Scale CLIP-Seq Data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Panda, A.; Martindale, J.; Gorospe, M. Affinity Pulldown of Biotinylated RNA for Detection of Protein-RNA Complexes. Bio-protocol 2016, 6, e2062. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Peinado, H.; Molina, P.; Olmeda, D.; Cubillo, E.; Santos, V.; Palacios, J.; Portillo, F.; Cano, A. The Morphological and Molecular Features of the Epithelial-to-Mesenchymal Transition. Nat. Protoc. 2009, 4, 1591–1613. [Google Scholar] [CrossRef]
- Dixon, D.A.; Tolley, N.D.; King, P.H.; Nabors, L.B.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Altered Expression of the MRNA Stability Factor HuR Promotes Cyclooxygenase-2 Expression in Colon Cancer Cells. J. Clin. Investig. 2001, 108, 1657–1665. [Google Scholar] [CrossRef]
- Fan, J.; Ishmael, F.T.; Fang, X.; Myers, A.; Cheadle, C.; Huang, S.-K.; Atasoy, U.; Gorospe, M.; Stellato, C. Chemokine Transcripts as Targets of the RNA-Binding Protein HuR in Human Airway Epithelium. J. Immunol. 2011, 186, 2482–2494. [Google Scholar] [CrossRef]
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A Pathway to Bone: Signaling Molecules and Transcription Factors Involved in Chondrocyte Development and Maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef]
- Wang, Q.-Y.; Zhou, C.-X.; Zhan, M.-N.; Tang, J.; Wang, C.-L.; Ma, C.-N.; He, M.; Chen, G.-Q.; He, J.-R.; Zhao, Q. MiR-133b Targets Sox9 to Control Pathogenesis and Metastasis of Breast Cancer. Cell Death Dis. 2018, 9, 752. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Dvir-Ginzberg, M. SOX9 and the Many Facets of Its Regulation in the Chondrocyte Lineage. Connect. Tissue Res. 2017, 58, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yin, W.; Yu, Z.-H.; Zhou, Y.-J.; Chi, J.-R.; Ge, J.; Cao, X.-C. MiR-190 Enhances Endocrine Therapy Sensitivity by Regulating SOX9 Expression in Breast Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 22. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.B.; Zhu, M.N.; Zhu, X.L. MiRNA-215-5p Suppresses the Aggressiveness of Breast Cancer Cells by Targeting Sox9. FEBS Open Bio 2019, 9, 1957–1967. [Google Scholar] [CrossRef]
- Wolfson, B. Roles of MicroRNA-140 in Stem Cell-Associated Early Stage Breast Cancer. World J. Stem Cells 2014, 6, 591. [Google Scholar] [CrossRef]
- Chao, T.-Y.; Kordaß, T.; Osen, W.; Eichmüller, S.B. SOX9 Is a Target of MiR-134-3p and MiR-224-3p in Breast Cancer Cell Lines. Mol. Cell. Biochem. 2022, 478, 305–315. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morillo-Bernal, J.; Pizarro-García, P.; Moreno-Bueno, G.; Cano, A.; Mazón, M.J.; Eraso, P.; Portillo, F. HuR (ELAVL1) Stabilizes SOX9 mRNA and Promotes Migration and Invasion in Breast Cancer Cells. Cancers 2024, 16, 384. https://doi.org/10.3390/cancers16020384
Morillo-Bernal J, Pizarro-García P, Moreno-Bueno G, Cano A, Mazón MJ, Eraso P, Portillo F. HuR (ELAVL1) Stabilizes SOX9 mRNA and Promotes Migration and Invasion in Breast Cancer Cells. Cancers. 2024; 16(2):384. https://doi.org/10.3390/cancers16020384
Chicago/Turabian StyleMorillo-Bernal, Jesús, Patricia Pizarro-García, Gema Moreno-Bueno, Amparo Cano, María J. Mazón, Pilar Eraso, and Francisco Portillo. 2024. "HuR (ELAVL1) Stabilizes SOX9 mRNA and Promotes Migration and Invasion in Breast Cancer Cells" Cancers 16, no. 2: 384. https://doi.org/10.3390/cancers16020384